
Time-Course Transcriptome Profiling Reveals Differential Resistance Responses of Tomato to a Phytotoxic Effector of the Pathogenic Oomycete Phytophthora cactorum

Abstract
:1. Introduction
2. Results
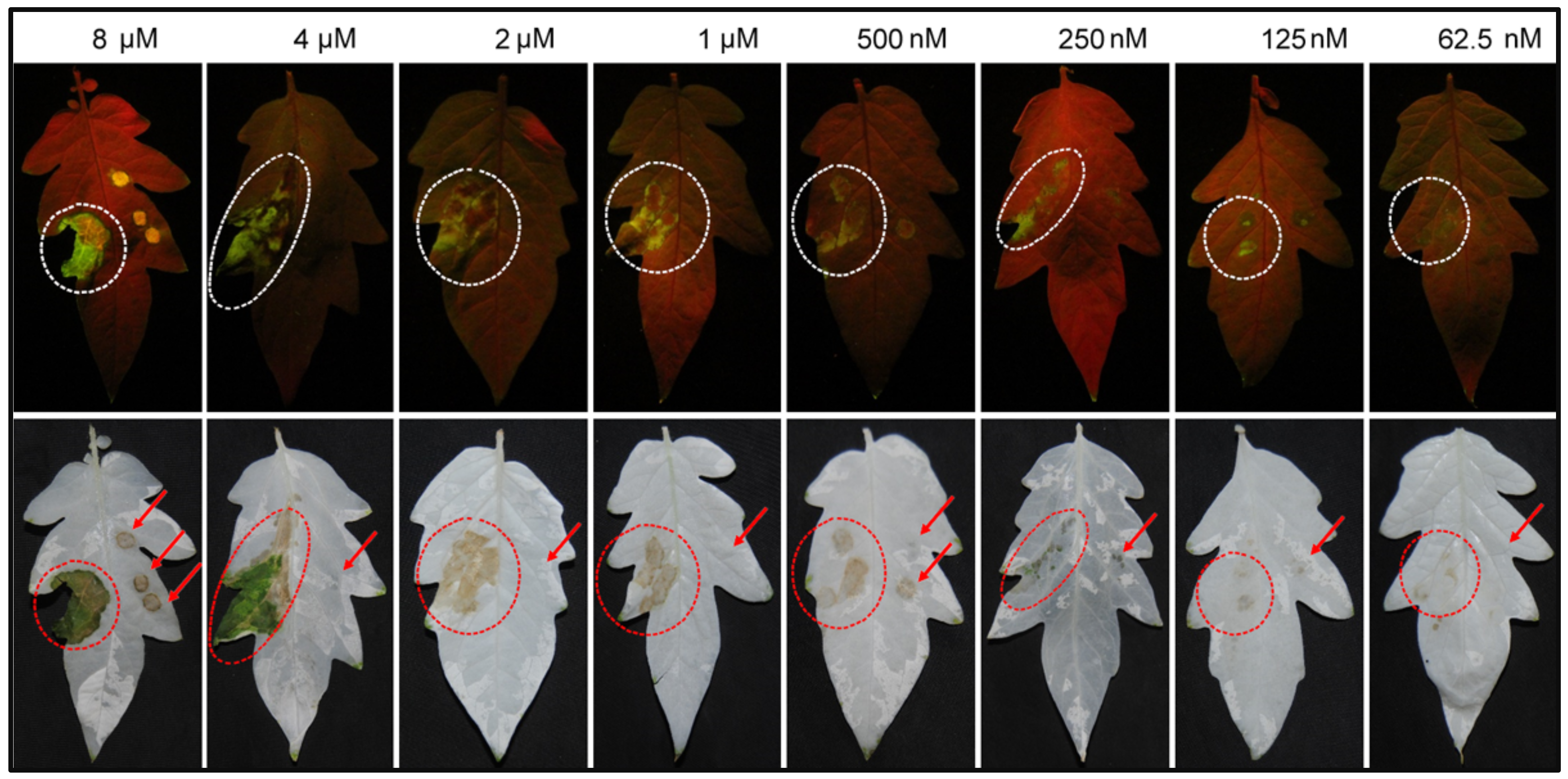
2.1. The Recombinant SCR96 Protein Secreted by Yeast Cells Can Cause Tomato Leaf Necrosis
2.2. RNA Sequencing and Mapping to the Reference Genome
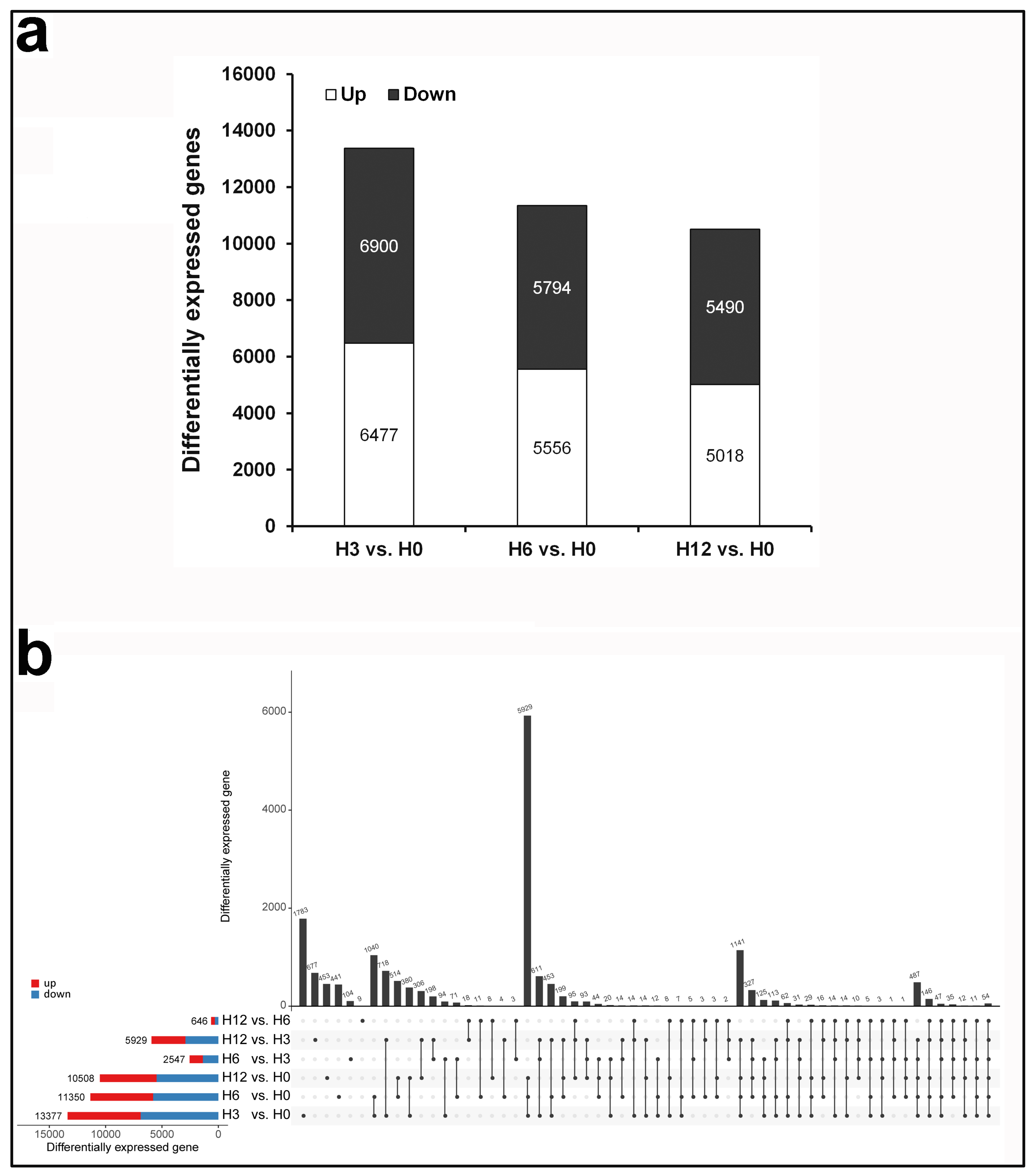
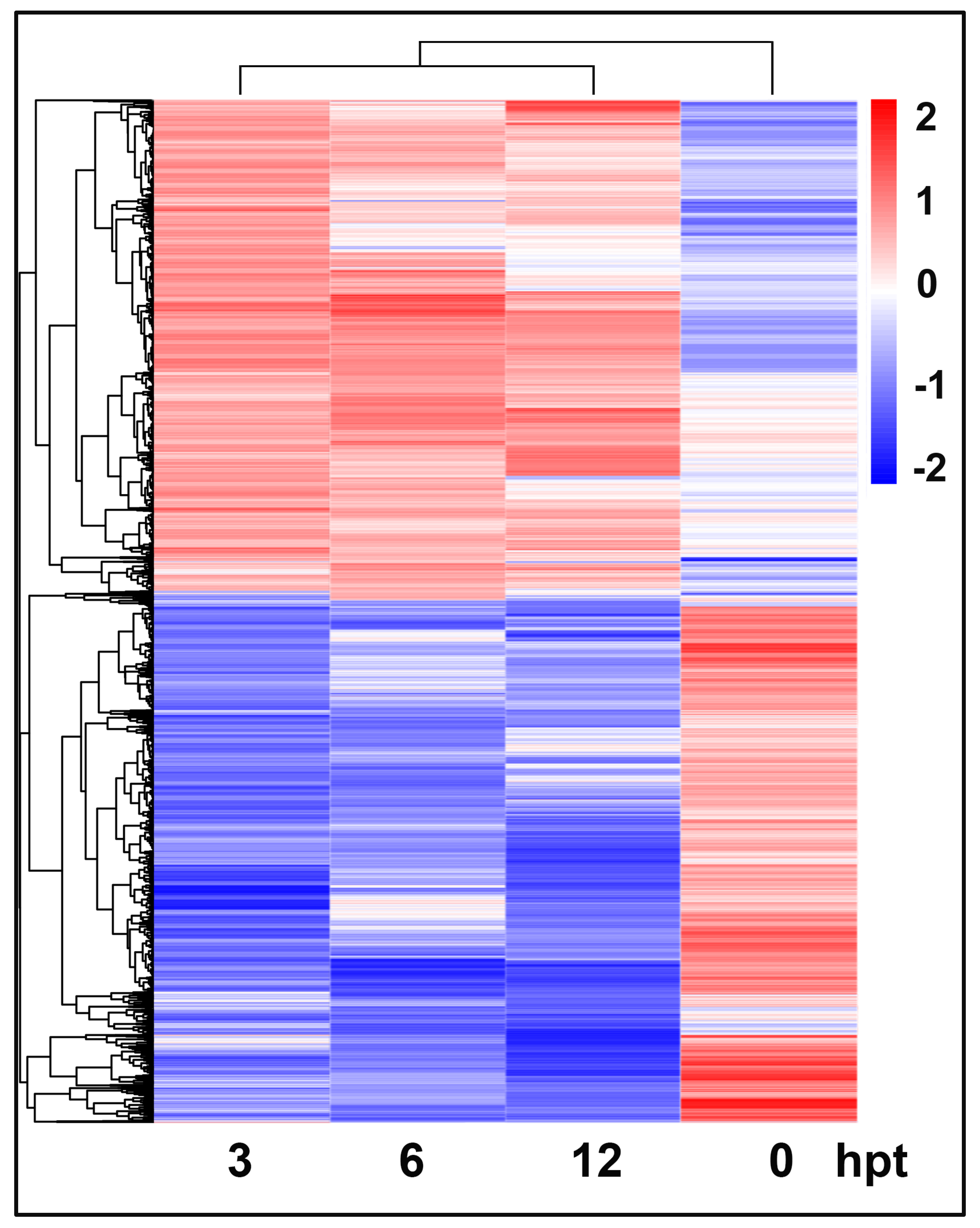
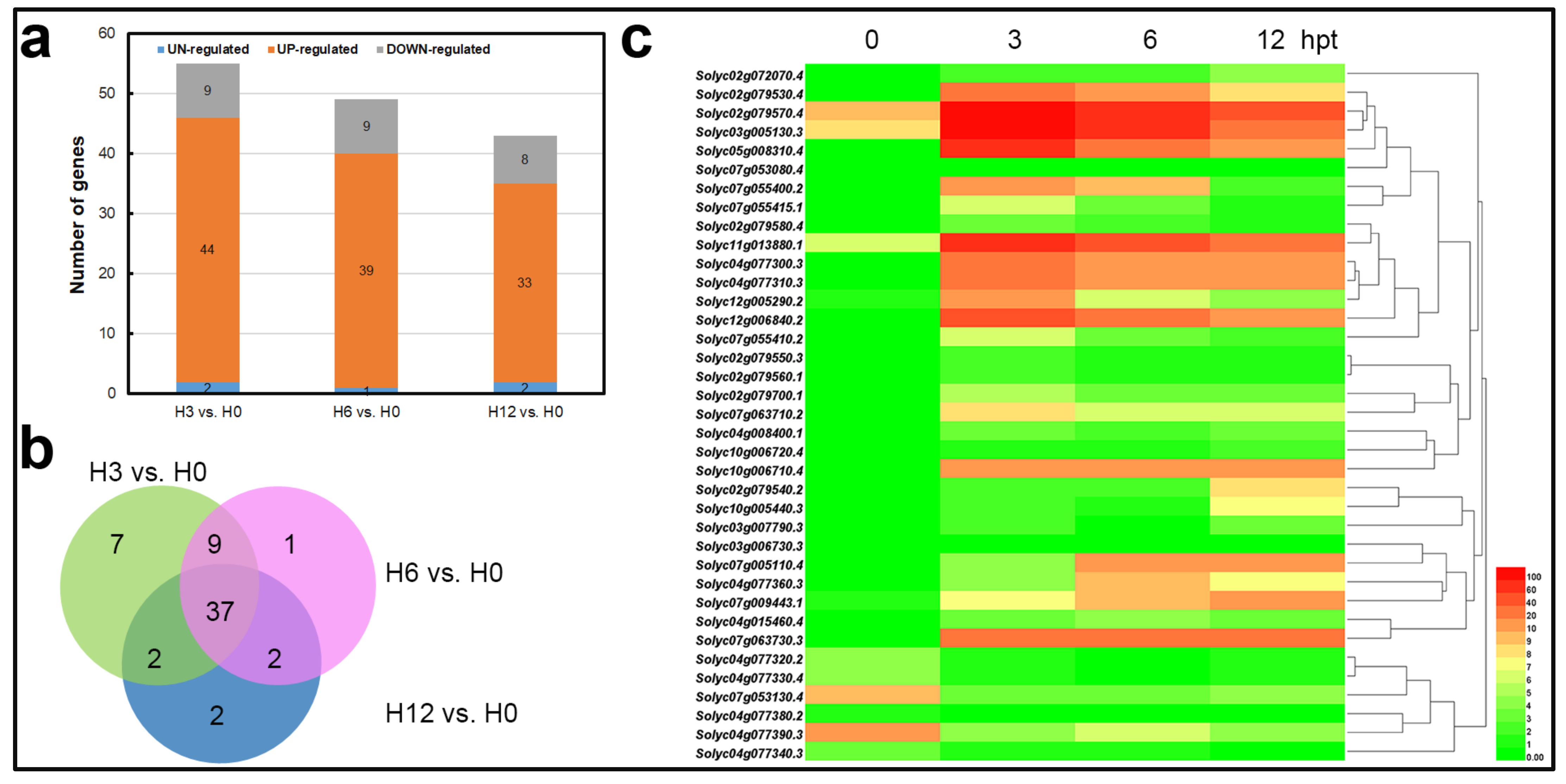
2.3. Identification of Differentially Expressed Genes
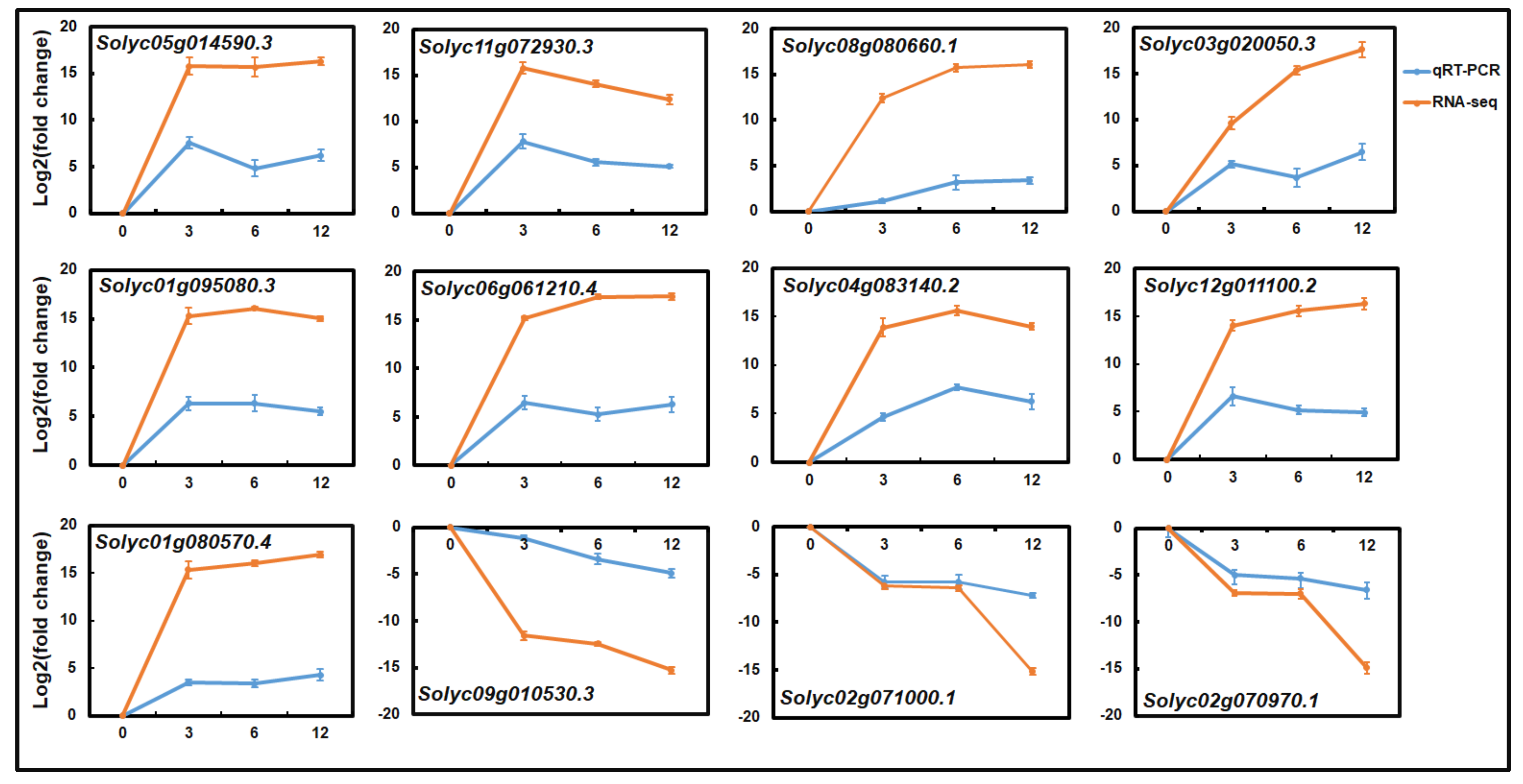
2.4. qRT-PCR Analysis of the Gene Expression Profiles
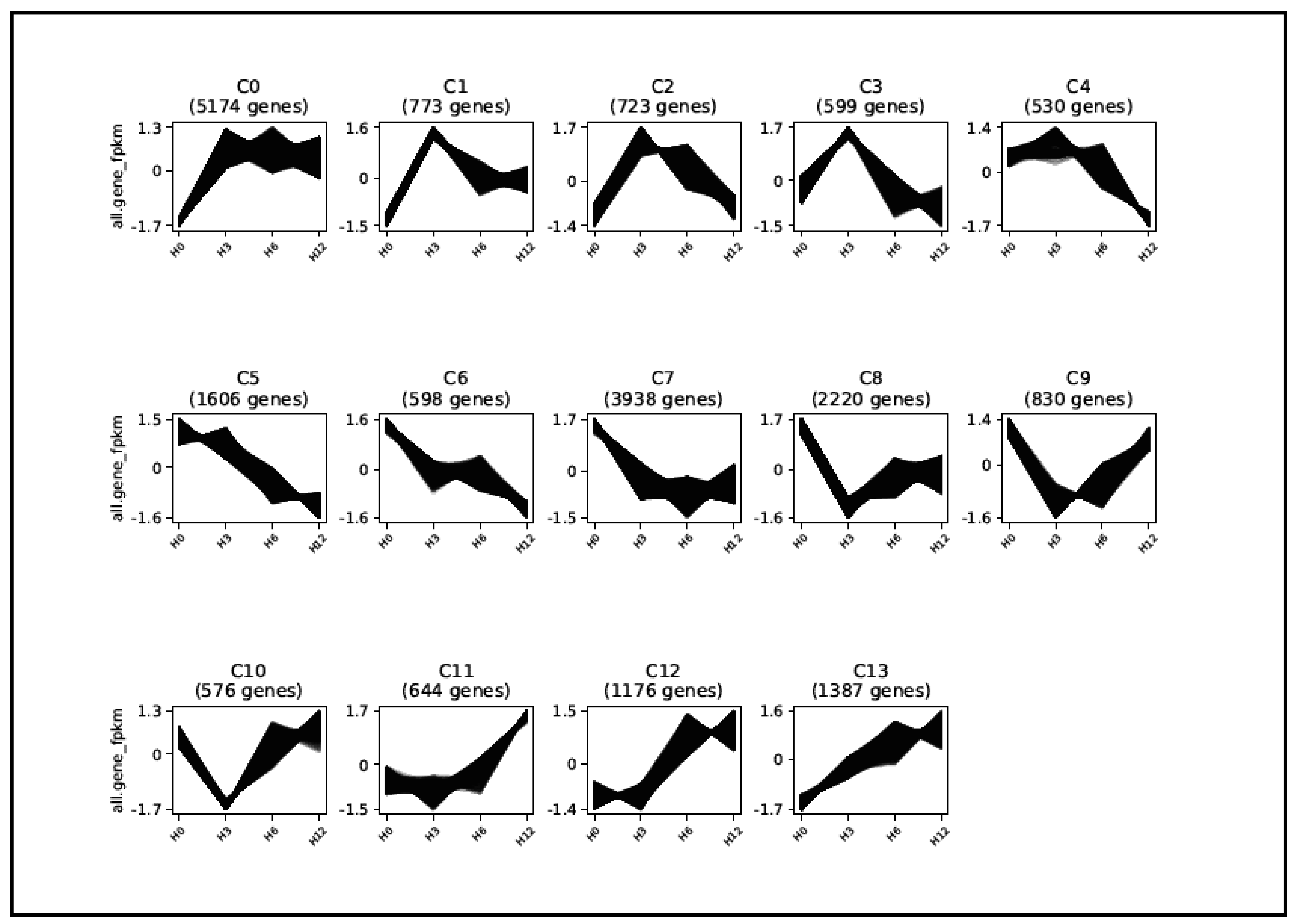
2.5. Temporal Clustering Analysis to Predict Putative Co-Regulated Genes
2.6. GO and KEGG Analyses Revealed Genes Involved in Plant Defense Responses and Metabolism
2.7. Genes Involved in Pathogen Detection and Signal Transduction
2.8. G-LecRK Gene Expression Changes
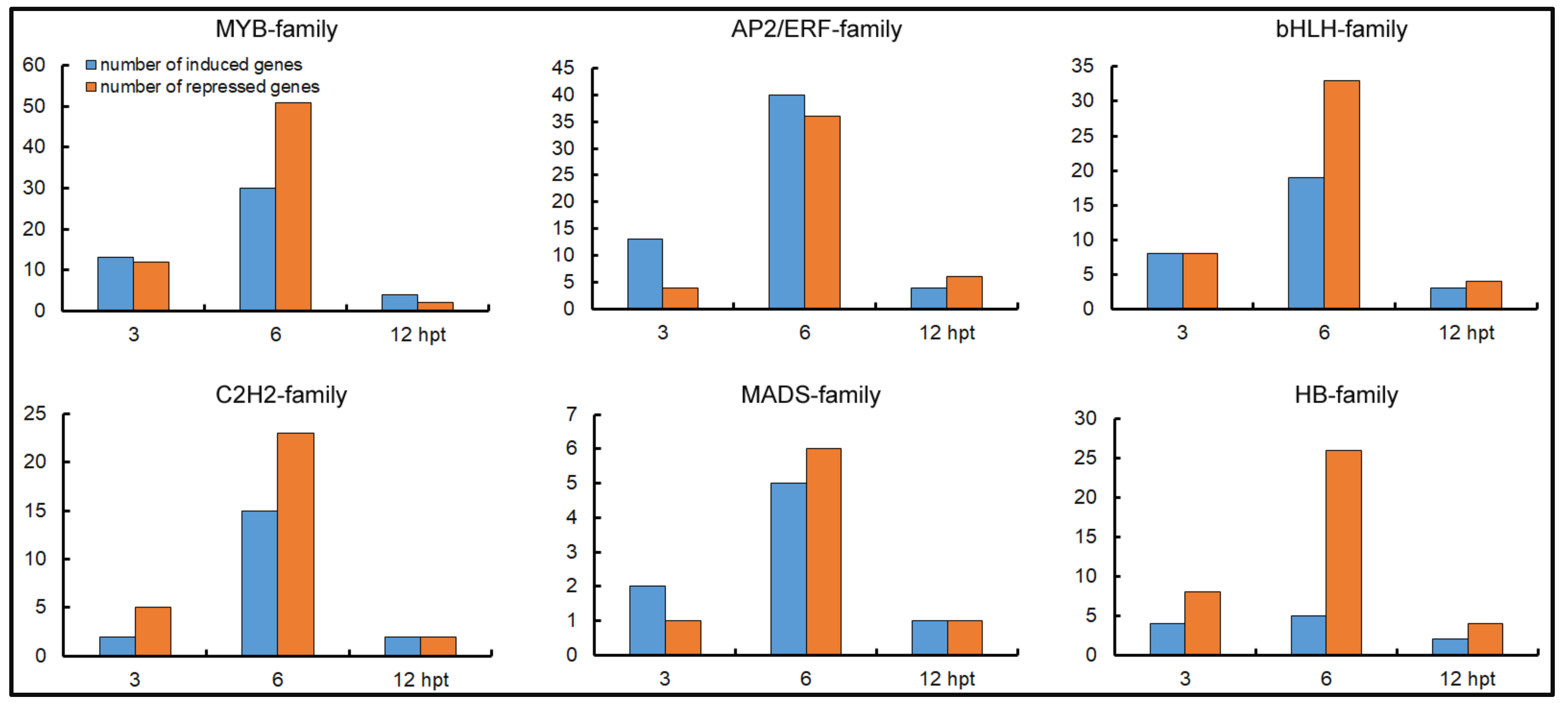
2.9. Activation of TF Genes
2.10. Genes Encoding PR Proteins
2.11. Genes Related to Transporter Activity
3. Discussion
3.1. Heterologous Expression of the Hydrophobic Effector Using a Newly Adapted Yeast System
3.2. SCR96 Induces Plant Defense Responses
3.3. Signaling Triggered by SCR96 Can Reprogram the Expression of Genes Encoding Proteins Involved in Regulation of Plant Immunity
3.4. Importance of Early Interaction in Determining Compatibility
4. Materials and Methods
4.1. Plant and Microbe Strains
4.2. Nucleic Acid Manipulation
4.3. Plasmid Construction
4.4. Yeast Expression and Protein Purification
4.5. Transcriptome Studies
4.6. Validation Analysis Using qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baldauf, S.L.; Roger, A.J.; Wenk-Siefert, I.; Doolittle, W.F. A kingdom-level phylogeny of eukaryotes based on combined protein data. Science 2000, 5493, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Erwin, D.C.; Ribeiro, O.K. Phytophthora Diseases Worldwide; American Phytopathological Society: St. Paul, MN, USA, 1996; pp. 245–440. [Google Scholar]
