
Discovering the Repeatome of Five Species Belonging to the Asteraceae Family: A Computational Study
 , and
, and
Abstract
:1. Introduction
2. Results
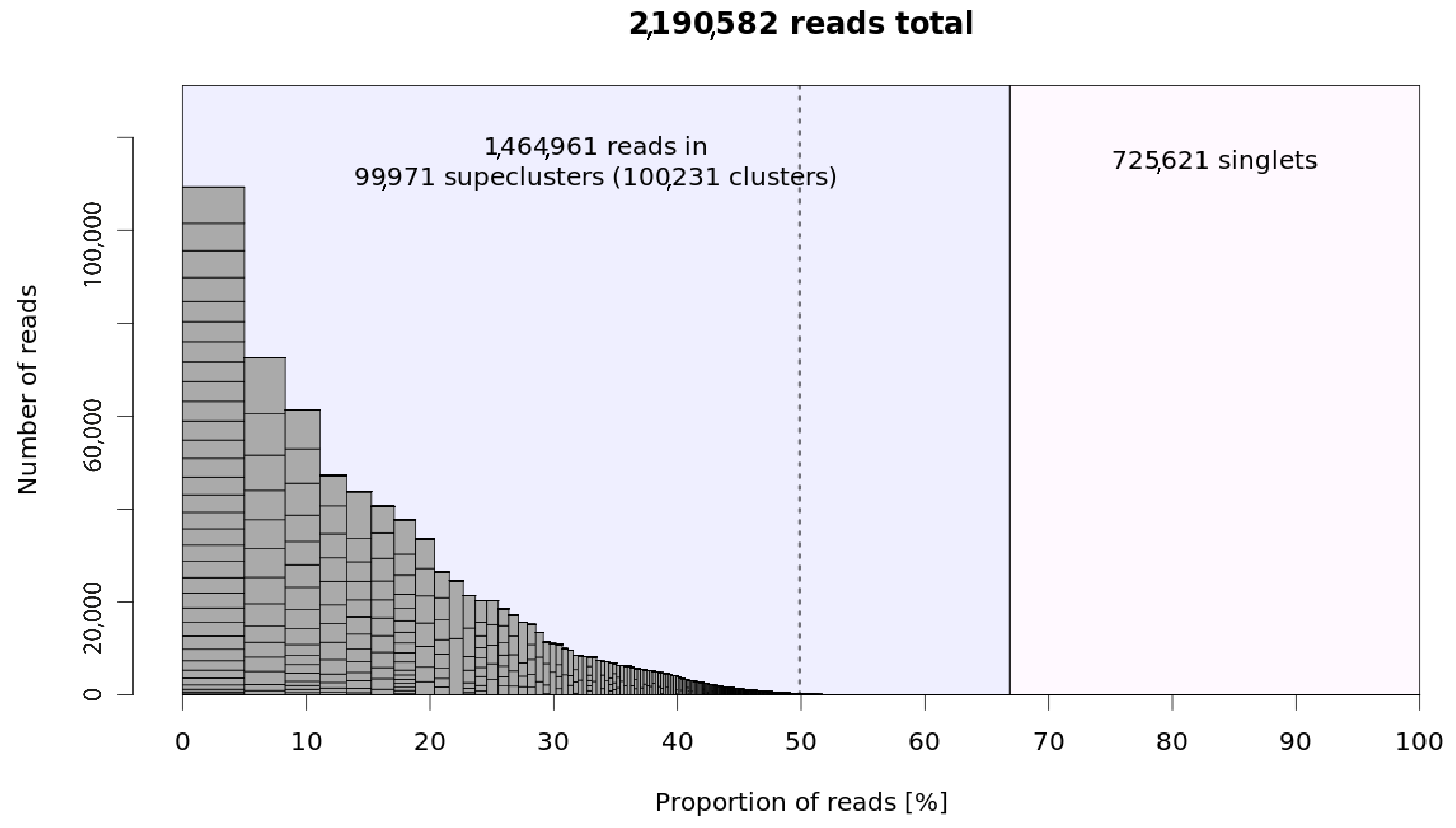
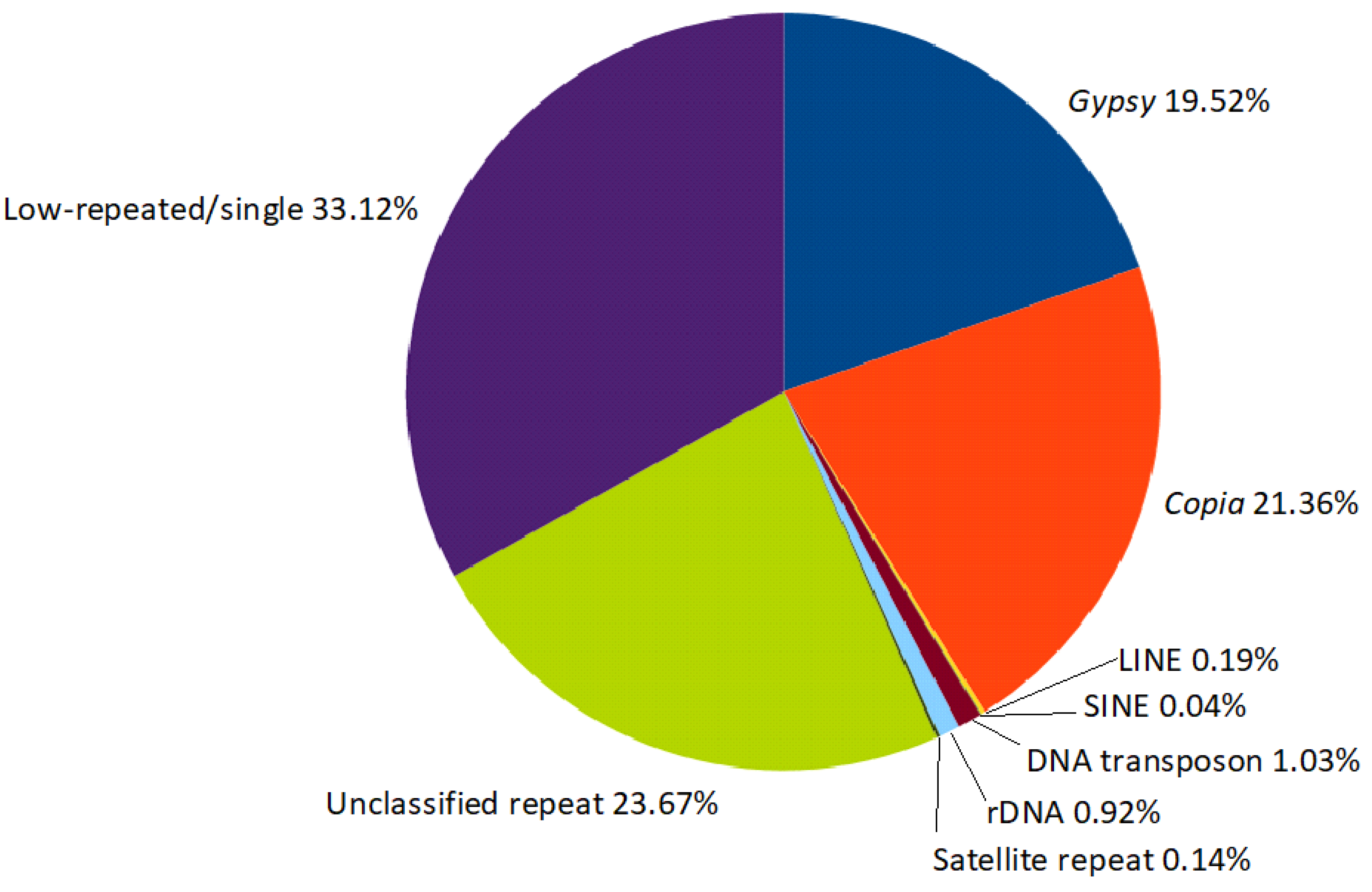
2.1. Metarepeatome Analysis of Asteraceae Species
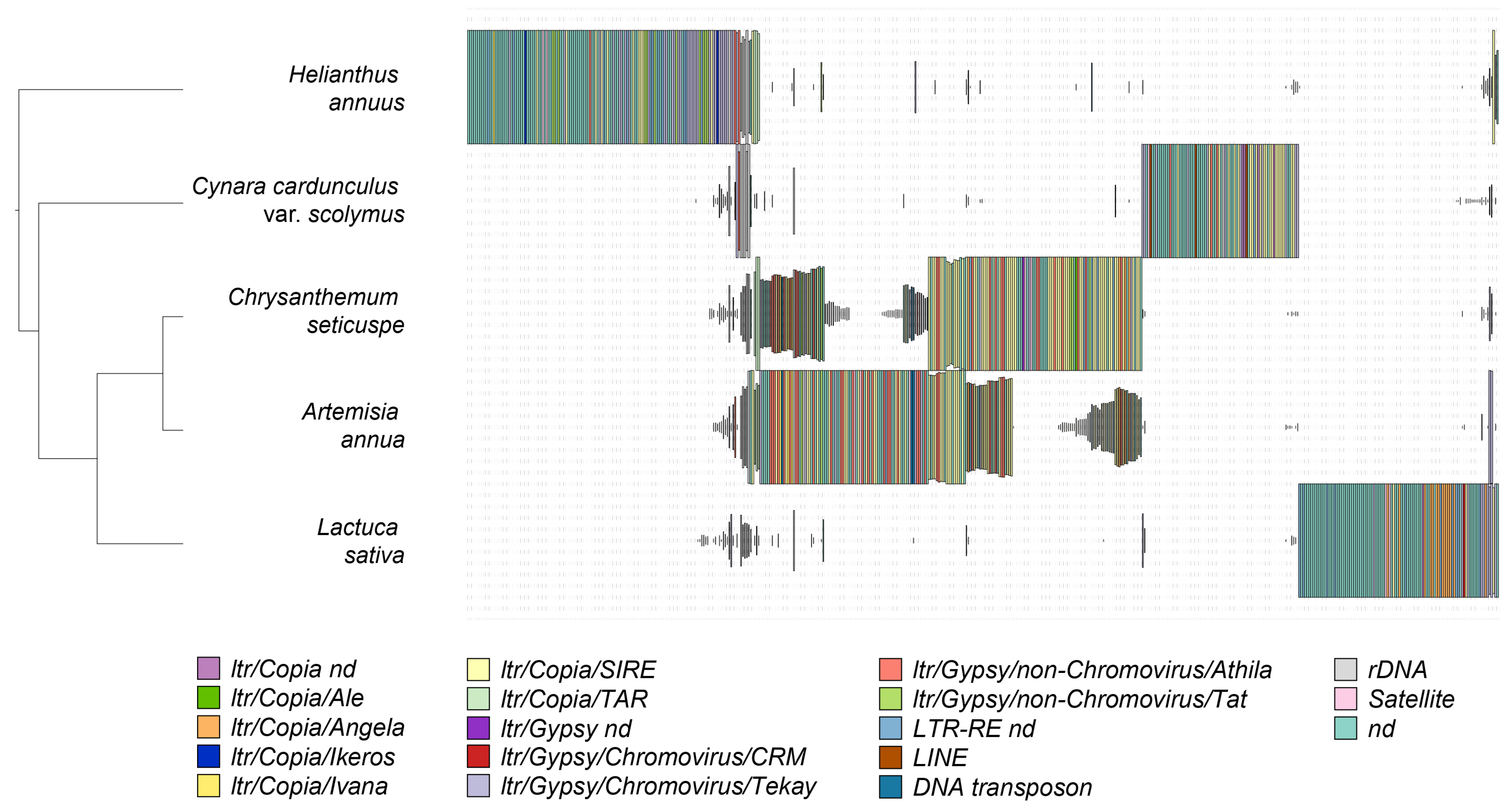
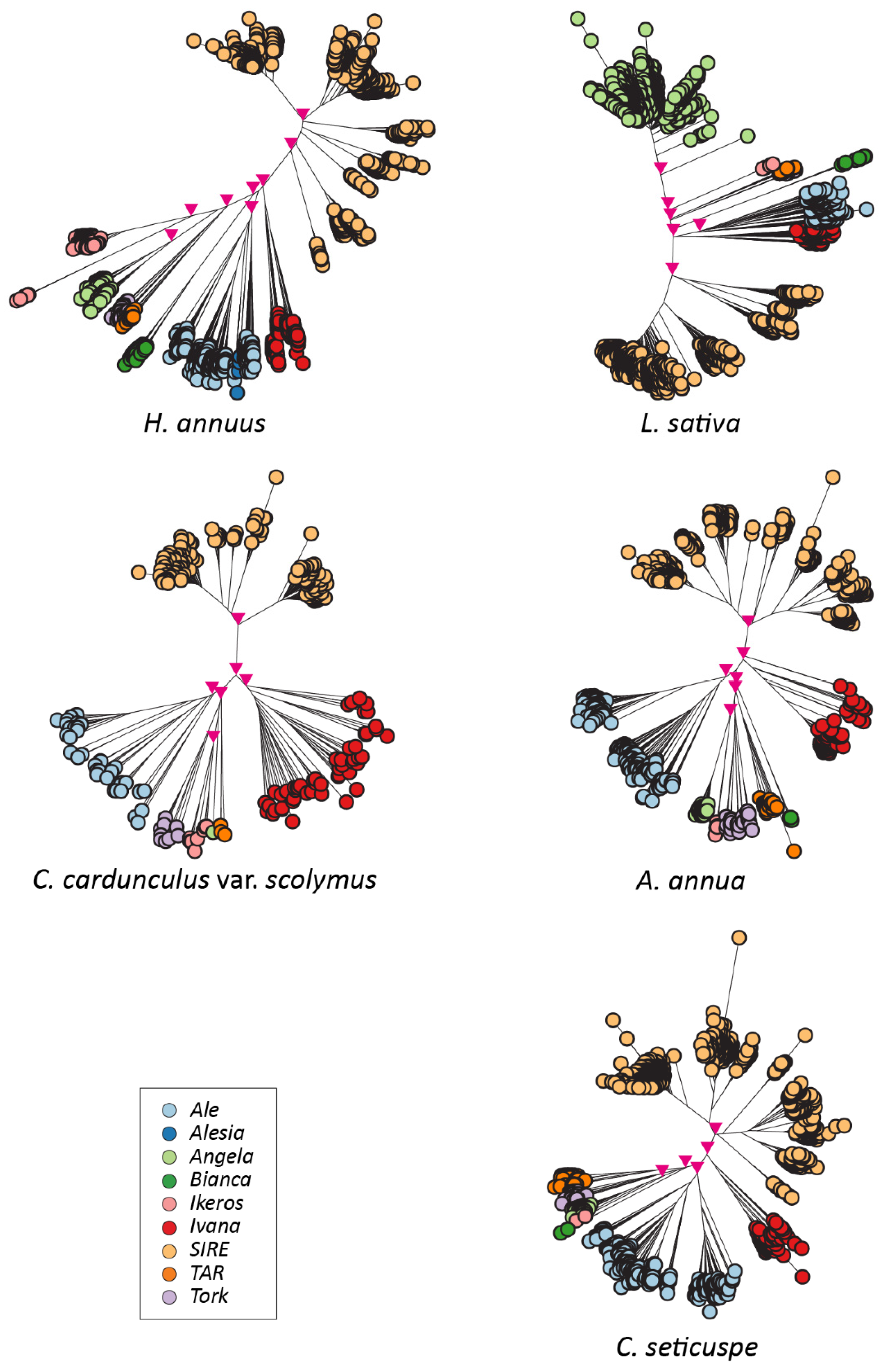
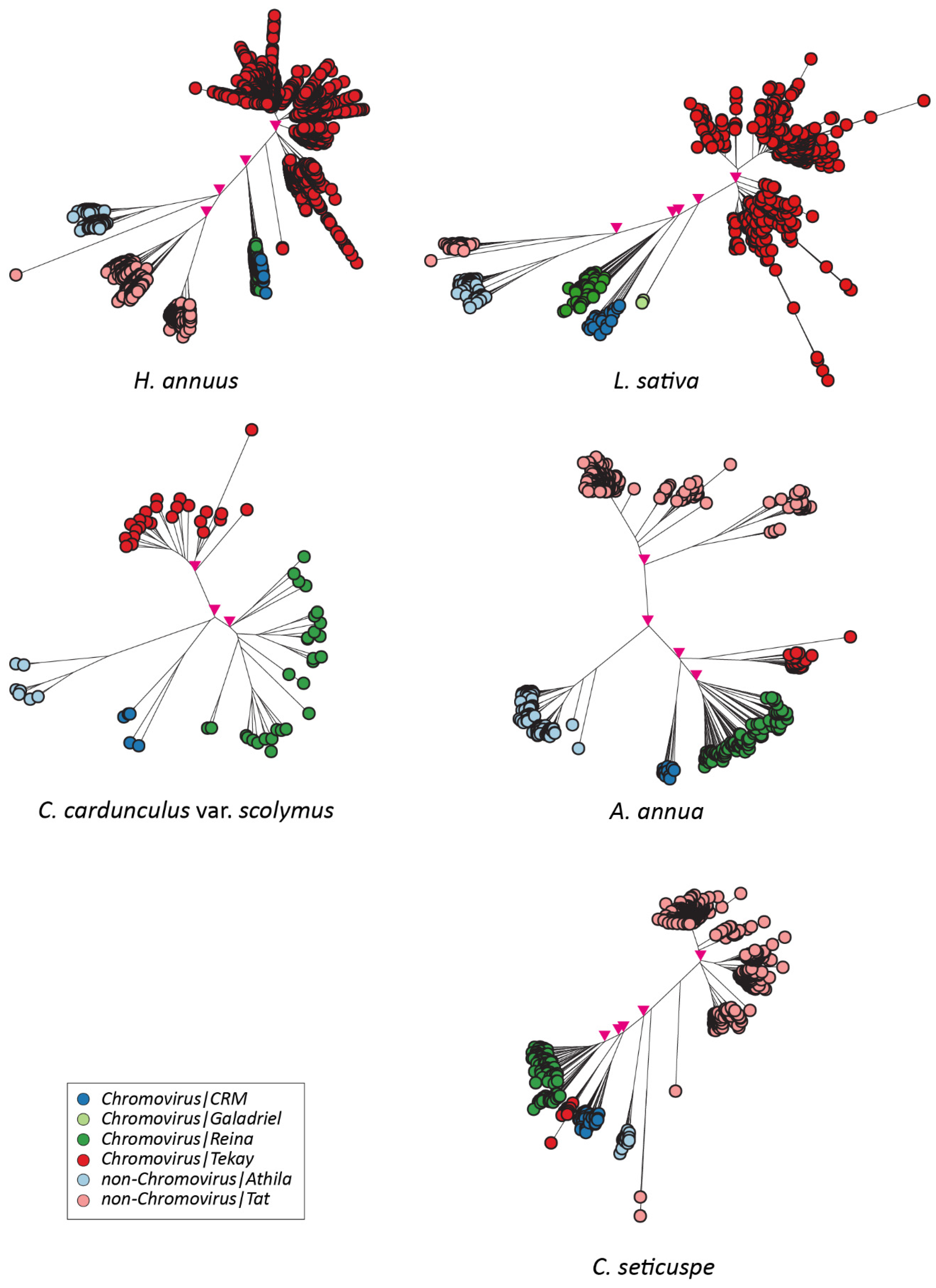
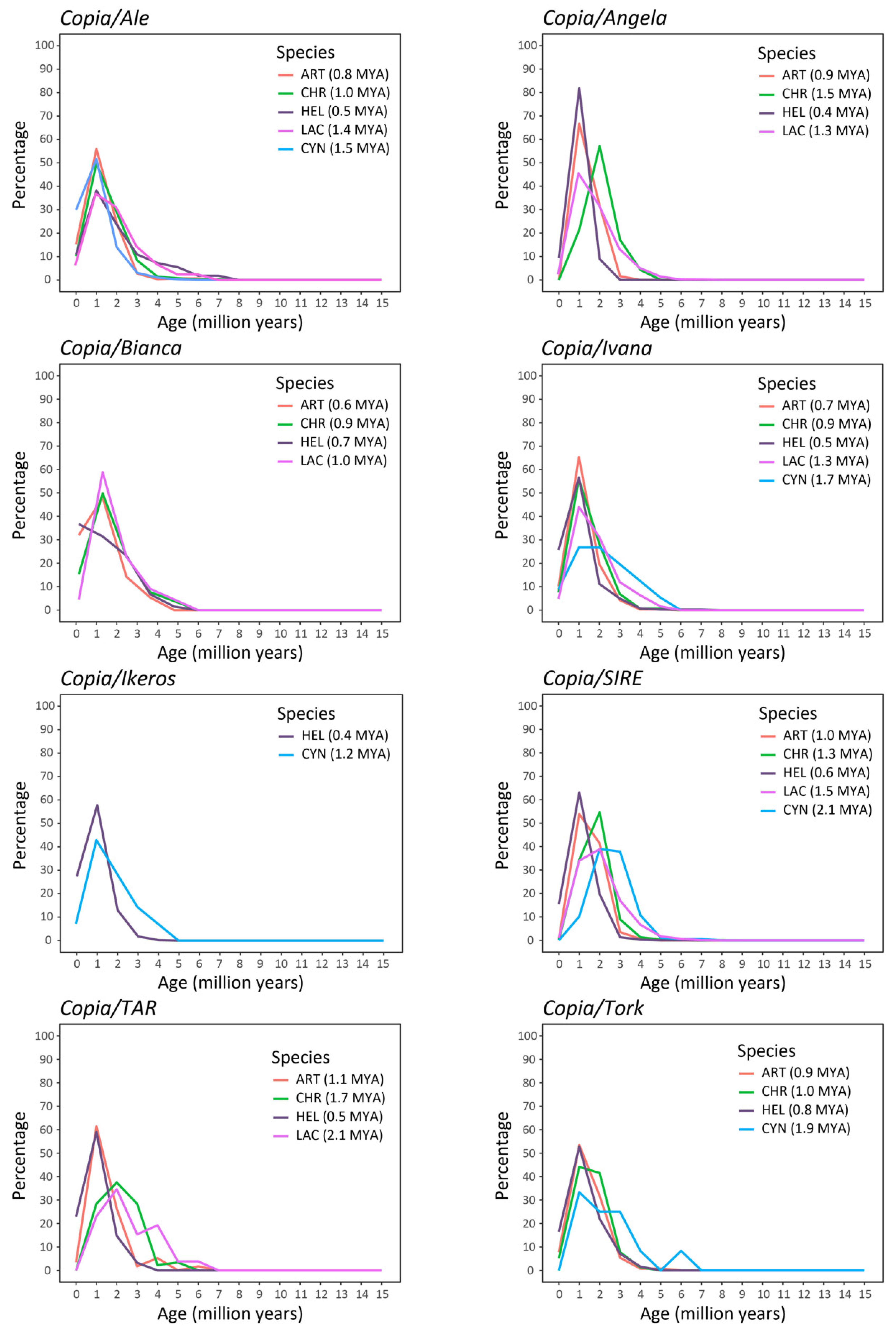
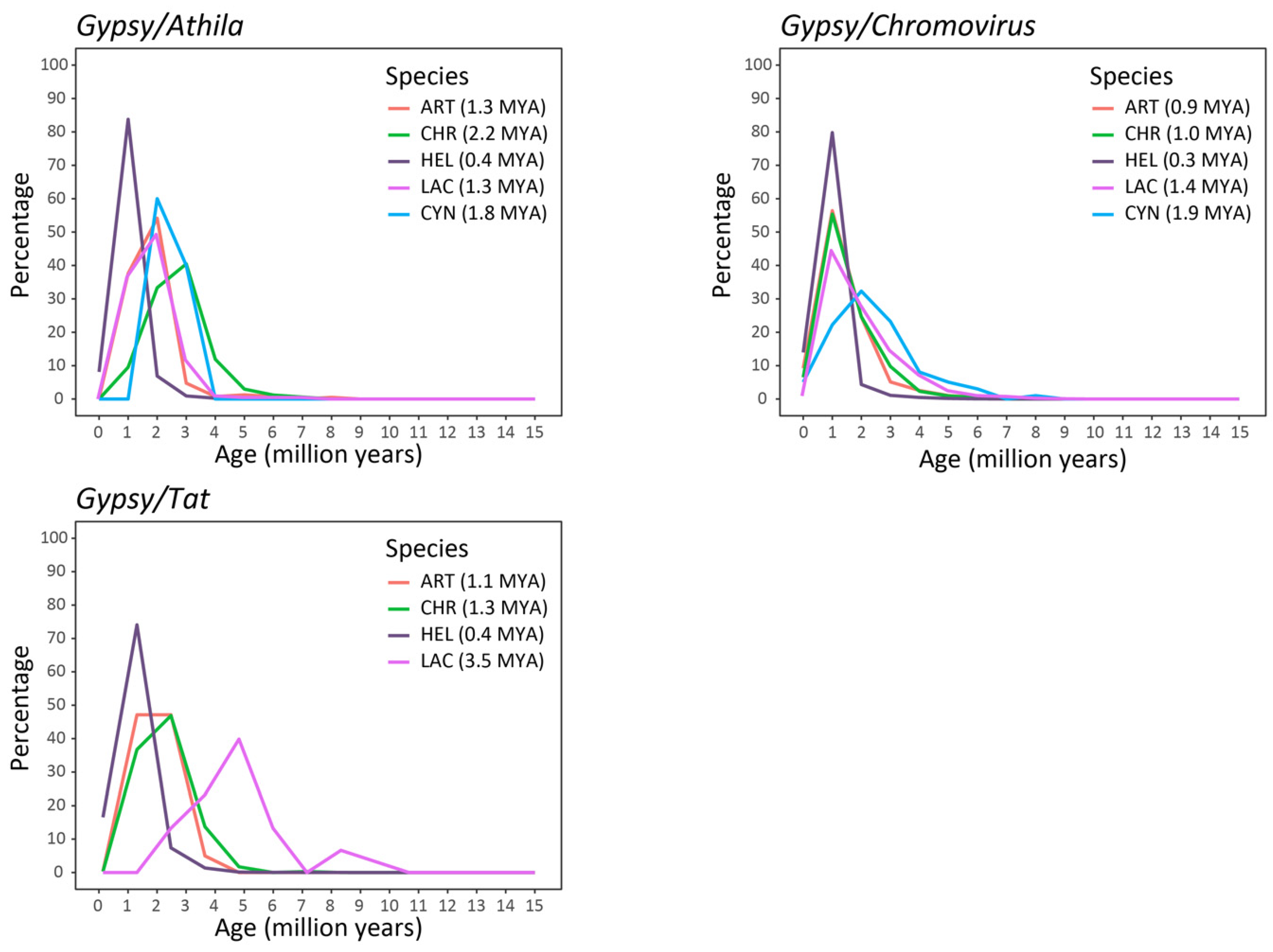
2.2. Isolation and Analysis of Full-Length LTR Retrotransposons
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Sequence Data Collection
5.2. Clustering Analyses with RepeatExplorer2
5.3. Identification and Characterisation of Full-Length LTR-REs
5.4. Phylogenetic Analysis of LTR-REs
5.5. Evaluation of the Insertion Time of LTR-REs
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Woo, T.H.; Hong, T.H.; Kim, S.S.; Chung, W.H.; Kang, H.J.; Kim, C.B.; Seo, J.M. Repeatome: A database for repeat element comparative analysis in human and chimpanzee. Genom. Inform. 2007, 5, 179–187. [Google Scholar]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. Mutable loci in maize. Carnegie Inst. Wash. Yearb. 1948, 47, 155–169. [Google Scholar]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef]
