
Transcriptional Memory in Taraxacum mongolicum in Response to Long-Term Different Grazing Intensities

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant materials and Treatments
2.2. RNA-Seq Library Preparation and ILLUMNA sequencing
2.3. Quality and De Novo Assembled
2.4. Functional Annotation and Identification of DEGs
2.5. RNA-Seq Result Validation by qRT-PCR
2.6. Statistical Analysis
3. Results
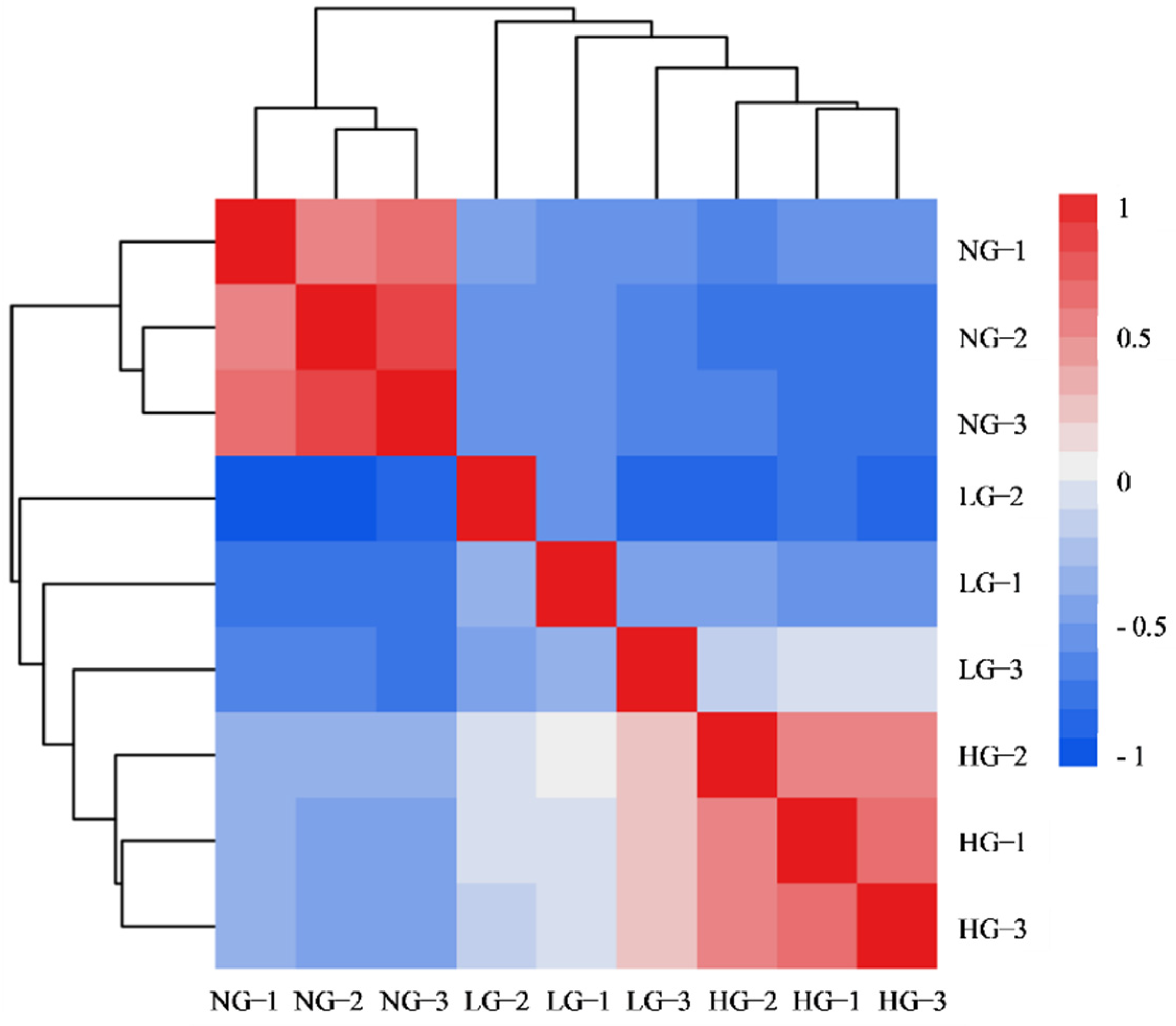
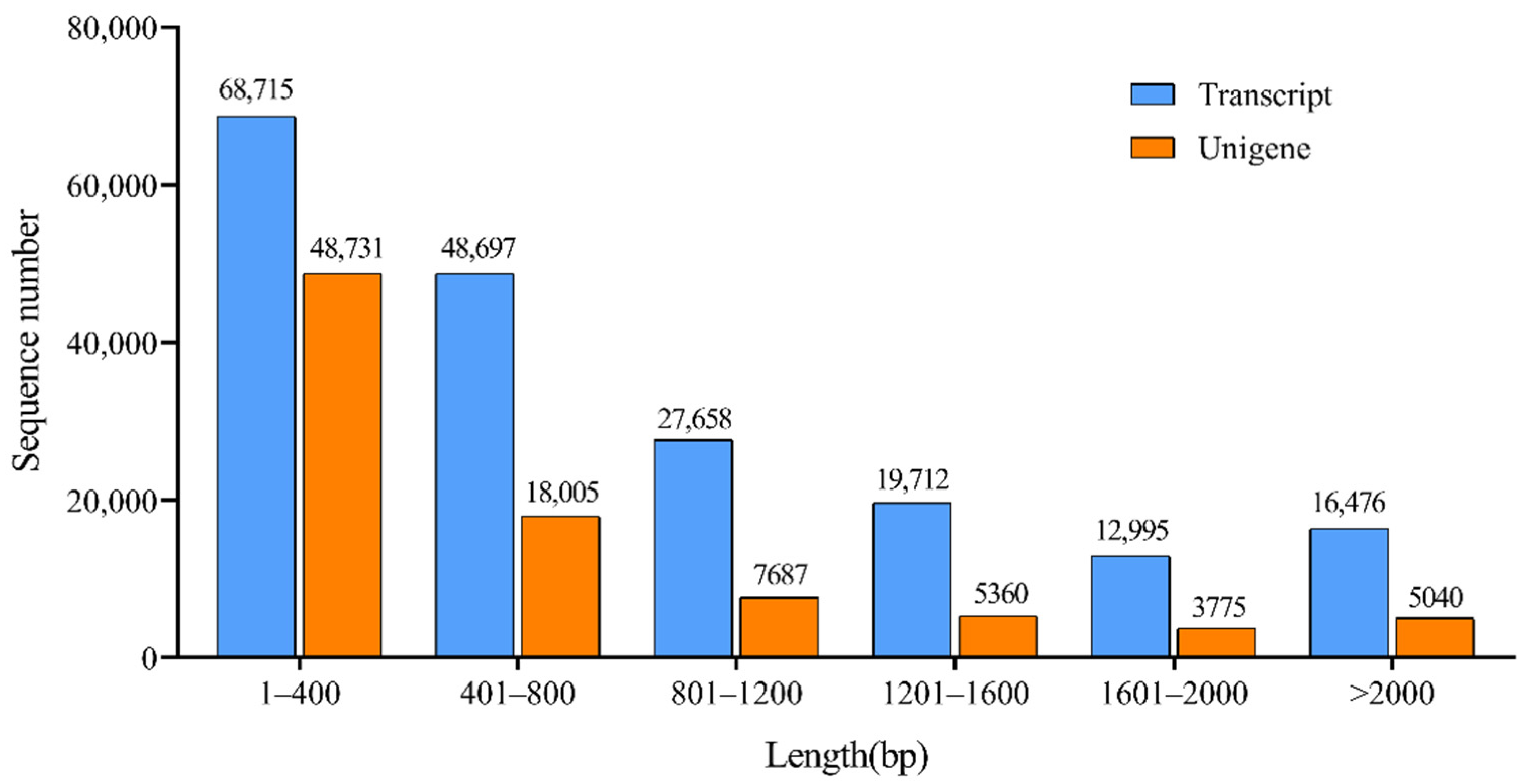
3.1. Illumina Sequencing and Reads Assembly
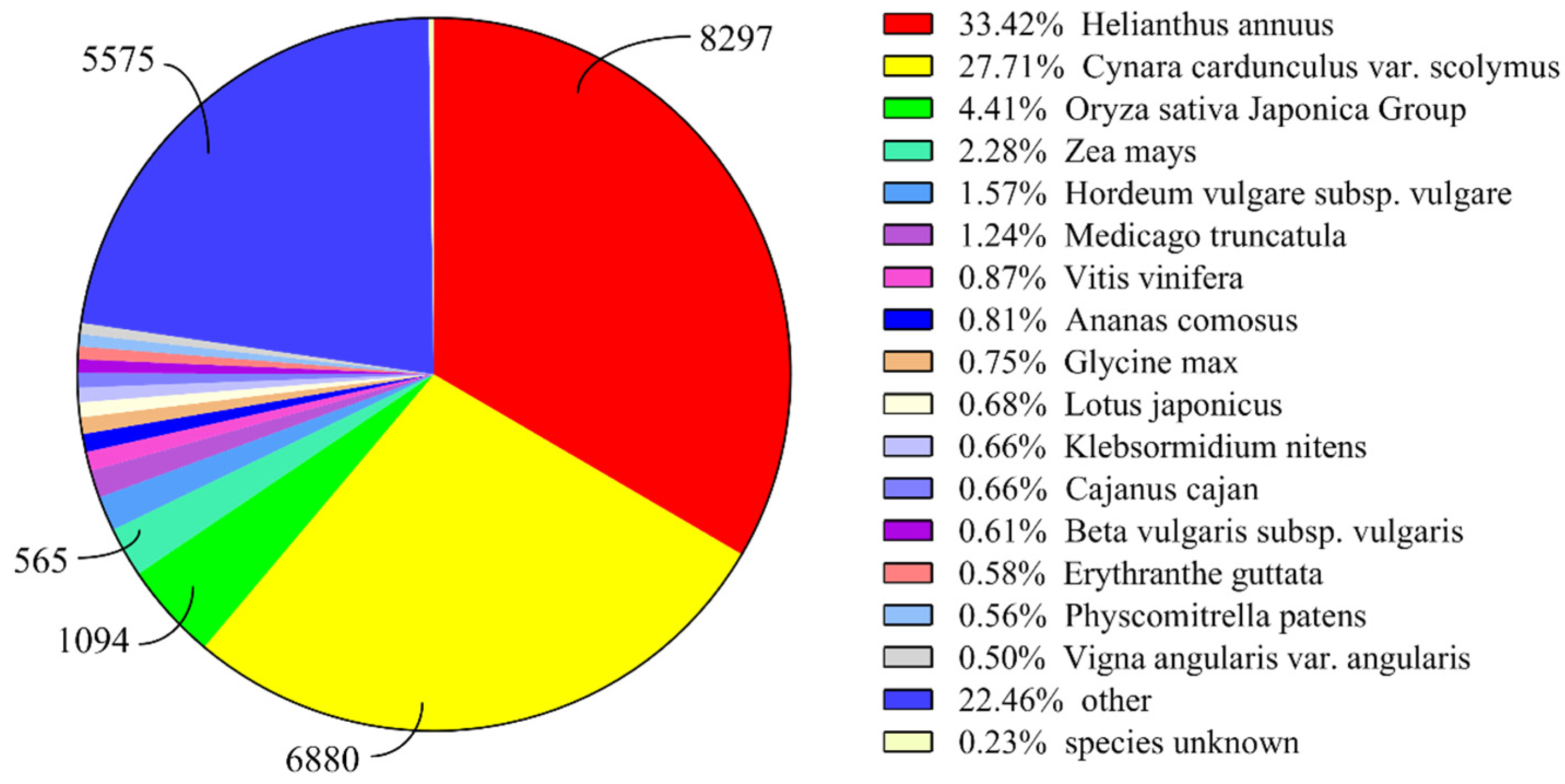
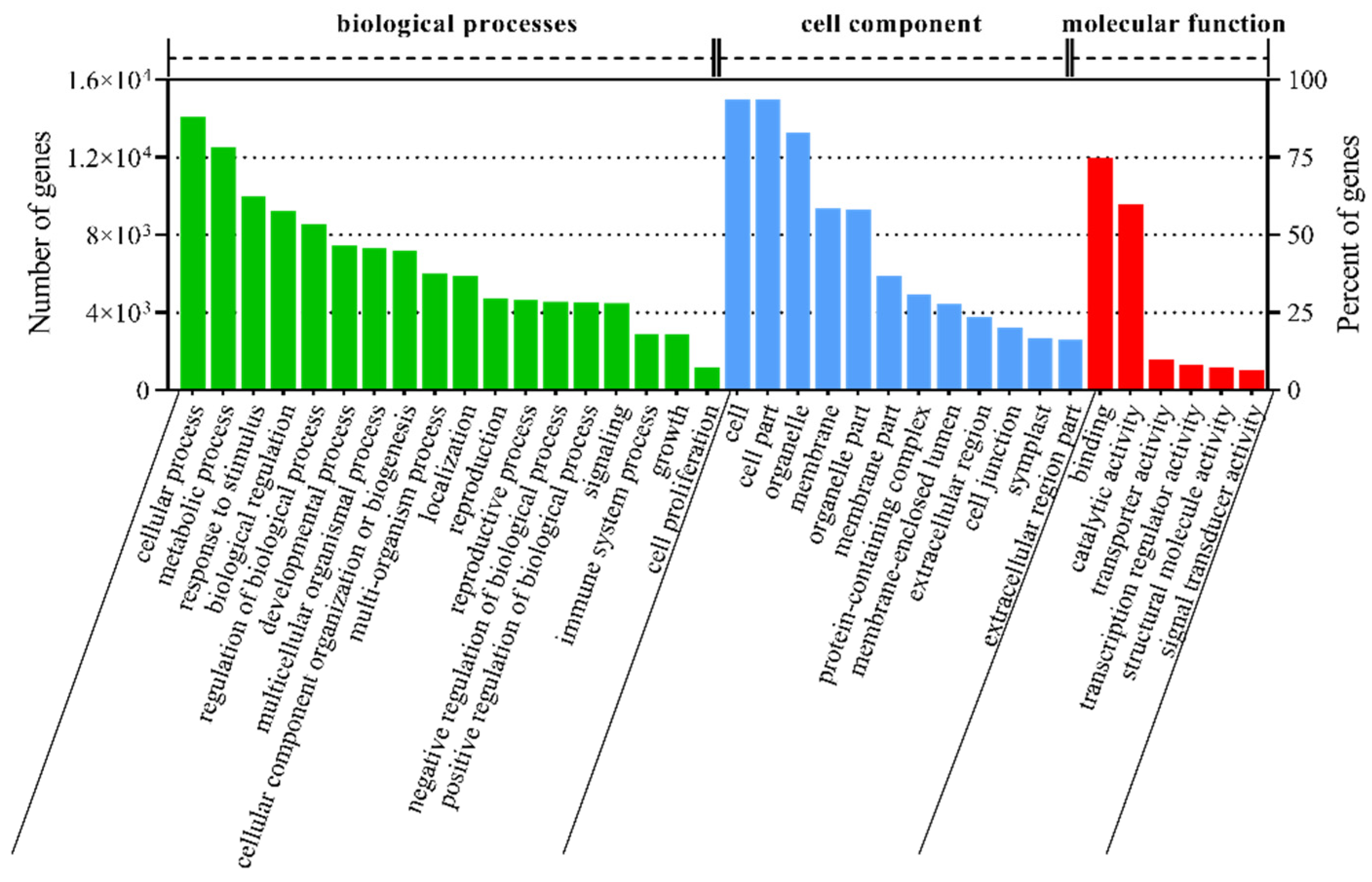
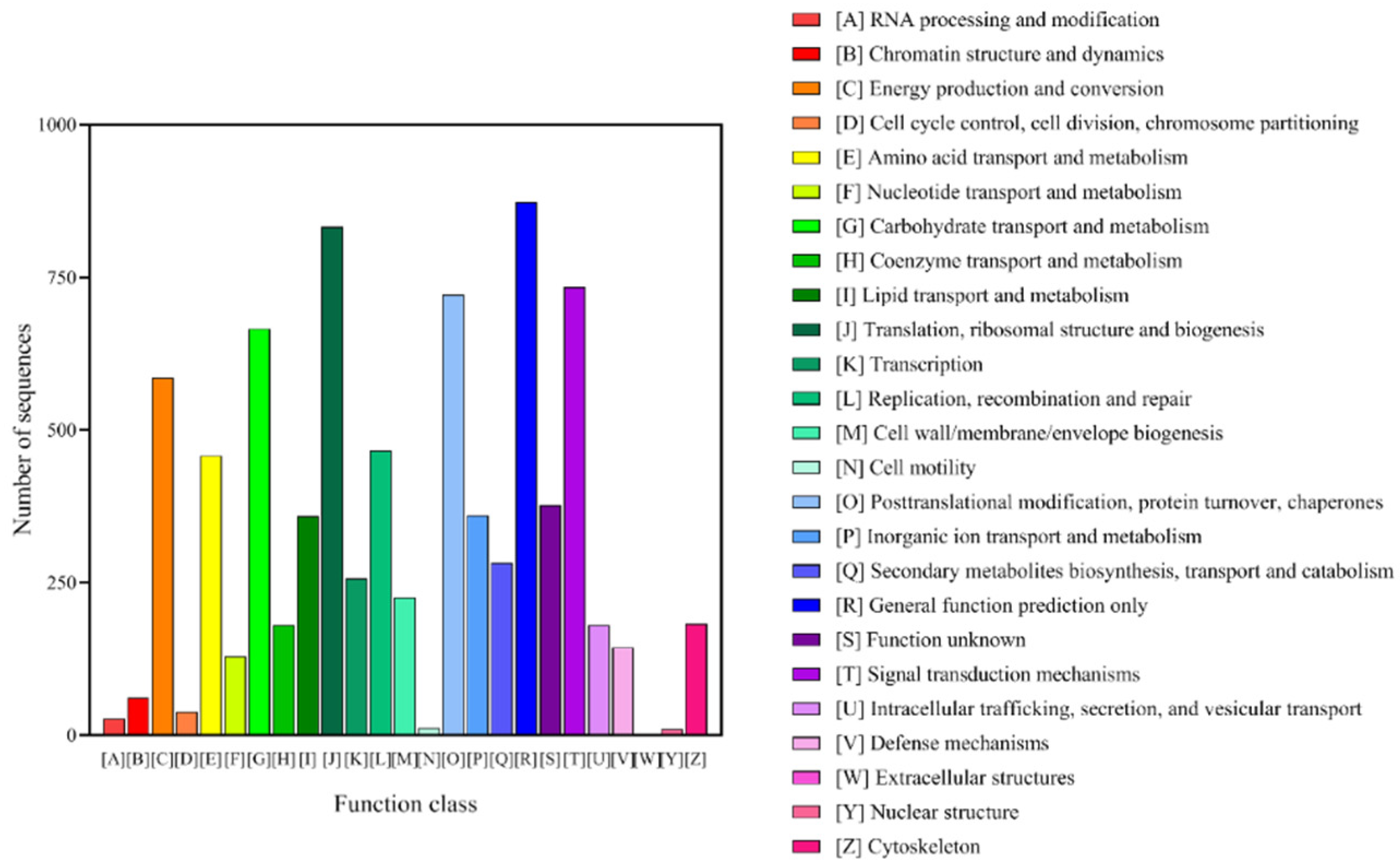
3.2. Annotation and Classification of T. mongolicum Unigenes
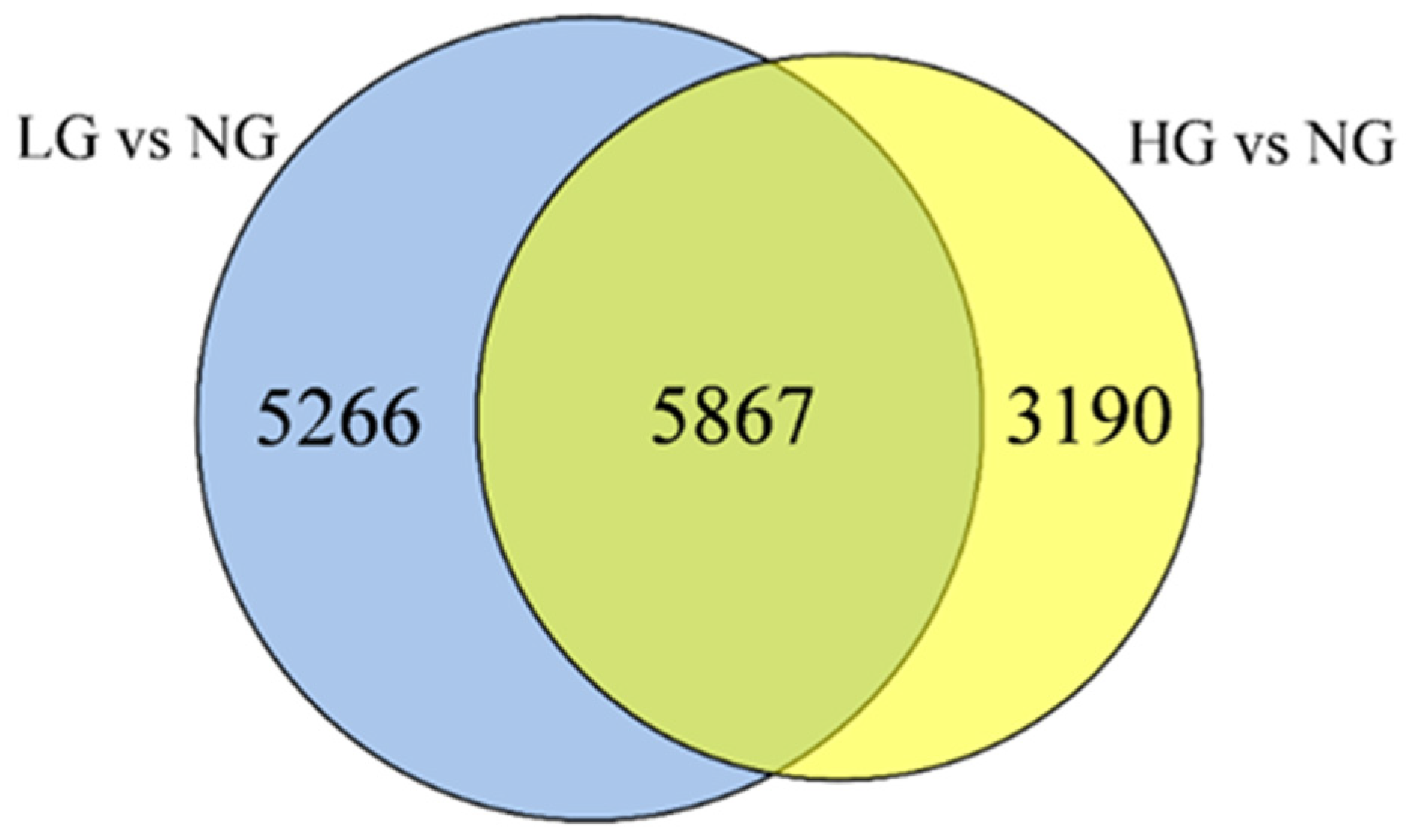
3.3. DEGs in Different Grazing Intensity
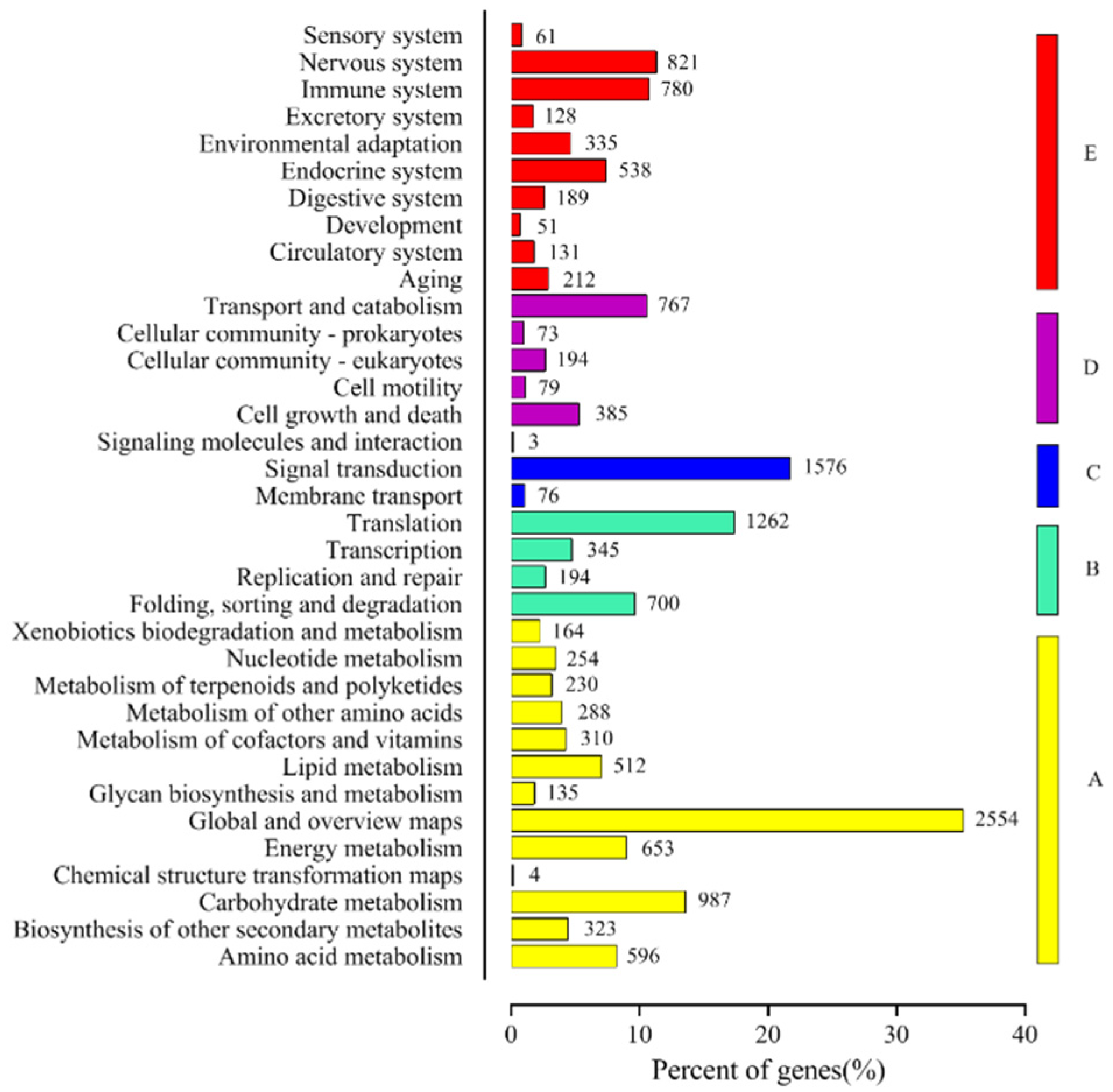
3.4. Pathways Enrichment Analysis of DEGs
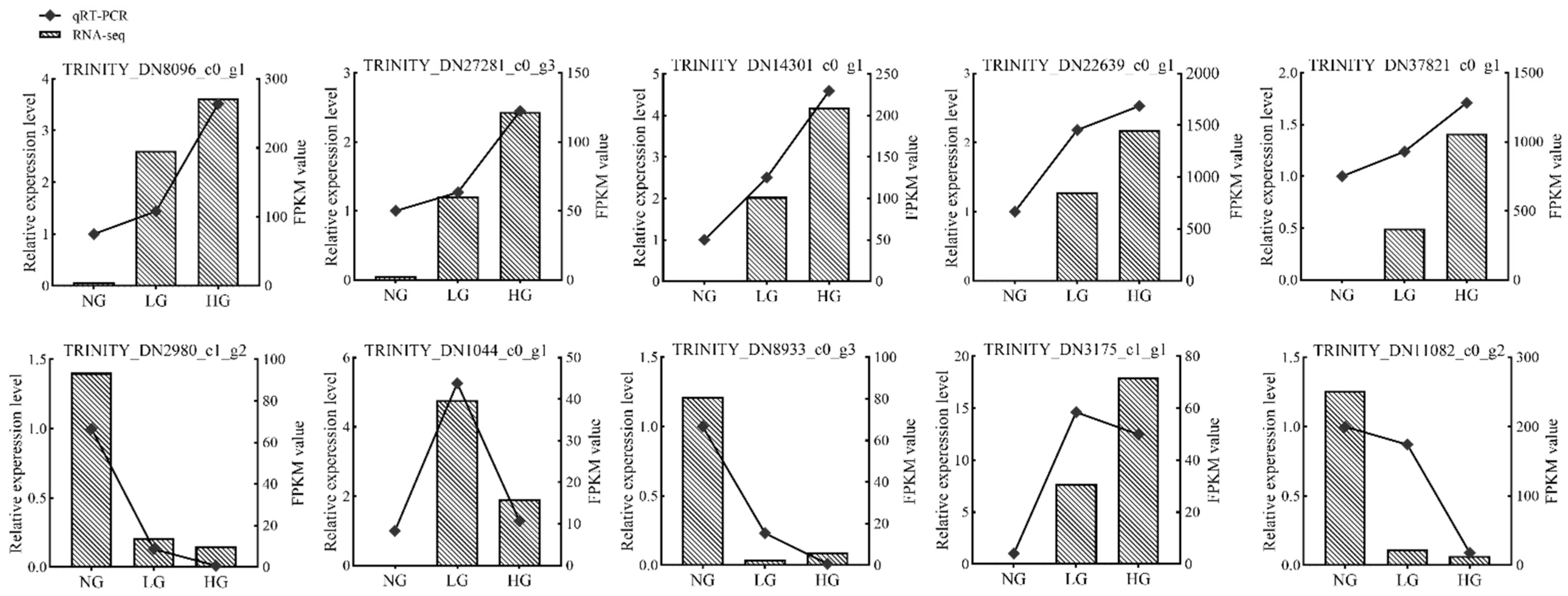
3.5. Validation of Gene Expression Profiles by qRT-PCR
3.6. Identification of Signal Transduction-Related Unigenes
3.7. Identification of Plant Hormone-Related Unigene
3.8. DEGs Involved in Metabolism and Biosynthesis
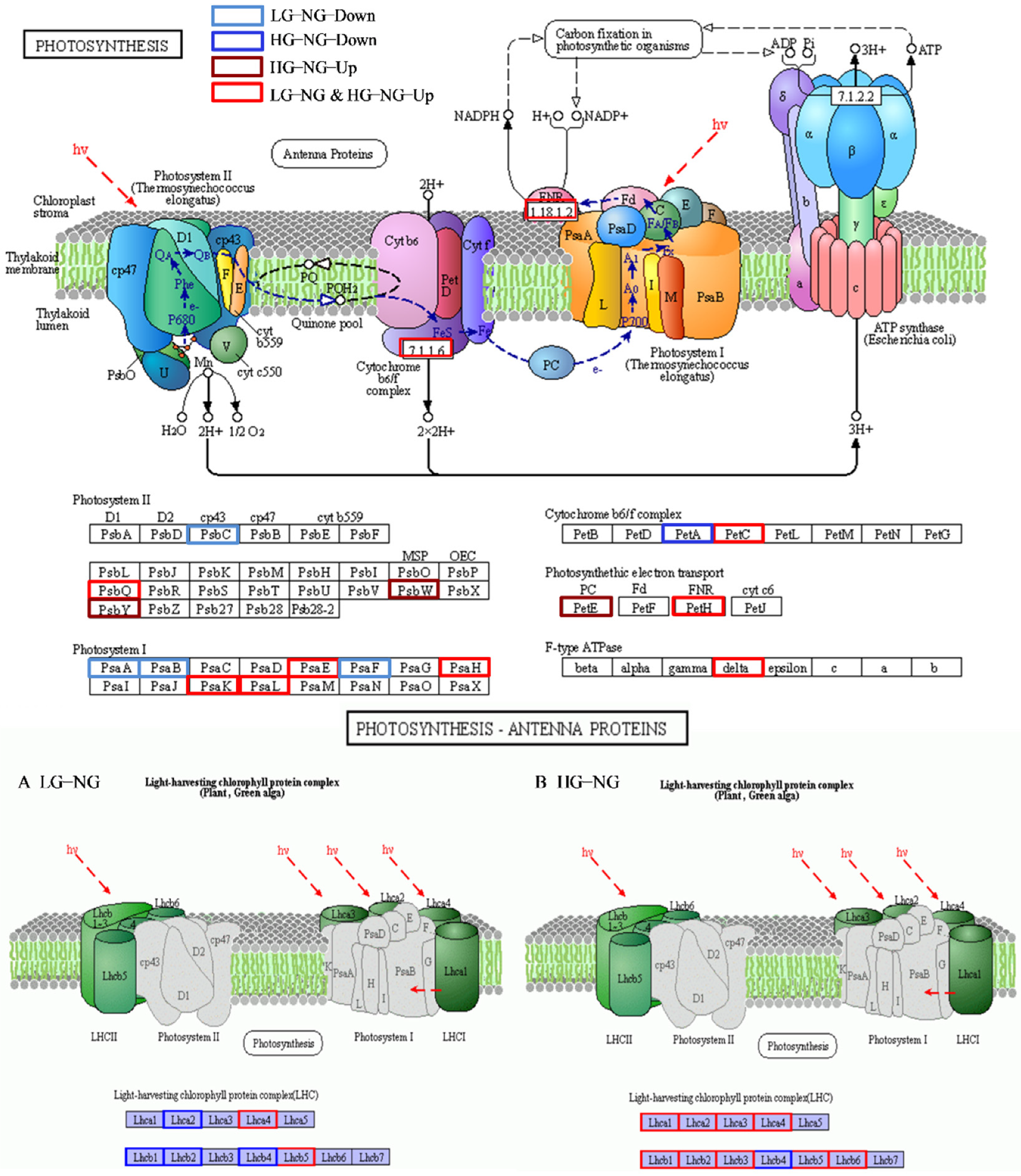
3.9. Identification of Photosynthesis Involved in the Response to Grazing Stress
4. Discussion
4.1. Signal Mediate Responses under Different Grazing Intensities
4.2. Phytohormone Signals under Different Grazing Intensities
4.3. Metabolism and Biosynthesis under Different Grazing Intensities
4.4. Photosynthesis under Different Grazing Intensities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petermann, J.S.; Buzhdygan, O.Y. Grassland biodiversity. Curr. Biol. 2021, 31, R1195–R1201. [Google Scholar] [CrossRef]
- Shirazi, S.; Ahmadi, A.; Abdi, N.; Toranj, H.; Khaleghi, M.R. Long-term grazing exclosure: Implications on water erosion and soil physicochemical properties (case study: Bozdaghin Rangelands, North Khorasan, Iran). Environ. Monit. Assess. 2021, 193, 51. [Google Scholar] [CrossRef]
- Li, W.; Li, X.; Zhao, Y.; Zheng, S.; Bai, Q. Ecosystem structure, functioning and stability under climate change and grazing in grasslands: Current status and future prospects. Curr. Opin. Environ. Sust. 2018, 33, 124–135. [Google Scholar] [CrossRef]
- Pavlu, V.; Hejcman, M.; Pavlu, L.; Gaisler, J.; Nezerkova, P. Effect of continuous grazing on forage quality, quantity and animal performance. Agric. Ecosyst. Environ. 2016, 113, 349–355. [Google Scholar] [CrossRef]
- Li, C.; Hao, X.; Willms, W.D.; Zhao, M.; Han, G. Seasonal response of herbage production and its nutrient and mineral contents to long-term cattle grazing on a rough fescue grassland. Agric. Ecosyst. Environ. 2009, 132, 32–38. [Google Scholar] [CrossRef]
