
Genome-Wide Analyses of MADS-Box Genes in Humulus lupulus L. Reveal Potential Participation in Plant Development, Floral Architecture, and Lupulin Gland Metabolism

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. MADS-Box Genes Encoded in the Hop Genome and Gene Ontology Annotation
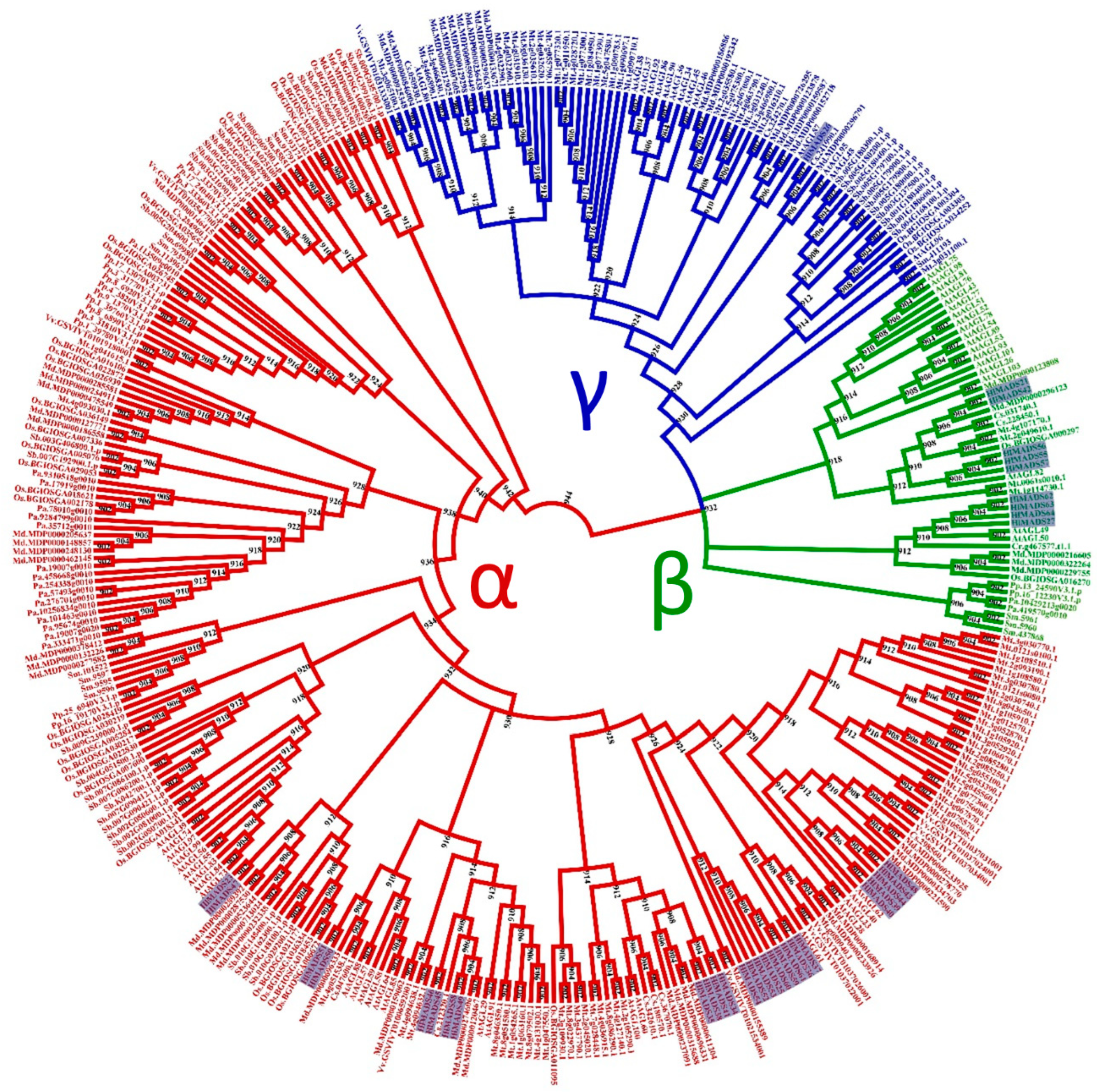
2.2. Phylogenetic Analyses Revealed Clades and Potential Functions of Hop MADS-Box Genes
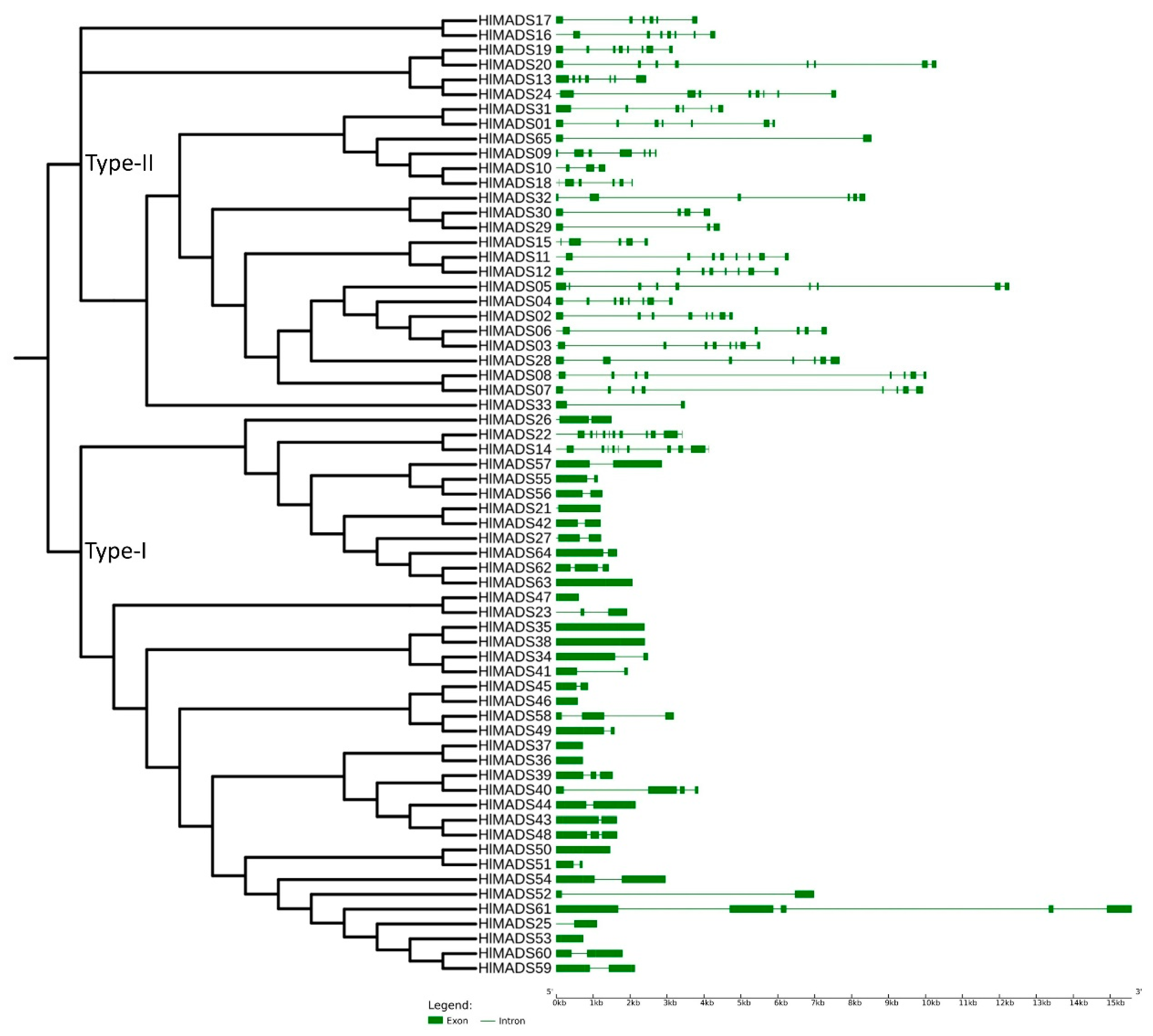
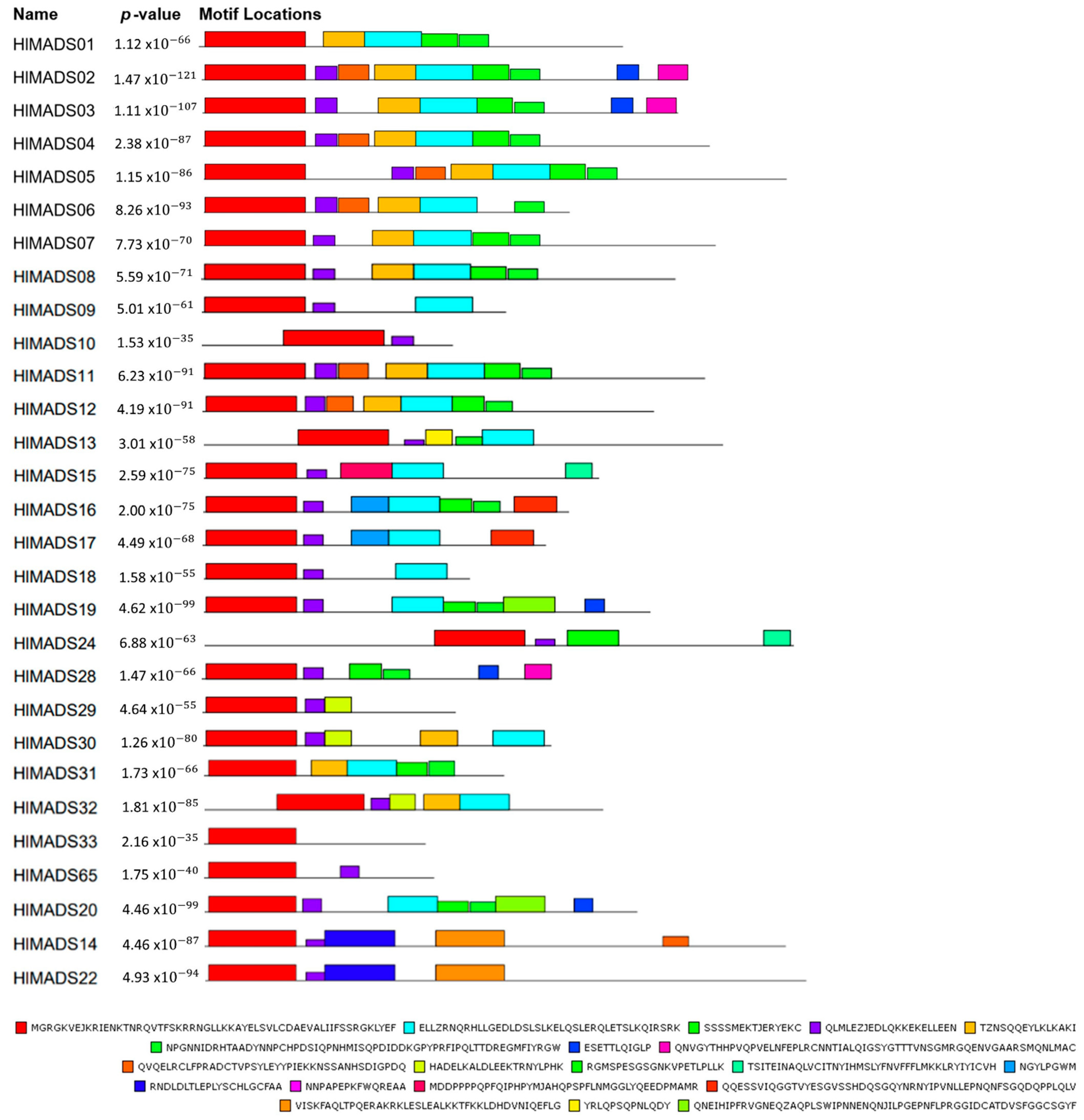
2.3. Structural and Motif Analyses of Hop MADS-Box Genes
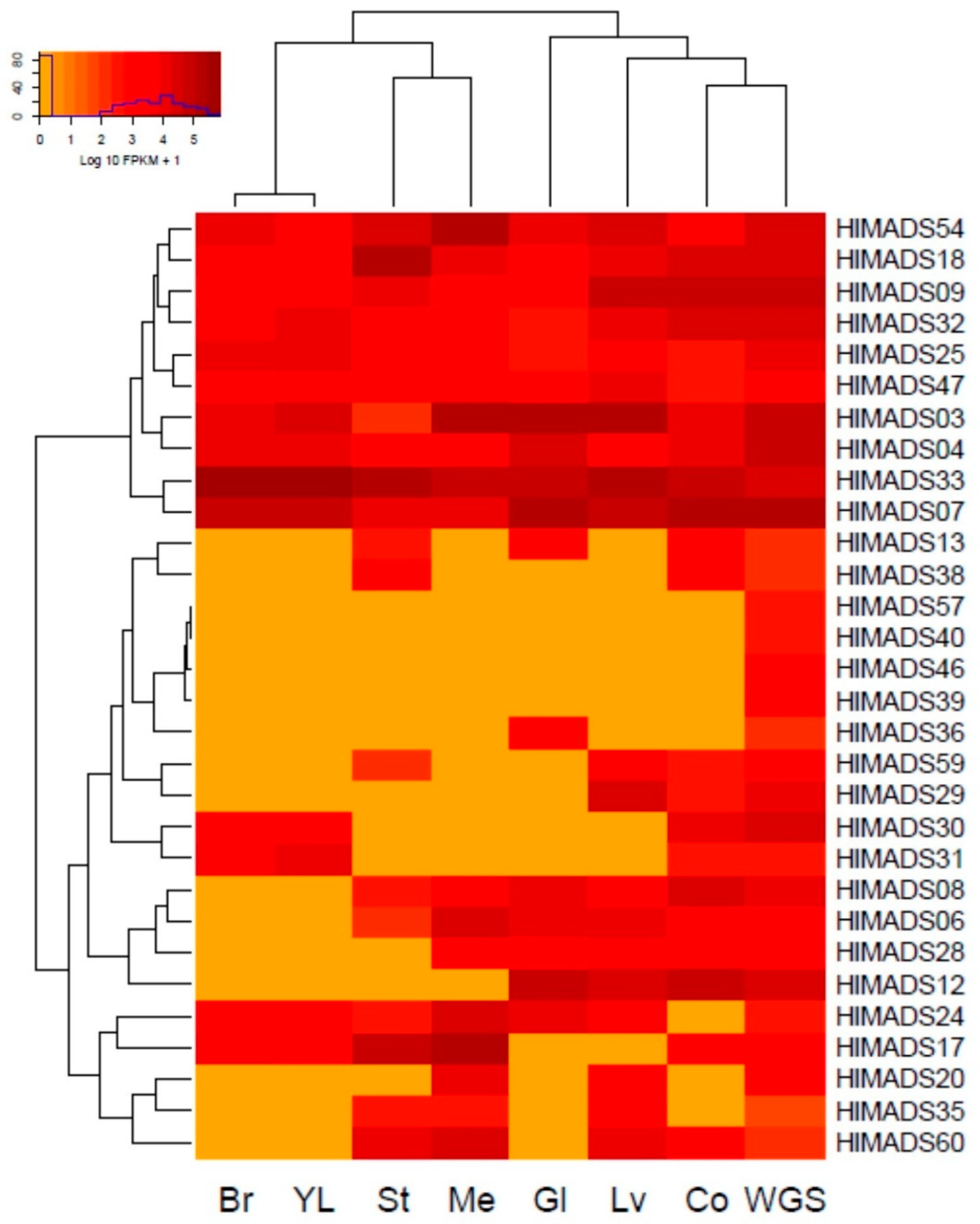
2.4. Transcriptional Profile of Hop MADS-Box Genes in Different Tissues
3. Discussion
4. Materials and Methods
4.1. Gene Prediction
4.2. Identification of MADS-Box Genes in the Hop Genome
4.3. Phylogenetic Analysis
4.4. MADS-Box Gene Structure and Conserved Protein Motif Analyses in Hop
4.5. Gene Ontology (GO) Annotation of the Hop MADS-Box Genes
4.6. Expression Analysis of MADS-Box Genes in Hop Tissues
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riechmann, J.L.; Krizek, B.A.; Meyerowitz, E.M. Dimerization Specificity of Arabidopsis MADS Domain Homeotic Proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. USA 1996, 93, 4793–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; Theißen, G. The Major Clades of MADS-Box Genes and Their Role in the Development and Evolution of Flowering Plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.H.; Angenent, G.C.; Kaufmann, K. Developmental and Evolutionary Diversity of Plant MADS-Domain Factors: Insights from Recent Studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daminato, M.; Masiero, S.; Resentini, F.; Lovisetto, A.; Casadoro, G. Characterization of TM8, a MADS-Box Gene Expressed in Tomato Flowers. BMC Plant Biol. 2014, 14, 319. [Google Scholar] [CrossRef] [Green Version]
- da Silveira Falavigna, V.; Severing, E.; Lai, X.; Estevan, J.; Farrera, I.; Hugouvieux, V.; Revers, L.F.; Zubieta, C.; Coupland, G.; Costes, E.; et al. Unraveling the Role of MADS Transcription Factor Complexes in Apple Tree Dormancy. New Phytol. 2021, 232, 2071–2088. [Google Scholar] [CrossRef]