- Kejnovsky, E.; Hawkins, J.S.; Feschotte, C. Plant transposable elements: Biology and evolution. In Plant Genome Diversity Volume 1; Springer: Vienna, Austria, 2012; pp. 17–34. [Google Scholar]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Vitte, C.; Fustier, M.A.; Alix, K.; Tenaillon, M.I. The bright side of transposons in crop evolution. Brief. Funct. Genom. 2014, 13, 276–295. [Google Scholar] [CrossRef] [Green Version]
- Orgel, L.E.; Crick, F.H. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Lisch, D. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef] [Green Version]
- Sinzelle, L.; Izsvak, Z.; Ivics, Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell. Mol. Life Sci. 2009, 66, 1073–1093. [Google Scholar] [CrossRef] [PubMed]
- Ventimiglia, M.; Marturano, G.; Vangelisti, A.; Usai, G.; Simoni, S.; Cavallini, A.; Giordani, T.; Natali, L.; Zuccolo, A.; Mascagni, F. Genome-wide identification and characterisation of exapted transposable elements in the large genome of sunflower (Helianthus annuus L.). Plant J. 2022, 113, 734–748. [Google Scholar] [CrossRef] [PubMed]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef]
- Viviani, A.; Ventimiglia, M.; Fambrini, M.; Vangelisti, A.; Mascagni, F.; Pugliesi, C.; Usai, G. Impact of transposable elements on the evolution of complex living systems and their epigenetic control. Biosystems 2021, 210, 104566. [Google Scholar] [CrossRef]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Mayer, K.F.; Waugh, R.; Langridge, P.; Close, T.J.; Wise, R.P.; Graner, A.; Matsumoto, T.; Sato, K.; Schulman, A.; Muehlbauer, G.J.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar]