- Kamoun, S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 2006, 44, 41–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.R.; Li, Y.P.; Li, Q.Y.; Xing, Y.P.; Liu, B.B.; Tong, Y.H.; Xu, J.Y. SCR96, a small cysteine-rich secretory protein of Phytophthora cactorum, can trigger cell death in the Solanaceae and is important for pathogenicity and oxidative stress tolerance. Mol. Plant Pathol. 2016, 17, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Zhang, Z.H.; Liu, W.; Tao, H.; Zhang, Y.; Shi, N.X.; Zhu, F.; Ji, Z.L.; Chen, X.R. Expression of the small cysteine-rich protein SCR96 from Phytophthora cactorum in mammalian cells: Phytotoxicity and exploitation of its polyclonal antibody. Biotechnol. Lett. 2020, 42, 125–133. [Google Scholar] [CrossRef]
- Nicastro, G.; Orsomando, G.; Ferrari, E.; Manconi, L.; Desario, F.; Amici, A.; Naso, A.; Carpaneto, A.; Pertinhez, T.A.; Ruggieri, S.; et al. Solution structure of the phytotoxic protein PcF: The first characterized member of the Phytophthora PcF toxin family. Protein Sci. 2009, 18, 1786–1791. [Google Scholar] [CrossRef] [Green Version]
- Böhm, H.; Albert, I.; Oome, S.; Raaymakers, T.M.; Van den Ackerveken, G.; Nürnberger, T. A conserved peptide pattern from a widespread microbial virulence factor triggers pattern-induced immunity in Arabidopsis. PLoS Pathog. 2014, 10, e1004491. [Google Scholar] [CrossRef] [Green Version]
- Albert, I.; Böhm, H.; Albert, M.; Feiler, C.E.; Imkampe, J.; Wallmeroth, N.; Brancato, C.; Raaymakers, T.M.; Oome, S.; Zhang, H.; et al. An RLP23–SOBIR1–BAK1 complex mediates NLP-triggered immunity. Nat. Plants 2015, 1, 15140. [Google Scholar] [CrossRef]
