
Insights from a Multi-Omics Integration (MOI) Study in Oil Palm (Elaeis guineensis Jacq.) Response to Abiotic Stresses: Part One—Salinity

 , ,
, , 
Abstract
:1. Introduction
2. Results
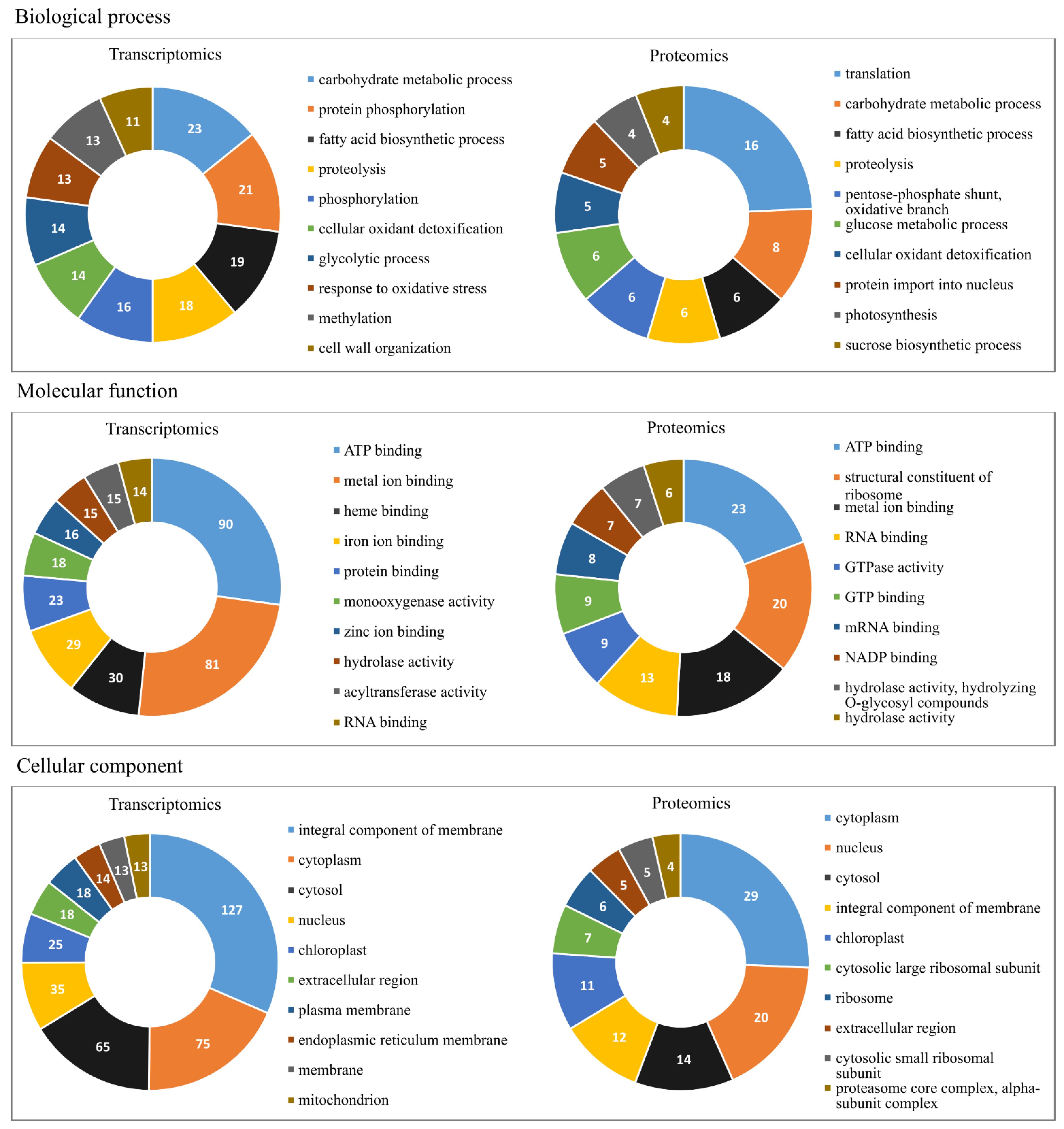
2.1. Oil Palm Transcriptome under Salinity Stress
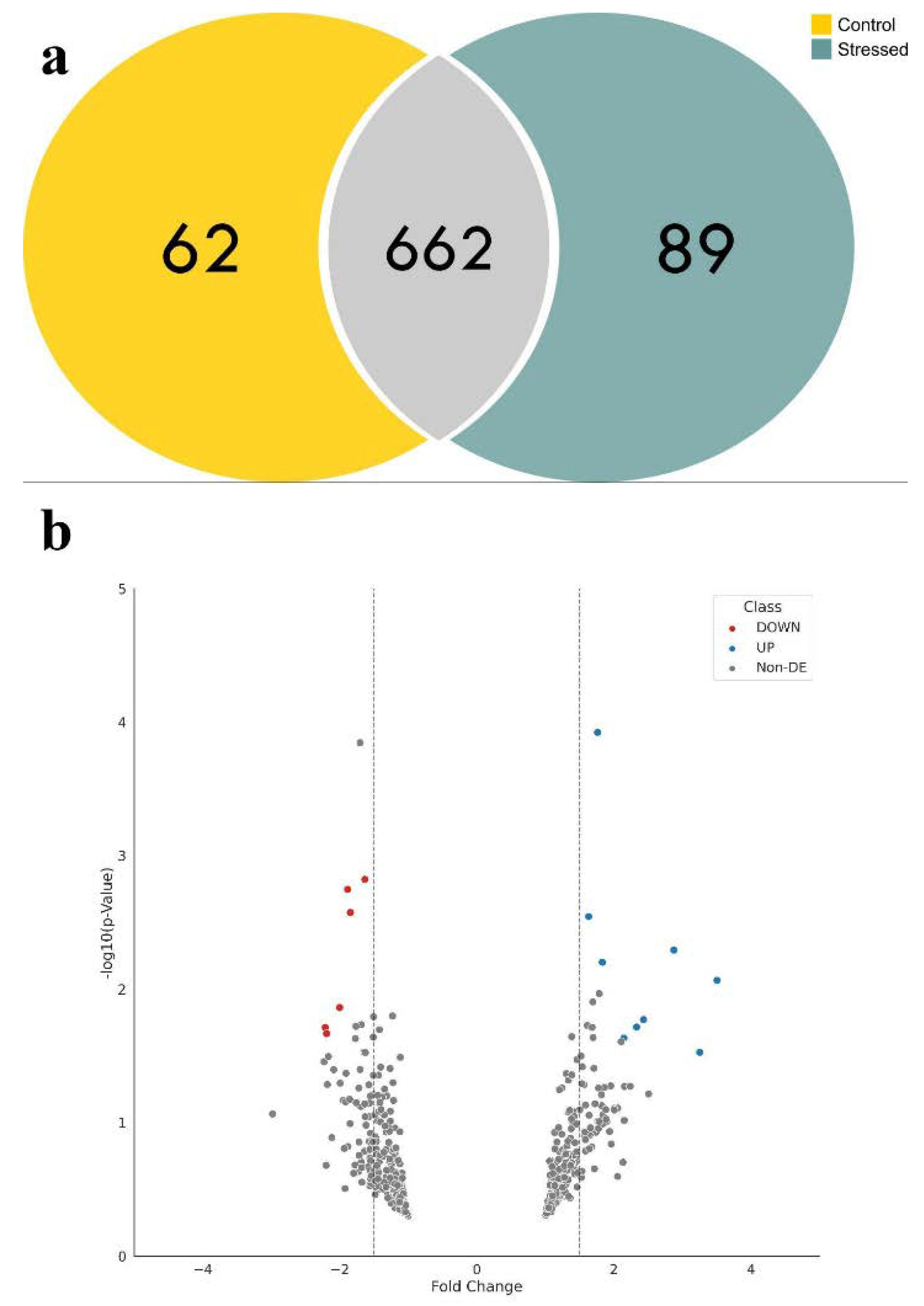
2.2. Oil Palm Proteome under Salinity Stress
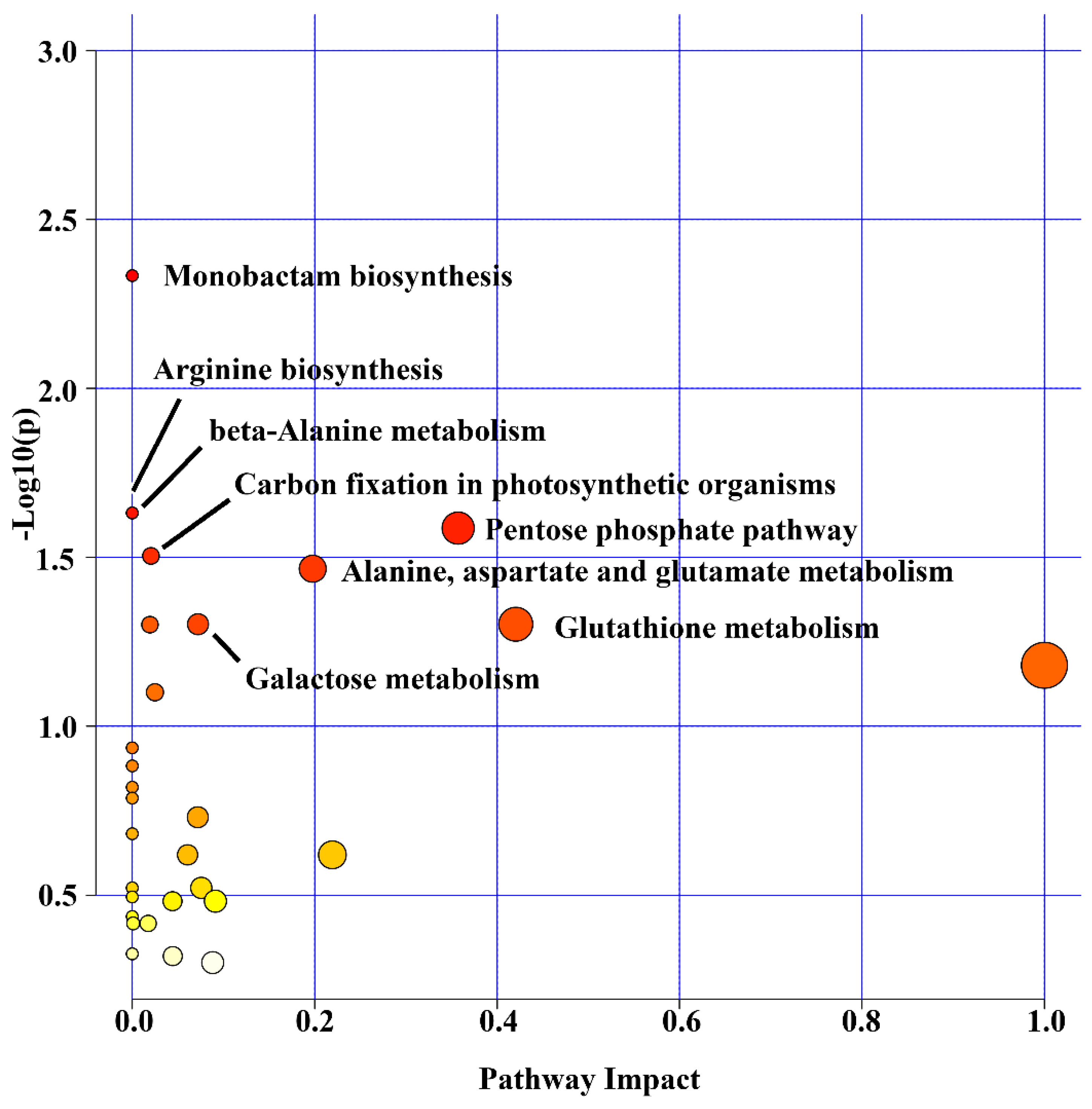
2.3. Oil Palm Metabolome under Salinity Stress
2.4. Integrating Oil Palm Transcriptome, Proteome and Metabolome
3. Discussion
4. Materials and Methods
4.1. Plant Material, Experimental Design and Saline Stress
4.2. Transcriptomics Data Analysis
4.3. Proteomics Data Analysis
4.4. Metabolomics Data Analysis
4.5. Functional Annotation and Itegratomics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Qaim, M.; Sibhatu, K.T.; Siregar, H.; Grass, I. Environmental, Economic, and Social Consequences of the Oil Palm Boom. Annu. Rev. Resour. Econ. 2020, 12, 321–344. [Google Scholar] [CrossRef]
- Shahbandeh, M. Production Volume of Palm Oil Worldwide from 2012/13 to 2021/22. Available online: https://www.statista.com/statistics/613471/palm-oil-production-volume-worldwide/ (accessed on 21 February 2022).