- Mascagni, F.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genom. 2017, 18, 634. [Google Scholar] [CrossRef] [Green Version]
- Staton, S.E.; Bakken, B.H.; Blackman, B.K.; Chapman, M.A.; Kane, N.C.; Tang, S.; Ungerer, M.C.; Knapp, S.J.; Rieseberg, L.H.; Burke, J.M. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012, 72, 142–153. [Google Scholar] [CrossRef]
- Natali, L.; Cossu, R.M.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.C.; Rieseberg, L.; et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genom. 2013, 14, 686. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Barghini, E.; Giordani, T.; Rieseberg, L.H.; Cavallini, A.; Natali, L. Repetitive DNA and plant domestication: Variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biol. Evol. 2015, 7, 3368–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staton, S.E. Transposable Elements Drive Lineage-Specific Patterns of Genome Evolution in the Asteraceae. Ph.D. Thesis, University of Georgia, Athens, GA, USA, 2014. [Google Scholar]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [Green Version]
- Cavallini, A.; Mascagni, F.; Giordani, T.; Natali, L. Genome skimming for plant retrotransposon identification and expression analysis. Agrochimica 2019, 63, 367–378. [Google Scholar] [CrossRef]
- Usai, G.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Comparative genome-wide analysis of repetitive DNA in the genus Populus L. Tree Genet. Genomes 2017, 13, 96. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Vangelisti, A.; Usai, G.; Giordani, T.; Cavallini, A.; Natali, L. A computational genome-wide analysis of long terminal repeats retrotransposon expression in sunflower roots (Helianthus annuus L.). Genetica 2020, 148, 13–23. [Google Scholar] [CrossRef]
- Phillippy, A.M.; Schatz, M.C.; Pop, M. Genome assembly forensics: Finding the elusive mis-assembly. Genome Biol. 2008, 9, R55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Chin-Wo, S.; Wang, Z.; Yang, X.; Kozik, A.; Arikit, S.; Song, C.; Xia, L.; Froenicke, L.; Lavelle, D.O.; Truco, M.J.; et al. Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat. Commun. 2017, 8, 14953. [Google Scholar] [CrossRef] [Green Version]
- Scaglione, D.; Reyes-Chin-Wo, S.; Acquadro, A.; Froenicke, L.; Portis, E.; Beitel, C.; Tirone, M.; Mauro, R.; Lo Monaco, A.; Mauromicale, G.; et al. The genome sequence of the outbreeding globe artichoke constructed de novo incorporating a phase-aware low-pass sequencing strategy of F 1 progeny. Sci. Rep. 2016, 6, 19427. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Zhang, L.; Liao, Z.; Wang, S.; Yan, T.; Shi, P.U.; Liu, M.; Fu, X.; Pan, Q.; Wang, Y.; et al. The genome of Artemisia annua provides insight into the evolution of Asteraceae family and artemisinin biosynthesis. Mol. Plant 2018, 11, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, H.; Sumitomo, K.; Hisamatsu, T.; Nagano, S.; Shirasawa, K.; Higuchi, Y.; Kusaba, M.; Koshioka, M.; Nakano, Y.; Yagi, M.; et al. De novo