- Du, J.; Verzaux, E.; Chaparro-Garcia, A.; Bijsterbosch, G.; Keizer, L.C.; Zhou, J.I.; Liebrand, T.W.; Xie, C.; Govers, F.; Robatzek, S.; et al. Elicitin recognition confers enhanced resistance to Phytophthora infestans in potato. Nat. Plants 2015, 1, 15034. [Google Scholar] [CrossRef]
- Bos, J.I.B.; Armstrong, M.; Whisson, S.C.; Torto, T.A.; Ochwo, M.; Birch, P.R.J.; Kamoun, S. Intraspecific comparative genomics to identify avirulence genes from Phytophthora. New Phytol. 2003, 159, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Orsomando, G.; Lorenzi, M.; Raffaelli, N.; Dalla Rizza, M.; Mezzetti, B.; Ruggieri, S. Phytotoxic protein PcF, purification, characterization, and cDNA sequencing of a novel hydroxyproline-containing factor secreted by the strawberry pathogen Phytophthora cactorum. J. Biol. Chem. 2001, 276, 21578–21584. [Google Scholar] [CrossRef] [Green Version]
- Stassen, J.H.; Van den Ackerveken, G. How do oomycete effectors interfere with plant life? Curr. Opin. Plant Biol. 2011, 14, 407–414. [Google Scholar] [CrossRef]
- Wawra, S.; Belmonte, R.; Löbach, L.; Saraiva, M.; Willems, A.; van West, P. Secretion, delivery and function of oomycete effector proteins. Curr. Opin. Microbiol. 2012, 15, 685–15691. [Google Scholar] [CrossRef] [Green Version]
- Orsomando, G.; Lorenzi, M.; Ferrari, E.; de Chiara, C.; Spisni, A.; Ruggieri, S. PcF protein from Phytophthora cactorum and its recombinant homologue elicit phenylalanine ammonia lyase activation in tomato. Cell. Mol. Life Sci. 2003, 60, 1470–1476. [Google Scholar] [CrossRef]
- Liu, Z.; Bos, J.I.; Armstrong, M.; Whisson, S.C.; da Cunha, L.; Torto-Alalibo, T.; Win, J.; Avrova, A.O.; Wright, F.; Birch, P.R.; et al. Patterns of diversifying selection in the phytotoxin-like scr74 gene family of Phytophthora infestans. Mol. Biol. Evol. 2005, 22, 659–672. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Jin, J.H.; Sheng, G.L.; Xing, Y.P.; Liu, W.; Zhou, X.; Liu, Y.Q.; Chen, X.R. A small cysteine-rich phytotoxic protein of Phytophthora capsici functions as both plant defense elicitor and virulence factor. Mol. Plant Microbe Interact. 2021, 34, 891–903. [Google Scholar] [CrossRef]