- Lei, L.; Zheng, H.; Bi, Y.; Yang, L.; Zou, D. Identification of a major QTL and candidate gene analysis of salt tolerance at the bud burst stage in rice (Oryza sativa L.) using QTL-seq and RNA-seq. Rice 2020, 13, 55. [Google Scholar] [CrossRef]
- Yates, S.A.; Swain, M.T.; Hegarty, M.J.; Chernukin, I.; Lowe, M.; Allison, G.G.; Ruttink, T.; Abberton, M.T.; Jenkins, G.; Skot, L. De Novo assembly of red clover transcriptome based on RNA-seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 453. [Google Scholar] [CrossRef]
- Cheong, Y.H. Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis. Plant Physiol. 2001, 129, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gong, J.; Wang, B.; Li, X.; Ding, Y.; Yang, B.; Zhu, C.; Liu, M.; Zhang, W. Regrowth strategies of Leymus chinensis in response to different grazing intensities. Ecol. Appl. 2020, 30, e02113. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Shankar, R.; Thakkar, B.; Kudapa, H.; Krishnamurthy, L.; Mantri, N.; Varshney, R.K.; Bhatia, S.; Jain, M. Transcriptome analyses reveal genotype- and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci. Rep. 2016, 6, 19228. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Huertas, R.; Torres-Jerez, I.; Liu, W.; Watson, B.; Scheible, W.R.; Udvardi, M. Transcriptional, metabolic, physiological and developmental responses of switchgrass to phosphorus limitation. Plant Cell Environ. 2021, 44, 186–202. [Google Scholar] [CrossRef]
- Dang, Z.; Jia, Y.; Tian, Y.; Li, J.; Zhang, Y.; Huang, L.; Liang, C.; Lockhart, P.J.; Matthew, C.; Li, F. Transcriptome-wide gene expression plasticity in Stipa grandis in response to grazing intensity differences. Int. J. Mol. Sci. 2021, 22, 11882. [Google Scholar] [CrossRef]
- Kenkel, C.D.; Matz, M.V. Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat. Ecol. Evol. 2016, 1, 14. [Google Scholar] [CrossRef]
- Watt, M.; Fiorani, F.; Usadel, B.; Rascher, U.; Muller, O.; Schurr, U. Phenotyping: New windows into the plant for breeders. Annu. Rev. Plant Biol. 2020, 71, 689–712. [Google Scholar] [CrossRef]
- Deng, S.; Ma, J.; Zhang, L.; Chen, F.; Sang, Z.; Jia, Z.; Ma, L. De Novo transcriptome sequencing and gene expression profiling of Magnolia wufengensis in response to cold stress. BMC Plant Biol. 2019, 19, 321. [Google Scholar] [CrossRef]
- Nikiforou, C.; Manetas, Y. Ecological stress memory: Evidence in two out of seven species through the examination of the relationship between leaf fluctuating asymmetry and photosynthesis. Ecol. Indic. 2017, 74, 530–534. [Google Scholar] [CrossRef]
- Amaral, M.N.; Arge, L.W.P.; Auler, P.A.; Rossatto, T.; Milech, C.; Magalhaes, A.M., Jr.; Braga, E.J.B. Long-term transcriptional memory in rice plants submitted to salt shock. Planta 2020, 251, 111. [Google Scholar] [CrossRef]