whole-genome assembly in Chrysanthemum seticuspe, a model species of Chrysanthemums, and its application to genetic and gene discovery analysis. DNA Res. 2019, 26, 195–203. [Google Scholar] [CrossRef] [Green Version]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Usai, G.; Natali, L.; Cavallini, A.; Giordani, T. A comparison of methods for LTR-retrotransposon insertion time profiling in the Populus trichocarpa genome. Caryologia 2018, 71, 85–92. [Google Scholar] [CrossRef]
- Rieseberg, L.H. Homoploid reticulate evolution in Helianthus (Asteraceae): Evidence from ribosomal genes. Am. J. Bot. 1991, 78, 1218–1237. [Google Scholar] [CrossRef]
- Novák, P.; Hřibová, E.; Neumann, P.; Koblížková, A.; Doležel, J.; Macas, J. Genome-wide analysis of repeat diversity across the family Musaceae. PLoS ONE 2014, 9, e98918. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Barghini, E.; Ceccarelli, M.; Baldoni, L.; Trapero, C.; Díez, C.M.; Natali, L.; Cavallini, A.; Giordani, T. The Singular Evolution of Olea Genome Structure. Front. Plant Sci. 2022, 13, 869048. [Google Scholar] [CrossRef]
- Giordani, T.; Cavallini, A.; Natali, L. The repetitive component of the sunflower genome. Curr. Plant Biol. 2014, 1, 45–54. [Google Scholar] [CrossRef]
- Liao, B.; Shen, X.; Xiang, L.; Guo, S.; Chen, S.; Meng, Y.; Liang, Y.; Ding, D.; Bai, J.; Zhang, D.; et al. Allele-aware chromosome-level genome assembly of Artemisia annua reveals the correlation between ADS expansion and artemisinin yield. Mol. Plant 2022, 15, 1310–1328. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.; Macas, J.; Novák, P.; Stuessy, T.F.; Villaseñor, J.L.; Weiss-Schneeweiss, H. Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 2020, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Zagorski, D.; Hartmann, M.; Bertrand, Y.J.; Paštová, L.; Slavíková, R.; Josefiová, J.; Fehrer, J. Characterization and dynamics of repeatomes in closely related species of Hieracium (Asteraceae) and their synthetic and apomictic hybrids. Front. Plant Sci. 2020, 11, 591053. [Google Scholar] [CrossRef]
- Fernández, P.; Hidalgo, O.; Juan, A.; Leitch, I.J.; Leitch, A.R.; Palazzesi, L.; Pegoraro, L.; Viruel, J.; Pellicer, J. Genome Insights into Autopolyploid Evolution: A Case Study in Senecio doronicum (Asteraceae) from the Southern Alps. Plants 2022, 11, 1235. [Google Scholar] [CrossRef]
- Tenaillon, M.I.; Hufford, M.B.; Gaut, B.S.; Ross-Ibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol. Evol. 2011, 3, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Neumann, P.; Koblizkova, A.; Navrátilová, A.; Macas, J. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 2006, 173, 1047–1056. [Google Scholar] [CrossRef] [Green Version]
- Christelová, P.; Valárik, M.; Hřibová, E.; De Langhe, E.; Doležel, J. A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol. Biol. 2011, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Staton, S.E.; Burke, J.M. Evolutionary transitions in the Asteraceae coincide with marked shifts in transposable element abundance. BMC Genom. 2015, 16, 623. [Google Scholar] [CrossRef] [Green Version]
- Simoni, S.; Clemente, C.; Usai, G.; Vangelisti, A.; Natali, L.; Tavarini, S.; Angelini, L.G.; Cavallini, A.; Mascagni, F.; Giordani, T. Characterisation of LTR-Retrotransposons of Stevia rebaudiana and Their Use for the Analysis of Genetic Variability. Int. J. Mol. Sci. 2022, 23, 6220. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.C.; Strakosh, S.C.; Stimpson, K.M. Proliferation of Ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 2009, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Ungerer, M.C.; Strakosh, S.C.; Zhen, Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr. Biol. 2006, 16, R872–R873. [Google Scholar] [CrossRef] [Green Version]
- Vukich, M.; Giordani, T.; Natali, L.; Cavallini, A. Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 2009, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 October 2022).
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxybased web server for genome-wide characterization of eukaryotic repetitive elements from next generation sequence read. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S.; Steinbiss, S.; Willhoeft, U.; Gremme, G.; Fine-grained, S.K. LTRdigest User’s Manual; University of Hamburg: Hamburg, Germany, 2010. [Google Scholar]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 1, 2–3. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 27 October 2022).