- Lin, X.; Armstrong, M.; Baker, K.; Wouters, D.; Visser, R.G.; Wolters, P.J.; Hein, I.; Vleeshouwers, V.G. RLP/K enrichment sequencing: A novel method to identify receptor-like protein (RLP) and receptor-like kinase (RLK) genes. New Phytol. 2020, 227, 1264–1276. [Google Scholar] [CrossRef] [Green Version]
- Coll, N.S.; Vercammen, D.; Smidler, A.; Clover, C.; Van Breusegem, F.; Dangl, J.L.; Epple, P. Arabidopsis type I metacaspases control cell death. Science 2010, 330, 1393–1397. [Google Scholar] [CrossRef]
- Wang, N.; Yin, Z.; Duan, W.; Zhang, X.; Pi, L.; Zhang, Y.; Dou, D. sORF-encoded polypeptide SEP1 is a novel virulence factor of Phytophthora pathogens. Mol. Plant Microbe Interact. 2021, 34, 157–167. [Google Scholar] [CrossRef]
- Lorang, J.; Kidarsa, T.; Bradford, C.S.; Gilbert, B.; Curtis, M.; Tzeng, S.C.; Maier, C.S.; Wolpert, T.J. Tricking the guard: Exploiting plant defense for disease susceptibility. Science 2012, 338, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Abu-Jamous, B.; Kelly, S. Clust: Automatic extraction of optimal co-expressed gene clusters from gene expression data. Genome Biol. 2018, 19, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant-Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadassery, J.; Oelmüller, R. Calcium signaling in pathogenic and beneficial plant microbe interactions: What can we learn from the interaction between Piriformospora indica and Arabidopsis thaliana. Plant Signal. Behav. 2009, 4, 1024–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Kissoudis, C.; Yan, Z.; Visser, R.G.F.; van der Linden, G. Plant behaviour under combined stress: Tomato responses to combined salinity and pathogen stress. Plant J. 2018, 93, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Wang, S.; De Rond, L.; Bertolin, N.; Wouters, R.H.; Wouters, D.; Domazakis, E.; Bitew, M.K.; Win, J.; Dong, S.; et al. Divergent evolution of PcF/SCR74 effectors in oomycetes is associated with distinct recognition patterns in solanaceous plants. mBio 2020, 11, e00947-20. [Google Scholar] [CrossRef]