- Li, G.; Kuijer, H.N.J.; Yang, X.; Liu, H.; Shen, C.; Shi, J.; Betts, N.; Tucker, M.R.; Liang, W.; Waugh, R.; et al. MADS1 Maintains Barley Spike Morphology at High Ambient Temperatures. Nat. Plants 2021, 7, 1093–1107. [Google Scholar] [CrossRef]
- Gómez-Soto, D.; Ramos-Sánchez, J.M.; Alique, D.; Conde, D.; Triozzi, P.M.; Perales, M.; Allona, I. Overexpression of a SOC1-Related Gene Promotes Bud Break in Ecodormant Poplars. Front. Plant Sci. 2021, 12, 670497. [Google Scholar] [CrossRef]
- Ayra, L.; Reyero-Saavedra, M.d.R.; Isidra-Arellano, M.C.; Lozano, L.; Ramírez, M.; Leija, A.; Fuentes, S.-I.; Girard, L.; Valdés-López, O.; Hernández, G. Control of the Rhizobia Nitrogen-Fixing Symbiosis by Common Bean MADS-Domain/AGL Transcription Factors. Front. Plant Sci. 2021, 12, 1061. [Google Scholar] [CrossRef]
- Hyun, Y.; Richter, R.; Vincent, C.; Martinez-Gallegos, R.; Porri, A.; Coupland, G. Multi-Layered Regulation of SPL15 and Cooperation with SOC1 Integrate Endogenous Flowering Pathways at the Arabidopsis Shoot Meristem. Dev. Cell 2016, 37, 254–266. [Google Scholar] [CrossRef]
- Immink, R.G.H.; Posé, D.; Ferrario, S.; Ott, F.; Kaufmann, K.; Valentim, F.L.; de Folter, S.; van der Wal, F.; van Dijk, A.D.J.; Schmid, M.; et al. Characterization of SOC1′s Central Role in Flowering by the Identification of Its Upstream and Downstream Regulators. Plant Physiol. 2012, 160, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, I. Regulation and Function of SOC1, a Flowering Pathway Integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, H.; Er, H.L.; Soo, H.M.; Kumar, P.P.; Han, J.-H.; Liou, Y.C.; Yu, H. Direct Interaction of AGL24 and SOC1 Integrates Flowering Signals in Arabidopsis. Dev. Camb. Engl. 2008, 135, 1481–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.; Fornara, F. AGL24 Acts in Concert with SOC1 and FUL during Arabidopsis Floral Transition. Plant Signal. Behav. 