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | GenBank Assembly Accession | Assembly Level | SRA ID | Raw Paired-End Reads | Trimmed Reads (100 bp) |
|---|---|---|---|---|---|---|
| Helianthus annuus | Sunflower | GCA_002127325.2 [31] | Chromosome | SRR5004633 | 124,824,626 | 82,204,512 |
| Lactuca sativa | Lettuce | GCA_002870075.2 [32] | Chromosome | SRR577192 | 187,005,846 | 117,409,692 |
| Cynara cardunculus var. scolymus | Globe artichocke | GCA_001531365.1 [33] | Chromosome | SRR1914381 | 91,528,290 | 73,595,420 |
| Artemisia annua | Annual mugwort | GCA_003112345.1 [34] | Scaffold | SRR5602595 | 1,330,400 | 1,076,116 |
| Chrysanthemum seticuspe | Chrysanthemum | GCA_004359105.1 [35] | Scaffold | DRR087118 | 382,227,342 | 330,102,622 |
| Species | Total Read Count [Nr] | Reads in Cluster [Nr] | Genome Proportion [%] | Genome Size [Gb] |
|---|---|---|---|---|
| Helianthus annuus | 438,456 | 343,922 | 78.44 | 3.6 |
| Lactuca sativa | 438,358 | 280,372 | 63.96 | 2.5 |
| Cynara cardunculus var. scolymus | 437,906 | 264,665 | 60.44 | 1.07 |
| Artemisia annua | 438,250 | 273,986 | 62.52 | 1.74 |
| Chrysanthemum seticuspe | 437,612 | 302,014 | 69.01 | 3.06 |
| LTR-RE | Superfamily | Lineage | Helianthus annuus | Lactuca sativa | Cynara cardunculus var. scolymus | Artemisia annua | Chrysantemum seticuspe | Maximum Difference |
|---|---|---|---|---|---|---|---|---|
| Copia | Ale | 0.00 | 0.00 | 0.00 | 0.00 | 0.17 | 0.17 | |
| Angela | 0.07 | 16.99 | 0.01 | 2.04 | 4.01 | 16.99 | ||
| Ikeros | 0.29 | 0.00 | 0.00 | 0.00 | 0.00 | 0.29 | ||
| Ivana | 0.00 | 0.00 | 0.00 | 0.23 | 0.10 | 0.23 | ||
| SIRE | 5.57 | 3.88 | 23.49 | 16.04 | 29.91 | 26.03 | ||
| TAR | 0.11 | 0.03 | 0.01 | 0.44 | 0.72 | 0.71 | ||
| Unknown | 0.31 | 0.95 | 0.00 | 1.05 | 0.38 | 1.05 | ||
| Total Copia | 6.35 | 21.86 | 23.50 | 19.80 | 35.28 | 28.93 | ||
| Gypsy | Chromovirus | 30.42 | 12.9 | 9.66 | 3.62 | 2.07 | 28.35 | |
| Athila | 3.05 | 0.55 | 0.95 | 15.46 | 9.38 | 14.90 | ||
| Tat | 5.25 | 0.01 | 0.00 | 0.90 | 0.80 | 5.25 | ||
| Unknown | 2.39 | 0.20 | 0.00 | 0.00 | 0.00 | 2.39 | ||
| Total Gypsy | 41.11 | 13.67 | 10.61 | 19.98 | 12.25 | 30.50 | ||
| Unknown | 4.24 | 12.59 | 1.15 | 3.06 | 4.79 | 11.44 | ||
| TOTAL | 51.70 | 48.12 | 35.27 | 42.84 | 52.32 | 17.05 | ||
| Gypsy/ Copia | 6.47 | 0.63 | 0.45 | 1.01 | 0.35 |
| Lineage | Helianthus annuus | Lactuca sativa | Cynara cardunculus var. scolymus | Artemisia annua | Chrysanthemum seticuspe |
|---|---|---|---|---|---|
| Ale | 674 | 208 | 55 | 288 | 630 |
| Alesia | 9 | 15 | 0 | 0 | 0 |
| Angela | 312 | 2278 | 1 | 63 | 70 |
| Bianca | 133 | 22 | 0 | 56 | 26 |
| Ikeros | 505 | 5 | 14 | 8 | 2 |
| Ivana | 400 | 125 | 56 | 323 | 304 |
| SIRE | 4711 | 2493 | 176 | 630 | 1284 |
| TAR | 61 | 26 | 4 | 57 | 88 |
| Tork | 182 | 8 | 12 | 129 | 77 |
| Copia unclassified | 656 | 246 | 19 | 380 | 602 |
| Copia total | 7643 | 5426 | 337 | 1934 | 3083 |
| Chromovirus|CRM | 119 | 46 | 5 | 30 | 47 |
| Chrommovirus|Galadriel | 3 | 3 | 0 | 0 | 0 |
| Chromovirus|Reina | 235 | 65 | 44 | 123 | 146 |
| Chromovirus|Tekay | 18,405 | 1027 | 48 | 35 | 21 |
| Chromovirus unclassified | 424 | 13 | - | 5 | 1 |
| non-Chromovirus|OTA|Athila | 2472 | 213 | 10 | 398 | 168 |
| non-Chromovirus|OTA|Tat | 5060 | 30 | 0 | 260 | 697 |
| non-Chromovirus|OTA unclassified | 7 | - | - | 4 | 1 |
| non-Chromovirus unclassified | - | - | - | - | - |
| Gypsy unclassified | 84 | - | - | 1 | - |
| Gypsy total | 26,809 | 1397 | 107 | 856 | 1081 |
| LTR-RE unclassified | 128 | 52 | 4 | 9 | 6 |
| TOTAL | 34,580 | 6875 | 448 | 2799 | 4170 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventimiglia, M.; Castellacci, M.; Usai, G.; Vangelisti, A.; Simoni, S.; Natali, L.; Cavallini, A.; Mascagni, F.; Giordani, T. Discovering the Repeatome of Five Species Belonging to the Asteraceae Family: A Computational Study. Plants 2023, 12, 1405. https://doi.org/10.3390/plants12061405
Ventimiglia M, Castellacci M, Usai G, Vangelisti A, Simoni S, Natali L, Cavallini A, Mascagni F, Giordani T. Discovering the Repeatome of Five Species Belonging to the Asteraceae Family: A Computational Study. Plants. 2023; 12(6):1405. https://doi.org/10.3390/plants12061405
Chicago/Turabian StyleVentimiglia, Maria, Marco Castellacci, Gabriele Usai, Alberto Vangelisti, Samuel Simoni, Lucia Natali, Andrea Cavallini, Flavia Mascagni, and Tommaso Giordani. 2023. "Discovering the Repeatome of Five Species Belonging to the Asteraceae Family: A Computational Study" Plants 12, no. 6: 1405. https://doi.org/10.3390/plants12061405