- Gonzalez, A.P.; Chrtek, J.; Dobrev, P.I.; Dumalasova, V.; Fehrer, J.; Mraz, P.; Latzel, V. Stress-induced memory alters growth of clonal offspring of white clover (Trifolium repens). Am. J. Bot. 2016, 103, 1567–1574. [Google Scholar] [CrossRef]
- Hilker, M.; Schmulling, T. Stress priming, memory, and signaling in plants. Plant Cell Environ. 2019, 42, 753–761. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Chen, J.; Wang, X.; Cai, J.; Zhou, Q.; Dai, T.; Cao, W.; Jiang, D. Parental drought-priming enhances tolerance to post-anthesis drought in offspring of wheat. Front. Plant Sci. 2018, 9, 261. [Google Scholar] [CrossRef]
- Baurle, I. Can’t remember to forget you: Chromatin-based priming of somatic stress responses. Semin. Cell Dev. Biol. 2018, 83, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Hou, X.; Wu, Z.; Kong, L.; Guo, H.; Hu, N.; Wan, D.; Zhang, J. De Novo transcriptomic profiling of the clonal Leymus chinensis response to long-term overgrazing-induced memory. Sci. Rep. 2018, 8, 17912. [Google Scholar] [CrossRef]
- Ritchie, M.E. Grazing management, forage production and soil carbon dynamics. Resources 2020, 9, 49. [Google Scholar] [CrossRef]
- Sitters, J.; Wubs, E.R.J.; Bakker, E.S.; Crowther, T.W.; Adler, P.B.; Bagchi, S.; Bakker, J.D.; Biederman, L.; Borer, E.T.; Cleland, E.E.; et al. Nutrient availability controls the impact of mammalian herbivores on soil carbon and nitrogen pools in grasslands. Glob. Chang. Biol. 2020, 26, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Döbert, T.F.; Bork, E.W.; Apfelbaum, S.; Carlyle, C.N.; Chang, S.X.; Khatri-Chhetri, U.; Silva Sobrinho, L.; Thompson, R.; Boyce, M.S. Adaptive multi-paddock grazing improves water infiltration in canadian grassland soils. Geoderma 2021, 401, 115314. [Google Scholar] [CrossRef]
- Nakano, T.; Bat-Oyun, T.; Shinoda, M. Responses of palatable plants to climate and grazing in semi-arid grasslands of Mongolia. Glob. Ecol. Conser. 2020, 24, e01231. [Google Scholar] [CrossRef]
- Liu, M.; Gong, J.; Li, Y.; Li, X.; Yang, B.; Zhang, Z.; Yang, L.; Hou, X. Growth-defense trade-off regulated by hormones in grass plants growing under different grazing intensities. Physiol. Plant. 2019, 166, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Kerchev, P.I.; Fenton, B.; Foyer, C.H.; Hancock, R.D. Plant responses to insect herbivory: Interactions between photosynthesis, reactive oxygen species and hormonal signaling pathways. Plant. Cell Environ. 2012, 35, 441–453. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Y.; Ray, I.; Song, M. Transcriptome responses in Alfalfa associated with tolerance to intensive animal grazing. Sci. Rep. 2016, 6, 19438. [Google Scholar] [CrossRef]