2012, 7, 1251–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, U.; Höhmann, S.; Nettesheim, K.; Wisman, E.; Saedler, H.; Huijser, P. Molecular Cloning of SVP: A Negative Regulator of the Floral Transition in Arabidopsis. Plant J. 2000, 21, 351–360. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. Flowering Locus C Encodes a Novel MADS Domain Protein That Acts as a Repressor of Flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Mateos, J.L.; Madrigal, P.; Tsuda, K.; Rawat, V.; Richter, R.; Romera-Branchat, M.; Fornara, F.; Schneeberger, K.; Krajewski, P.; Coupland, G. Combinatorial Activities of Short Vegetative Phase and Flowering Locus C Define Distinct Modes of Flowering Regulation in Arabidopsis. Genome Biol. 2015, 16, 31. [Google Scholar] [CrossRef] [Green Version]
- Madrid, E.; Chandler, J.W.; Coupland, G. Gene Regulatory Networks Controlled by FLOWERING LOCUS C That Confer Variation in Seasonal Flowering and Life History. J. Exp. Bot. 2021, 72, 4–14. [Google Scholar] [CrossRef]
- Kim, D.-H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the Timing of Flowering in Plants. Annu. Rev. Cell Dev. Biol. 2009, 25, 277–299. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.A.; He, Y.; Schmitz, R.J.; Amasino, R.M.; Panella, L.W.; Richards, C.M. Evolutionary Conservation of the FLOWERING LOCUS C-Mediated Vernalization Response: Evidence From the Sugar Beet (Beta Vulgaris). Genetics 2007, 176, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Ruelens, P.; de Maagd, R.A.; Proost, S.; Theißen, G.; Geuten, K.; Kaufmann, K. Flowering Locus C in Monocots and the Tandem Origin of Angiosperm-Specific MADS-Box Genes. Nat. Commun. 2013, 4, 2280. [Google Scholar] [CrossRef] [Green Version]
- Kagaya, H.; Ito, N.; Shibuya, T.; Komori, S.; Kato, K.; Kanayama, Y. Characterization of FLOWERING LOCUS C Homologs in Apple as a Model for Fruit Trees. Int. J. Mol. Sci. 2020, 21, 4562. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.R.; Cesarino, I.; Mazzafera, P.; Dornelas, M.C. Flower Development in Coffea arabica L.: New Insights into MADS-Box Genes. Plant Reprod. 