- van Loon, L.C.; Rep, M.; Pieterse, C.M. Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 2006, 44, 135–162. [Google Scholar] [CrossRef] [Green Version]
- Theodoulou, F.L.; Kerr, I.D. ABC transporter research: Going strong 40 years on. Biochem. Soc. Trans. 2015, 43, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Fass, D. Disulfide bonding in protein biophysics. Annu. Rev. Biophys. 2012, 41, 63–79. [Google Scholar] [CrossRef]
- Orsomando, G.; Brunetti, L.; Pucci, K.; Ruggeri, B.; Ruggieri, S. Comparative structural and functional characterization of putative protein effectors belonging to the PcF toxin family from Phytophthora spp. Protein Sci. 2011, 20, 2047–2059. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Huang, S.X.; Tao, H.; Zhang, Y.; Zhao, Y.; Ji, Z.L.; Chen, X.R. Gene transcriptional pattern, prokaryotic expression and functional analysis of an apoplastic, hydrophobic and small effector SCR82 from Phytophthora capsici. Acta Microbiol. Sin. 2019, 59, 1586–1599. [Google Scholar]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.R.; Huang, S.X.; Zhang, Y.; Sheng, G.L.; Zhang, B.Y.; Li, Q.Y.; Zhu, F.; Xu, J.Y. Transcription profiling and identification of infection-related genes in Phytophthora cactorum. Mol. Genet. Genom. 2018, 293, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Song, T.; Zhu, L.; Ye, W.; Wang, Y.; Shao, Y.; Dong, S.; Zhang, Z.; Dou, D.; Zheng, X.; et al. A Phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP. Plant Cell 2015, 27, 2057–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Zhu, L.; Song, T.; Wang, Y.; Zhang, Q.; Xia, Y.; Qiu, M.; Lin, Y.; Li, H.; Kong, L.; et al. A paralogous decoy protects Phytophthora sojae apoplastic effector PsXEG1 from a host inhibitor. Science 2017, 355, 710–714. [Google Scholar] [CrossRef]