- Ahmad, F.B.; Zhang, Z.; Doherty, W.O.S.; O’Hara, I.M. The Outlook of the Production of Advanced Fuels and Chemicals from Integrated Oil Palm Biomass Biorefinery. Renew. Sustain. Energy Rev. 2019, 109, 386–411. [Google Scholar] [CrossRef]
- Furumo, P.R.; Aide, T.M. Characterizing Commercial Oil Palm Expansion in Latin America: Land Use Change and Trade. Environ. Res. Lett. 2017, 12, 024008. [Google Scholar] [CrossRef]
- Bentes, E.d.S.; Homma, A.K.O. Importação e Exportação de Óleo e Palmiste de Dendezeiro no Brasil (2010–2015); Embrapa Amazônia Oriental: Belém, Brazil, 2016. [Google Scholar]
- Vieira, L.R.; Silva, V.N.B.; Casari, R.A.d.C.N.; Carmona, P.A.O.; Sousa, C.A.F.d.; Souza Júnior, M.T. Morphophysiological Responses of Young Oil Palm Plants to Salinity Stress. Pesqui. Agropecuária Bras. 2020, 55, e01835. [Google Scholar] [CrossRef]
- Bittencourt, C.B.; Lins, P.d.C.; Boari, A.d.J.; Quirino, B.F.; Teixeira, W.G.; Souza Júnior, M.T. Oil Palm Fatal Yellowing (FY), a Disease with an Elusive Causal Agent. In Elaeis Guineensis; IntechOpen: London, UK, 2022. [Google Scholar]
- Costa, S.J.d.; Erasmo, E.A.L.; Tavares, T.C.d.O.; Silva, J. Respostas Fisiológicas de Dendê Submetidas ao Estresse Hídrico em Condições do Cerrado. Rev. Bras. Agropecuária Sustentável 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, J.A.d.; Junqueira, N.T.V.; Braga, M.F.; de Sá, M.A.C. Parâmetros de Irrigação Durante o Período Seco em Plantas Jovens de Dendê Cultivadas no Cerrado; Comunicado Técnico; Embrapa Cerrados: Brasília, Brazil, 2008. [Google Scholar]
- Antonini, A.J.C.d.A.; Oliveira, A.D.d. Potencial de Cultivo da Palma de Óleo Irrigada nas Condições do Cerrado; Documentos/Embrapa Cerrados; Empresa Brasileira de Pesquisa Agropecuária: Brasília, Brazil, 2021. [Google Scholar]
- Shahid, S.A.; Zaman, M.; Heng, L. Soil Salinity: Historical Perspectives and a World Overview of the Problem. In Guideline for Salinity Assessment, Mitigation and Adaptation Using Nuclear and Related Techniques; Springer International Publishing: Cham, Switzerland, 2018; pp. 43–53. [Google Scholar]
- Carillo, P.; Grazia, M.; Pontecorvo, G.; Fuggi, A.; Woodrow, P. Salinity Stress and Salt Tolerance. In Abiotic Stress in Plants-Mechanisms and Adaptations; InTech: Rijeka, Croatia, 2011; pp. 21–38. [Google Scholar]
- Isayenkov, S.V.; Maathuis, F.J.M. Plant Salinity Stress: Many Unanswered Questions Remain. Front. Plant Sci. 2019, 10, 80. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant Salt-Tolerance Mechanism: A Review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef]
- Salgado, F.F.; Vieira, L.R.; Silva, V.N.B.; Leão, A.P.; Grynberg, P.; do Carmo Costa, M.M.; Togawa, R.C.; de Sousa, C.A.F.; Júnior, M.T.S. Expression Analysis of MiRNAs and Their Putative Target Genes Confirm a Preponderant Role of Transcription Factors in the Early Response of Oil Palm Plants to Salinity Stress. BMC Plant Biol. 2021, 21, 518. [Google Scholar] [CrossRef]
- Rodrigues-Neto, J.C.R.; Vieira, L.R.; de Aquino Ribeiro, J.A.; de Sousa, C.A.F.; Júnior, M.T.S.; Abdelnur, P.V. Metabolic Effect of Drought Stress on the Leaves of Young Oil Palm (Elaeis guineensis) Plants Using UHPLC–MS and Multivariate Analysis. Sci. Rep. 2021, 11, 18271. [Google Scholar] [CrossRef]