2014, 27, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, A.; Lloyd, A.; Hepworth, J.; Tudor, E.H.; Jones, D.M.; Woodhouse, S.; Bilham, L.; Chinoy, C.; Williams, K.; Corke, F.; et al. Total FLC Transcript Dynamics from Divergent Paralogue Expression Explains Flowering Diversity in Brassica Napus. New Phytol. 2021, 229, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Geuten, K. The Role of Flowering Locus C Relatives in Cereals. Front. Plant Sci. 2020, 11, 617340. [Google Scholar] [CrossRef]
- Sharma, N.; Ruelens, P.; D’hauw, M.; Maggen, T.; Dochy, N.; Torfs, S.; Kaufmann, K.; Rohde, A.; Geuten, K. A Flowering Locus C Homolog Is a Vernalization-Regulated Repressor in Brachypodium and Is Cold Regulated in Wheat. Plant Physiol. 2017, 173, 1301–1315. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; dePamphilis, C. The ABCs of Floral Evolution. Cell 2000, 101, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. Genes Directing Flower Development in Arabidopsis. Plant Cell 1989, 1, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Benedito, V.A.; Visser, P.B.; van Tuyl, J.M.; Angenent, G.C.; de Vries, S.C.; Krens, F.A. Ectopic Expression of LLAG1, an Agamous Homologue from Lily (Lilium longiflorum Thunb.) Causes Floral Homeotic Modifications in Arabidopsis. J. Exp. Bot. 2004, 55, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Thomson, B.; Wellmer, F. Chapter Eight—Molecular Regulation of Flower Development. In Current Topics in Developmental Biology; Grossniklaus, U., Ed.; Plant Development and Evolution; Academic Press: Cambridge, MA, USA, 2019; Volume 131, pp. 185–210. [Google Scholar]
- Coen, E.S.; Meyerowitz, E.M. The War of the Whorls: Genetic Interactions Controlling Flower Development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef]
- Theißen, G.; Saedler, H. Floral Quartets. Nature 2001, 409, 469–471. [Google Scholar] [CrossRef]
- Irish, V.F.; Sussex, I.M. Function of the Apetala-1 Gene during Arabidopsis Floral Development. Plant Cell 1990, 2, 741–753. [Google Scholar] [PubMed] [Green Version]