- Wang, Y.; Tyler, B.M.; Wang, Y. Defense and counterdefense during plant-pathogenic oomycete infection. Annu. Rev. Microbiol. 2019, 73, 667–696. [Google Scholar] [CrossRef]
- Naveed, Z.A.; Wei, X.; Chen, J.; Mubeen, H.; Ali, G.S. The PTI to ETI continuum in Phytophthora-plant interactions. Front. Plant Sci. 2020, 11, 593905. [Google Scholar] [CrossRef]
- Ngou, B.P.M.; Ahn, H.K.; Ding, P.; Jones, J.D.G. Mutual potentiation of plant immunity by cell-surface and intracellular receptors. Nature 2021, 592, 110–115. [Google Scholar] [CrossRef]
- Yuan, M.; Jiang, Z.; Bi, G.; Nomura, K.; Liu, M.; Wang, Y.; Cai, B.; Zhou, J.M.; He, S.Y.; Xin, X.F. Pattern-recognition receptors are required for NLR-mediated plant immunity. Nature 2021, 592, 105–109. [Google Scholar] [CrossRef]
- Stergiopoulos, I.; de Wit, P.J.G.M. Fungal effector proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef] [Green Version]
- Amorim, L.L.B.; da Fonseca Dos Santos, R.; Neto, J.P.B.; Guida-Santos, M.; Crovella, S.; Benko-Iseppon, A.M. Transcription factors involved in plant resistance to pathogens. Curr. Protein Pept. Sci. 2017, 18, 335–351. [Google Scholar] [CrossRef]
- Ng, D.W.; Abeysinghe, J.K.; Kamali, M. Regulating the regulators: The control of transcription factors in plant defense signaling. Int. J. Mol. Sci. 2018, 19, 3737. [Google Scholar] [CrossRef] [Green Version]
- Livaja, M.; Zeidler, D.; von Rad, U.; Durner, J. Transcriptional responses of Arabidopsis thaliana to the bacteria-derived PAMPs harpin and lipopolysaccharide. Immunobiology 2008, 213, 161–171. [Google Scholar] [CrossRef]
- Meng, Q.; Gupta, R.; Kwon, S.J.; Wang, Y.; Agrawal, G.K.; Rakwal, R.; Park, S.R.; Kim, S.T. Transcriptomic analysis of Oryza sativa leaves reveals key changes in response to Magnaporthe oryzae MSP1. Plant Pathol. J. 2018, 34, 257–268. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Meyerowitz, E.M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633–646. [Google Scholar]