- Zhang, Z.; Gong, J.; Shi, J.; Li, X.; Song, L.; Zhang, W.; Li, Y.; Zhang, S.; Dong, J.; Liu, Y. Multiple herbivory pressures lead to different carbon assimilation and allocation strategies: Evidence from a perennial grass in a typical steppe in northern China. Agric. Ecosyst. Environ. 2022, 326, 107776. [Google Scholar] [CrossRef]
- Agrawal, A.A. Overcompensation of plants in response to herbivory and the by-product benefits of mutualism. Trends Plant Sci. 2003, 5, 309–313. [Google Scholar] [CrossRef]
- Sun, J.; Ma, B.; Lu, X. Grazing enhances soil nutrient effects: Trade-offs between Aboveground and belowground biomass in alpine grasslands of the Tibetan plateau. Land Degrad. Dev. 2018, 29, 337–348. [Google Scholar] [CrossRef]
- Li, W.; Lee, C.; Kim, Y.H.; Ma, J.Y.; Shim, S.H. Chemical constituents of the aerial part of Taraxacum mongolicum and their chemotaxonomic significance. Nat. Prod. Res. 2017, 31, 2303–2307. [Google Scholar] [CrossRef]
- Li, Y.; Lv, M.; Wang, J.; Tian, Z.; Yu, B.; Wang, B.; Liu, J.; Liu, H. Dandelion (Taraxacum mongolicum Hand.-Mazz.) supplementation-enhanced rumen fermentation through the interaction between ruminal microbiome and metabolome. Microorganisms 2020, 9, 83. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, L.; Zhao, X.; Liu, W.; Li, Y.; Gu, S.; Zhou, X. Stability of alpine meadow ecosystem on the Qinghai-Tibetan plateau. Sci. Bull. 2006, 51, 320–327. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Koncz, P.; Vadász-Besnyői, V.; Csathó, A.I.; Nagy, J.; Szerdahelyi, T.; Tóth, Z.; Pintér, K.; Fóti, S.; Papp, M.; Balogh, J. Carbon uptake changed but vegetation composition remained stable during transition from grazing to mowing grassland management. Agric. Ecosyst. Environ. 2020, 304, 107161. [Google Scholar] [CrossRef]
- Mendes, C.; Dias, E.; Rochefort, L.; Azevedo, J. Regenerative succession of Azorean peatlands after grazing: Vegetation path to self-recovery. Wetl. Ecol. Manag. 2020, 28, 177–190. [Google Scholar] [CrossRef]
- Chen, S.; Cai, Y.; Zhang, L.; Yan, X.; Cheng, L.; Qi, D.; Zhou, Q.; Li, X.; Liu, G. Transcriptome analysis reveals common and distinct mechanisms for Sheepgrass (Leymus Chinensis) responses to defoliation compared to mechanical wounding. PLoS ONE 2014, 9, e89495. [Google Scholar] [CrossRef] [PubMed]