- Goto, K.; Meyerowitz, E.M. Function and Regulation of the Arabidopsis Floral Homeotic Gene Pistillata. Genes Dev. 1994, 8, 1548–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, T.; Brockman, L.L.; Meyerowitz, E.M. The Homeotic Gene APETALA3 of Arabidopsis Thaliana Encodes a MADS Box and Is Expressed in Petals and Stamens. Cell 1992, 68, 683–697. [Google Scholar] [CrossRef]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The Protein Encoded by the Arabidopsis Homeotic Gene Agamous Resembles Transcription Factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef]
- Savidge, B.; Rounsley, S.D.; Yanofsky, M.F. Temporal Relationship between the Transcription of Two Arabidopsis MADS Box Genes and the Floral Organ Identity Genes. Plant Cell 1995, 7, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C Floral Organ Identity Functions Require SEPALLATA MADS-Box Genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Kyozuka, J.; Kobayashi, T.; Morita, M.; Shimamoto, K. Spatially and Temporally Regulated Expression of Rice MADS Box Genes with Similarity to Arabidopsis Class A, B and C Genes. Plant Cell Physiol. 2000, 41, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Shi, X.; Lin, X.; Liu, Y.; Chong, K.; Theißen, G.; Meng, Z. The ABCs of Flower Development: Mutational Analysis of AP1/FUL-like Genes in Rice Provides Evidence for a Homeotic (A)-Function in Grasses. Plant J. 2017, 89, 310–324. [Google Scholar] [CrossRef]
- Kuijer, H.N.J.; Shirley, N.J.; Khor, S.F.; Shi, J.; Schwerdt, J.; Zhang, D.; Li, G.; Burton, R.A. Transcript Profiling of MIKCc MADS-Box Genes Reveals Conserved and Novel Roles in Barley Inflorescence Development. Front. Plant Sci. 2021, 12, 1834. [Google Scholar] [CrossRef]
- Chi, Y.; Huang, F.; Liu, H.; Yang, S.; Yu, D. An APETALA1-like Gene of Soybean Regulates Flowering Time and Specifies Floral Organs. J. Plant Physiol. 2011, 168, 2251–2259. [Google Scholar] [CrossRef]