- Jin, J.H.; Zhang, H.X.; Ali, M.; Wei, A.M.; Luo, D.X.; Gong, Z.H. The CaAP2/ERF064 regulates dual functions in pepper: Plant cell death and resistance to Phytophthora capsici. Genes 2019, 10, 541. [Google Scholar] [CrossRef] [Green Version]
- Gutterson, N.; Reuber, T.L. Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr. Opin. Plant Biol. 2004, 7, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.H.; Wang, M.; Zhang, H.X.; Khan, A.; Wei, A.M.; Luo, D.X.; Gong, Z.H. Genome-wide identification of the AP2/ERF transcription factor family in pepper (Capsicum annuum L.). Genome 2018, 61, 663–674. [Google Scholar] [CrossRef]
- Benny, J.; Pisciotta, A.; Caruso, T.; Martinelli, F. Identification of key genes and its chromosome regions linked to drought responses in leaves across different crops through meta-analysis of RNA-Seq data. BMC Plant Biol. 2019, 19, 194. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Long, L.; Xu, L.; Lindsey, K.; Zhang, X.; Zhu, L. Suppression of the homeobox gene HDTF1 enhances resistance to Verticillium dahliae and Botrytis cinerea in cotton. J. Integr. Plant Biol. 2016, 58, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Wang, L.; Zhou, X.; Ren, X.; Dai, X.; Liu, H. PopW activates PAMP-triggered immunity in controlling tomato bacterial spot disease. Biochem. Biophys. Res. Commun. 2015, 463, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Lamour, K.H.; Stam, R.; Jupe, J.; Huitema, E. The oomycete broad-host-range pathogen Phytophthora capsici. Mol. Plant Pathol. 2012, 13, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Letchworth, G.J. High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. Biotechniques 2004, 36, 152–154. [Google Scholar] [CrossRef]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641.