- Billker, O.; Lourido, S.; Sibley, L.D. Calcium-dependent signaling and kinases in Apicomplexan parasites. Cell Host Microbe. 2009, 5, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Valmonte, G.R.; Arthur, K.; Higgins, C.M.; MacDiarmid, R.M. Calcium-dependent protein kinases in plants: Evolution, expression and function. Plant Cell Physiol. 2014, 55, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Li, R.; Zou, B.; Zhang, X.; Cong, J.; Wang, R.; Xia, Y.; Li, G. Calmodulin-binding protein CBP60g is a positive regulator of both disease resistance and drought tolerance in Arabidopsis. Plant Cell Rep. 2012, 31, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Grabarek, Z. Structural basis for diversity of the EF-hand calcium-binding proteins. J. Mol. Biol. 2006, 359, 509–525. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Yang, L.; Deng, X.; Wu, H.; Zhang, M.; Liu, Y.; Zhang, S.; Xu, J. Mitogen-activated protein kinases and calcium-dependent protein kinases are involved in wounding-induced ethylene biosynthesis in Arabidopsis Thaliana. Plant Cell Environ. 2018, 41, 134–147. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, S. Mitogen-activated protein kinase cascades in signaling plant growth and development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. Mitogen-activated protein kinase cascades in plant signaling. J. Integr. Plant Biol. 2022, 64, 301–341. [Google Scholar] [CrossRef]
- Ye, Y.; Ding, Y.; Jiang, Q.; Wang, F.; Sun, J.; Zhu, C. The role of receptor-like protein kinases (RLKs) in abiotic stress response in plants. Plant Cell Rep. 2017, 36, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, S.; Suzuki, N.; Miller, G.; Choi, W.-G.; Toyota, M.; Devireddy, A.R.; Mittler, R. A tidal wave of signals: Calcium and ROS at the forefront of rapid systemic signaling. Trends Plant Sci. 2014, 19, 623–630. [Google Scholar] [CrossRef]
- Nambara, E.; Van Wees, S.C.M. Plant hormone functions and interactions in biological systems. Plant J. 2021, 105, 287–289. [Google Scholar] [CrossRef]
- Wang, K.L.-C.; Li, H.; Ecker, J.R. Ethylene biosynthesis and signaling networks. Plant Cell 2002, 14, S131–S151. [Google Scholar] [CrossRef]
- Chen, H.; Bullock, D.A.; Alonso, J.M.; Stepanova, A.N. To fight or to grow: The Balancing role of ethylene in plant abiotic stress responses. Plants 2021, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-L.; Ma, G.-J.; Zhang, M.-L.; Xiong, H.; Wu, H.; Zhao, C.-Z.; Liu, C.-S.; Jia, H.-X.; Chen, L.; Kjorven, J.O.; et al. The ARF7 and ARF19 transcription factors positively regulate PHOSPHATE STARVATION RESPONSE1 in Arabidopsis Roots. Plant Physiol. 2018, 178, 413–427. [Google Scholar] [CrossRef]
- Liu, N.; Avramova, Z. Molecular mechanism of the priming by jasmonic acid of specific dehydration stress response genes in Arabidopsis. Epigenet. Chromatin. 2016, 9, 8. [Google Scholar] [CrossRef]