- Huang, F.; Xu, G.; Chi, Y.; Liu, H.; Xue, Q.; Zhao, T.; Gai, J.; Yu, D. A Soybean MADS-Box Protein Modulates Floral Organ Numbers, Petal Identity and Sterility. BMC Plant Biol. 2014, 14, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Clemens, J.; Jameson, P.E. Expression of Floral Identity Genes in Clianthus Maximus during Mass Inflorescence Abortion and Floral Development. Ann. Bot. 2011, 107, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The Emerging Importance of Type I MADS Box Transcription Factors for Plant Reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauerle, W.L. Disentangling Photoperiod from Hop Vernalization and Dormancy for Global Production and Speed Breeding. Sci. Rep. 2019, 9, 16003. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.G.; Schwabe, W.W. Factors Controlling Flowering in the Hop (Humulus lupulus L.). Ann. Bot. 1969, 33, 781–793. [Google Scholar] [CrossRef]
- Benkherouf, A.Y.; Logrén, N.; Somborac, T.; Kortesniemi, M.; Soini, S.L.; Yang, B.; Salo-Ahen, O.M.H.; Laaksonen, O.; Uusi-Oukari, M. Hops Compounds Modulatory Effects and 6-Prenylnaringenin Dual Mode of Action on GABAA Receptors. Eur. J. Pharmacol. 2020, 873, 172962. [Google Scholar] [CrossRef]
- Jiang, C.-H.; Sun, T.-L.; Xiang, D.-X.; Wei, S.-S.; Li, W.-Q. Anticancer Activity and Mechanism of Xanthohumol: A Prenylated Flavonoid From Hops (Humulus lupulus L.). Front. Pharmacol. 2018, 9, 530. [Google Scholar] [CrossRef]
- Yamashita, M.; Fukizawa, S.; Nonaka, Y. Hop-Derived Prenylflavonoid Isoxanthohumol Suppresses Insulin Resistance by Changing the Intestinal Microbiota and Suppressing Chronic Inflammation in High Fat Diet-Fed Mice. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1537–1547. [Google Scholar] [CrossRef]
- Parenicová, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and Phylogenetic Analyses of the Complete MADS-Box Transcription Factor Family in Arabidopsis: New Openings to the MADS World. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-Box Gene Family in Rice: Genome-Wide Identification, Organization and Expression Profiling during Reproductive Development and Stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Grimplet, J.; Martínez-Zapater, J.M.; Carmona, M.J. Structural and Functional Annotation of the MADS-Box Transcription Factor Family in Grapevine. BMC Genom. 