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. Package “gplots”: Various R Programming Tools for Plotting Data; R Package Version 2.17.0; ScienceOpen: Berlin, Germany, 2016. [Google Scholar]
- Chen, X.R.; Xing, Y.P.; Li, Y.P.; Tong, Y.H.; Xu, J.Y. RNA-seq reveals infection-related gene expression changes in Phytophthora capsici. PLoS ONE 2013, 8, e74588. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Kim, J.G.; Li, X.; Roden, J.A.; Taylor, K.W.; Aakre, C.D.; Su, B.; Lalonde, S.; Kirik, A.; Chen, Y.; Baranage, G.; et al. Xanthomonas T3S effector XopN suppresses PAMP-triggered immunity and interacts with a tomato atypical receptor-like kinase and TFT1. Plant Cell 2009, 21, 1305–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | GO Term | Functional Description | False Discovery Rate (FDR) a | ||
|---|---|---|---|---|---|
| H3 vs. H0 | H6 vs. H0 | H12 vs. H0 | |||
| Biological processes | |||||
| GO:0006952 | Defense response | 0.0005 | 2.84 × 10−5 | 0.0008 | |
| GO:0051179 | Localization | 0.017 | 0.010 | 0.007 | |
| GO:0009607 | Response to biotic stimulus | 0.061 | 0.044 | 0.027 | |
| GO:0060154 | Cellular process regulating host cell cycle in response to virus | 0.036 | 0.031 | ― | |
| GO:0002376 | Immune system process | 0.036 | 0.029 | 0.029 | |
| GO:0055114 | Oxidation–reduction process | 0.078 | 0.043 | 0.022 | |
| GO:0008152 | Metabolic process | 0.026 | 0.042 | 0.026 | |
| Cellular component | |||||
| GO:0016020 | Membrane | 0.0002 | 7.71 × 10−5 | 0.0003 | |
| GO:0005576 | Extracellular region | 0.099 | 0.099 | 0.088 | |
| GO:0043226 | Organelle | 0.035 | 0.031 | ― | |
| GO:0043234 | Protein-containing complex | 0.047 | 0.034 | 0.091 | |
| GO:0030054 | Cell junction | 0.019 | 0.017 | 0.015 | |
| Molecular function | |||||
| GO:0000989 | Transcription factor binding | 0.063 | 0.028 | 0.066 | |
| GO:0005102 | Signaling receptor binding | 0.059 | 0.053 | 0.049 | |
| GO:0030674 | Protein binding and bridging | 0.044 | ― | 0.058 | |
| GO:0001664 | G-protein-coupled receptor binding | 0.005 | 0.034 | 0.014 | |
| GO:0003676 | Nucleic acid binding | 0.013 | 0.077 | 0.0091 | |
| GO:0003824 | Catalytic activity | 0.027 | 0.07 | 0.02 | |
| GO:0005215 | Transporter activity | 0.039 | 0.043 | 0.032 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Wen, K.; Huang, S.-X.; Lu, Y.; Liu, Y.; Jin, J.-H.; Kale, S.D.; Chen, X.-R. Time-Course Transcriptome Profiling Reveals Differential Resistance Responses of Tomato to a Phytotoxic Effector of the Pathogenic Oomycete Phytophthora cactorum. Plants 2023, 12, 883. https://doi.org/10.3390/plants12040883
Zhou X, Wen K, Huang S-X, Lu Y, Liu Y, Jin J-H, Kale SD, Chen X-R. Time-Course Transcriptome Profiling Reveals Differential Resistance Responses of Tomato to a Phytotoxic Effector of the Pathogenic Oomycete Phytophthora cactorum. Plants. 2023; 12(4):883. https://doi.org/10.3390/plants12040883
Chicago/Turabian StyleZhou, Xue, Ke Wen, Shen-Xin Huang, Yi Lu, Yang Liu, Jing-Hao Jin, Shiv D. Kale, and Xiao-Ren Chen. 2023. "Time-Course Transcriptome Profiling Reveals Differential Resistance Responses of Tomato to a Phytotoxic Effector of the Pathogenic Oomycete Phytophthora cactorum" Plants 12, no. 4: 883. https://doi.org/10.3390/plants12040883