- Lyons, R.; Manners, J.M.; Kazan, K. Jasmonate biosynthesis and signaling in monocots: A comparative overview. Plant Cell Rep. 2013, 32, 815–827. [Google Scholar] [CrossRef]
- Afrin, S.; Huang, J.; Luo, Z. JA-mediated transcriptional regulation of secondary metabolism in medicinal plants. Sci. Bull. 2015, 60, 1062–1072. [Google Scholar] [CrossRef]
- Campos, M.L.; Yoshida, Y.; Major, I.T.; Ferreira, D.D.; Weraduwage, S.M.; Froehlich, J.E.; Johnson, B.F.; Kramer, D.M.; Jander, G.; Sharkey, T.D.; et al. Rewiring of Jasmonate and Phytochrome B signalling uncouples plant growth-defense tradeoffs. Nat. Commun. 2016, 7, 12570. [Google Scholar] [CrossRef] [PubMed]
- Barto, E.K.; Cipollini, D. Testing the optimal defense theory and the growth-differentiation balance hypothesis in Arabidopsis Thaliana. Oecologia 2005, 146, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kou, S.-M.; Jin, R.; Wu, Y.-Y.; Huang, J.-W.; Zhang, Q.-Y.; Sun, N.-J.; Yang, Y.; Guan, C.-F.; Wang, W.-Q.; Zhu, C.-Q.; et al. Transcriptome analysis revealed the roles of carbohydrate metabolism on differential Acetaldehyde production capacity in persimmon fruit in response to high-CO2 treatment. J. Agric. Food Chem. 2021, 69, 836–845. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, F.; Marshall, R.S.; Ding, X.; Chatt, E.C.; Kirkpatrick, L.D.; Augustine, R.C.; Li, F.; Otegui, M.S.; Vierstra, R.D. Autophagy plays prominent roles in amino acid, nucleotide, and carbohydrate metabolism during fixed-carbon starvation in maize. Plant Cell 2020, 32, 2699–2724. [Google Scholar] [CrossRef] [PubMed]
- Couée, I.; Sulmon, C.; Gouesbet, G.; El Amrani, A. Involvement of soluble sugars in reactive oxygen species balance and responses to oxidative stress in plants. J. Exp. Bot. 2006, 57, 449–459. [Google Scholar] [CrossRef]
- Plaxton, W.C. The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 185–214. [Google Scholar] [CrossRef]
- Ventura, I.; Brunello, L.; Iacopino, S.; Valeri, M.C.; Novi, G.; Dornbusch, T.; Perata, P.; Loreti, E. Arabidopsis phenotyping reveals the importance of Alcohol Dehydrogenase and Pyruvate Decarboxylase for aerobic plant growth. Sci. Rep. 2020, 10, 16669. [Google Scholar] [CrossRef]
- Kato-Noguchi, H. Wounding stress induces Alcohol Dehydrogenase in maize and lettuce seedlings. Plant Growth Regul. 2001, 35, 285–288. [Google Scholar] [CrossRef]
- Tesniere, C.; Torregrosa, L.; Pradal, M.; Souquet, J.-M.; Gilles, C.; Dos Santos, K.; Chatelet, P.; Gunata, Z. Effects of genetic manipulation of Alcohol Dehydrogenase levels on the response to stress and the synthesis of secondary metabolites in grapevine leaves. J. Exp. Bot. 2006, 57, 91–99. [Google Scholar] [CrossRef]