- Santos, M.D.M.; Lima, D.B.; Fischer, J.S.G.; Clasen, M.A.; Kurt, L.U.; Camillo-Andrade, A.C.; Monteiro, L.C.; de Aquino, P.F.; Neves-Ferreira, A.G.C.; Valente, R.H.; et al. Simple, Efficient and Thorough Shotgun Proteomic Analysis with PatternLab V. Nat. Protoc. 2022, 17. [Google Scholar] [CrossRef]
- Cha-Um, S.; Takabe, T.; Kirdmanee, C. Ion Contents, Relative Electrolyte Leakage, Proline Accumulation, Photosynthetic Abilities and Growth Characters of Oil Palm Seedlings in Response to Salt Stress. Pak. J. Bot. 2010, 42, 2191–2200. [Google Scholar]
- Ferreira, T.M.M.; Leão, A.P.; Sousa, C.A.F.d.; Souza Júnior, M.T. Genes Highly Overexpressed in Salt-Stressed Young Oil Palm (Elaeis guineensis) Plants. Rev. Bras. Eng. Agrícola Ambient. 2021, 25, 813–818. [Google Scholar] [CrossRef]
- Jamil, I.N.; Remali, J.; Azizan, K.A.; Nor Muhammad, N.A.; Arita, M.; Goh, H.-H.; Aizat, W.M. Systematic Multi-Omics Integration (MOI) Approach in Plant Systems Biology. Front. Plant Sci. 2020, 11, 944. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, J.; Pan, C. Integrated Physiological, Proteomic, and Metabolomic Analyses of Pecan Cultivar ‘Pawnee’ Adaptation to Salt Stress. Sci. Rep. 2022, 12, 1841. [Google Scholar] [CrossRef]
- Mostek, A.; Börner, A.; Badowiec, A.; Weidner, S. Alterations in Root Proteome of Salt-Sensitive and Tolerant Barley Lines under Salt Stress Conditions. J. Plant Physiol. 2015, 174, 166–176. [Google Scholar] [CrossRef]
- Omidbakhshfard, M.A.; Omranian, N.; Ahmadi, F.S.; Nikoloski, Z.; Mueller-Roeber, B. Effect of Salt Stress on Genes Encoding Translation-Associated Proteins in Arabidopsis thaliana. Plant Signal. Behav. 2012, 7, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Chong, P.; Zhao, M.; Liu, H. Physiological Response and Proteomics Analysis of Reaumuria soongorica under Salt Stress. Sci. Rep. 2022, 12, 2539. [Google Scholar] [CrossRef]
- Chauhan, J.S.; Mishra, N.K.; Raghava, G.P. Identification of ATP Binding Residues of a Protein from Its Primary Sequence. BMC Bioinform. 2009, 10, 434. [Google Scholar] [CrossRef] [Green Version]
- Dahuja, A.; Kumar, R.R.; Sakhare, A.; Watts, A.; Singh, B.; Goswami, S.; Sachdev, A.; Praveen, S. Role of ATP-binding Cassette Transporters in Maintaining Plant Homeostasis under Abiotic and Biotic Stresses. Physiol. Plant. 2021, 171, 785–801. [Google Scholar] [CrossRef]
- Wang, L.; Lee, M.; Ye, B.; Yue, G.H. Genes, Pathways and Networks Responding to Drought Stress in Oil Palm Roots. Sci. Rep. 2020, 10, 21303. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, L.; Barkla, B.J. Membrane Lipid Remodeling in Response to Salinity. Int. J. Mol. Sci. 2019, 20, 4264. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.M.F. Plasma Membrane Permeability as an Indicator of Salt Tolerance in Plants. Biol. Plant 2013, 57, 1–10. [Google Scholar] [CrossRef]
- Couturier, J.; Chibani, K.; Jacquot, J.-P.; Rouhier, N. Cysteine–Based Redox Regulation and Signaling in Plants. Front. Plant Sci. 2013, 4, 105. [Google Scholar] [CrossRef] [Green Version]