2016, 17, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, D.; Cao, Y.; Chen, T.; Abdullah, M.; Jin, Q.; Fan, H.; Lin, Y.; Cai, Y. Evolution and Functional Divergence of MADS-Box Genes in Pyrus. Sci. Rep. 2019, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.T.; Sudarsanam, R.; Henning, J.; Hendrix, D. HopBase: A Unified Resource for Humulus Genomics. Database 2017, 2017, bax009. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.-H.; Ju, Y.; Seo, P.J.; Lee, J.-H.; Park, C.-M. The SOC1-SPL Module Integrates Photoperiod and Gibberellic Acid Signals to Control Flowering Time in Arabidopsis. Plant J. 2012, 69, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Hamilton, M.; Reddy, A.S.N.; Ben-Hur, A. Exploring the Relationship between Intron Retention and Chromatin Accessibility in Plants. BMC Genom. 2018, 19, 21. [Google Scholar] [CrossRef] [Green Version]
- Adamczyk, B.J.; Lehti-Shiu, M.D.; Fernandez, D.E. The MADS Domain Factors AGL15 and AGL18 Act Redundantly as Repressors of the Floral Transition in Arabidopsis. Plant J. Cell Mol. Biol. 2007, 50, 1007–1019. [Google Scholar] [CrossRef]
- Serivichyaswat, P.; Ryu, H.-S.; Kim, W.; Kim, S.; Chung, K.S.; Kim, J.J.; Ahn, J.H. Expression of the Floral Repressor MiRNA156 Is Positively Regulated by the AGAMOUS-like Proteins AGL15 and AGL18. Mol. Cells 2015, 38, 259–266. [Google Scholar] [CrossRef]
- Gustafson-Brown, C.; Savidge, B.; Yanofsky, M.F. Regulation of the Arabidopsis Floral Homeotic Gene APETALA1. Cell 1994, 76, 131–143. [Google Scholar] [CrossRef]
- Shephard, H.L.; Parker, J.S.; Darby, P.; Ainsworth, C.C. Sexual Development and Sex Chromosomes in Hop. New Phytol. 2000, 148, 397–411. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Feng, C.; Liu, M.; Wang, J.; Hu, Y. Genome-Wide Identification, Characterization of the MADS-Box Gene Family in Chinese Jujube and Their Involvement in Flower Development. Sci. Rep. 2017, 7, 1025. [Google Scholar] [CrossRef]