- Alsamadany, H. De Novo leaf transcriptome assembly of Bougainvillea spectabilis for the identification of genes involves in the secondary metabolite pathways. Gene 2020, 746, 144660. [Google Scholar] [CrossRef]
- Obata, T. Metabolons in plant primary and secondary metabolism. Phytochem. Rev. 2019, 18, 1483–1507. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Luo, W.; Tanaka, G.; Konishi, Y.; Matsuura, H.; Takahashi, K. Wounding stress induces Phenylalanine Ammonia Lyases, leading to the accumulation of Phenylpropanoids in the model Liverwort Marchantia Polymorpha. Phytochemistry 2018, 155, 30–36. [Google Scholar] [CrossRef]
- Guan, Y.; Hu, W.; Xu, Y.; Yang, X.; Ji, Y.; Feng, K.; Sarengaowa. Metabolomics and physiological analyses validates previous findings on the mechanism of response to wounding stress of different intensities in broccoli. Food Res. Int. 2021, 140, 110058. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, X. Recent advances in Polyphenol Oxidase-mediated plant stress responses. Phytochemistry 2021, 181, 112588. [Google Scholar] [CrossRef]
- Liu, M.; Gong, J.; Yang, B.; Ding, Y.; Zhang, Z.; Wang, B.; Zhu, C.; Hou, X. Differences in the photosynthetic and physiological responses of Leymus Chinensis to different levels of grazing intensity. BMC Plant Biol. 2019, 19, 558. [Google Scholar] [CrossRef]
- Bag, P.; Chukhutsina, V.; Zhang, Z.; Paul, S.; Ivanov, A.G.; Shutova, T.; Croce, R.; Holzwarth, A.R.; Jansson, S. Direct energy transfer from photosystem II to photosystem I confers winter sustainability in scots pine. Nat. Commun. 2020, 11, 6388. [Google Scholar] [CrossRef]
- Ren, W.; Hu, N.; Hou, X.; Zhang, J.; Guo, H.; Liu, Z.; Kong, L.; Wu, Z.; Wang, H.; Li, X. Long-term overgrazing-induced memory decreases photosynthesis of clonal offspring in a perennial grassland plant. Front. Plant Sci. 2017, 8, 419. [Google Scholar] [CrossRef]
- Khorobrykh, S.; Havurinne, V.; Mattila, H.; Tyystjärvi, E. Oxygen and ROS in photosynthesis. Plants 2020, 9, 91. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Assembled Bases | Q20 (%) | GC (%) |
|---|---|---|---|---|---|
| LG−1 | 34,877,558 | 34,826,890 | 5,179,996,953 | 96.94 | 46.51 |
| LG−2 | 37,462,782 | 37,404,946 | 5,562,407,266 | 96.81 | 46.48 |
| LG−3 | 42,165,774 | 42,080,700 | 6,247,968,369 | 96.94 | 46.02 |
| HG−1 | 50,461,110 | 50,385,728 | 7,483,079,145 | 97.13 | 48.55 |
| HG−2 | 41,660,330 | 41,560,876 | 6,177,856,152 | 96.56 | 47.49 |
| HG−3 | 44,534,628 | 44,395,988 | 6,593,904,050 | 96.9 | 48.35 |
| NG−1 | 38,969,368 | 38,908,870 | 5,795,654,929 | 97.06 | 48.16 |
| NG−2 | 39,309,852 | 39,241,864 | 5,835,824,125 | 96.91 | 46.58 |
| NG−3 | 48,385,668 | 48,308,606 | 7,164,002,702 | 97.24 | 46.76 |
| All−unigene | 377,827,065 | 377,114,472 | 56,040,693,687 | 96.94 | 47.21 |
| Type | Unigene | Transcript |
|---|---|---|
| Total sequence number | 88,598 | 194,253 |
| Total sequence base | 58,417,702 | 168,900,058 |
| Largest length (bp) | 10,562 | 10,562 |
| Smallest length (bp) | 201 | 186 |
| Average length (bp) | 659.36 | 869.48 |
| N50 length (bp) | 1118 | 1341 |
| N90 length (bp) | 259 | 371 |
| Type | Unigene | Transcript |
|---|---|---|
| Nr | 24,825 | 69,093 |
| SwissProt | 16,215 | 43,153 |
| String | 14,402 | 35,730 |
| GO | 16,039 | 42,629 |
| KEGG | 10,437 | 28,358 |
| Pfam | 10,761 | 33,095 |
| ID | Pathways | Q-Value | No. of Genes |
|---|---|---|---|
| LG vs. NG | |||
| ko04626 | Plant–pathogen interaction | 0.001 | 20 |
| ko04962 | Vasopressin-regulated water reabsorption | 0.003 | 6 |
| ko00010 | Glycolysis/Gluconeogenesis | 0.008 | 19 |
| ko00030 | Pentose phosphate pathway | 0.010 | 11 |
| ko04016 | MAPK signaling pathway-plant | 0.019 | 23 |
| ko03018 | RNA degradation | 0.022 | 21 |
| ko00511 | Other glycan degradation | 0.033 | 6 |
| ko00500 | Starch and sucrose metabolism | 0.034 | 16 |
| ko04712 | Circadian rhythm-plant | 0.050 | 11 |
| HG vs. NG | |||
| ko00196 | Photosynthesis-antenna proteins | 0.000 | 10 |
| ko00500 | Starch and sucrose metabolism | 0.003 | 17 |
| ko00010 | Glycolysis/Gluconeogenesis | 0.004 | 18 |
| ko04016 | MAPK signaling pathway-plant | 0.008 | 22 |
| ko04020 | Calcium signaling pathway | 0.017 | 7 |
| ko04712 | Circadian rhythm-plant | 0.019 | 11 |
| ko04626 | Plant–pathogen interaction | 0.021 | 16 |
| ko00680 | Methane metabolism | 0.022 | 10 |
| ko04922 | Glucagon signaling pathway | 0.022 | 10 |
| ko01110 | Biosynthesis of secondary metabolites | 0.030 | 128 |
| ko04391 | Hippo signaling pathway-fly | 0.041 | 5 |
| ko04915 | Estrogen signaling pathway | 0.041 | 5 |
| ko00030 | Pentose phosphate pathway | 0.042 | 9 |
| ko00710 | Carbon fixation in photosynthetic organisms | 0.047 | 13 |
| ko04075 | Plant hormone signal transduction | 0.051 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhu, W.; Ren, F.; Zhao, N.; Xu, S.; Sun, P. Transcriptional Memory in Taraxacum mongolicum in Response to Long-Term Different Grazing Intensities. Plants 2022, 11, 2251. https://doi.org/10.3390/plants11172251
Wang Y, Zhu W, Ren F, Zhao N, Xu S, Sun P. Transcriptional Memory in Taraxacum mongolicum in Response to Long-Term Different Grazing Intensities. Plants. 2022; 11(17):2251. https://doi.org/10.3390/plants11172251
Chicago/Turabian StyleWang, Yalin, Wenyan Zhu, Fei Ren, Na Zhao, Shixiao Xu, and Ping Sun. 2022. "Transcriptional Memory in Taraxacum mongolicum in Response to Long-Term Different Grazing Intensities" Plants 11, no. 17: 2251. https://doi.org/10.3390/plants11172251