- Okada, Y.; Ito, K. Cloning and Analysis of Valerophenone Synthase Gene Expressed Specifically in Lupulin Gland of Hop (Humulus lupulus L.). Biosci. Biotechnol. Biochem. 2001, 65, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using Native and Syntenically Mapped CDNA Alignments to Improve de Novo Gene Finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinform. 2019, 65, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis Genome Annotation Using Maximal Transcript Alignment Assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A Portal for the Functional and Evolutionary Study of Plant Transcription Factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate Detection of Unreliable Alignment Regions Accounting for the Uncertainty of Multiple Parameters. Nucleic Acids Res. 2015, 43, W7–W14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A Web Tool for Analyzing and Plotting GO Annotations, 2018 Update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. bioRxiv 2014. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Márquez Gutiérrez, R.; Cherubino Ribeiro, T.H.; de Oliveira, R.R.; Benedito, V.A.; Chalfun-Junior, A. Genome-Wide Analyses of MADS-Box Genes in Humulus lupulus L. Reveal Potential Participation in Plant Development, Floral Architecture, and Lupulin Gland Metabolism. Plants 2022, 11, 1237. https://doi.org/10.3390/plants11091237
Márquez Gutiérrez R, Cherubino Ribeiro TH, de Oliveira RR, Benedito VA, Chalfun-Junior A. Genome-Wide Analyses of MADS-Box Genes in Humulus lupulus L. Reveal Potential Participation in Plant Development, Floral Architecture, and Lupulin Gland Metabolism. Plants. 2022; 11(9):1237. https://doi.org/10.3390/plants11091237
Chicago/Turabian StyleMárquez Gutiérrez, Robert, Thales Henrique Cherubino Ribeiro, Raphael Ricon de Oliveira, Vagner Augusto Benedito, and Antonio Chalfun-Junior. 2022. "Genome-Wide Analyses of MADS-Box Genes in Humulus lupulus L. Reveal Potential Participation in Plant Development, Floral Architecture, and Lupulin Gland Metabolism" Plants 11, no. 9: 1237. https://doi.org/10.3390/plants11091237