- Hell, R.; Wirtz, M. Metabolism of Cysteine in Plants and Phototrophic Bacteria. In Sulfur Metabolism in Phototrophic Organisms; Springer: Berlin/Heidelberg, Germany, 2008; Volume 27, pp. 59–91. [Google Scholar]
- Blaszczyk, A.; Brodzik, R.; Sirko, A. Increased Resistance to Oxidative Stress in Transgenic Tobacco Plants Overexpressing Bacterial Serine Acetyltransferase. Plant J. 1999, 20, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.N.; Mobin, M.; Abbas, Z.K.; Siddiqui, M.H. Nitric Oxide-Induced Synthesis of Hydrogen Sulfide Alleviates Osmotic Stress in Wheat Seedlings through Sustaining Antioxidant Enzymes, Osmolyte Accumulation and Cysteine Homeostasis. Nitric Oxide 2017, 68, 91–102. [Google Scholar] [CrossRef]
- Amir, R.; Hacham, Y. Methionine Metabolism in Plants. In Sulfur: A Missing Link between Soils, Crops, and Nutrition; American Society of Agronomy, Inc.: Madison, WI, USA, 2008; Volume 50, pp. 251–279. [Google Scholar]
- Qi, Y.-C.; Wang, F.-F.; Zhang, H.; Liu, W.-Q. Overexpression of Suadea salsa S-Adenosylmethionine Synthetase Gene Promotes Salt Tolerance in Transgenic Tobacco. Acta Physiol. Plant. 2010, 32, 263–269. [Google Scholar] [CrossRef]
- Noctor, G.; Mhamdi, A.; Chaouch, S.; Han, Y.; Neukermans, J.; Marquez-Garcia, B.; Queval, G.; Foyer, C.H. Glutathione in Plants: An Integrated Overview. Plant Cell Environ. 2012, 35, 454–484. [Google Scholar] [CrossRef]
- Ong, A.-L.; Teh, C.-K.; Mayes, S.; Massawe, F.; Appleton, D.R.; Kulaveerasingam, H. An Improved Oil Palm Genome Assembly as a Valuable Resource for Crop Improvement and Comparative Genomics in the Arecoideae Subfamily. Plants 2020, 9, 1476. [Google Scholar] [CrossRef]
- Singh, R.; Ong-Abdullah, M.; Low, E.-T.L.; Manaf, M.A.A.; Rosli, R.; Nookiah, R.; Ooi, L.C.-L.; Ooi, S.; Chan, K.-L.; Halim, M.A.; et al. Oil Palm Genome Sequence Reveals Divergence of Interfertile Species in Old and New Worlds. Nature 2013, 500, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wen, Z.; Zhang, J.; Chen, X.; Cui, J.; Xu, W.; Liu, H. Exogenous Glutathione Alleviates Salt-Induced Oxidative Stress in Tomato Seedlings by Regulating Glutathione Metabolism, Redox Status, and the Antioxidant System. Sci. Hortic. 2017, 220, 90–101. [Google Scholar] [CrossRef]
- Walker, R.P.; Chen, Z.-H.; Famiani, F. Gluconeogenesis in Plants: A Key Interface between Organic Acid/Amino Acid/Lipid and Sugar Metabolism. Molecules 2021, 26, 5129. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Yuan, Y.; Shu, S.; Sun, J.; Guo, S.; Yuan, R.; Tang, Y. Effects of Exogenous Putrescine on Glycolysis and Krebs Cycle Metabolism in Cucumber Leaves Subjected to Salt Stress. Plant Growth Regul. 2016, 79, 319–330. [Google Scholar] [CrossRef]
- Suzuki, M.; Hashioka, A.; Mimura, T.; Ashihara, H. Salt Stress and Glycolytic Regulation in Suspension-Cultured Cells of the Mangrove Tree, Bruguiera sexangula. Physiol. Plant. 2005, 123, 246–253. [Google Scholar] [CrossRef]
- Guo, R.; Shi, L.; Yan, C.; Zhong, X.; Gu, F.; Liu, Q.; Xia, X.; Li, H. Ionomic and Metabolic Responses to Neutral Salt or Alkaline Salt Stresses in Maize (Zea mays L.) Seedlings. BMC Plant Biol. 2017, 17, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Tian, Y.; Cang, J.; Yu, J.; Wang, D.; He, F.; Jiao, H.; Tan, Y. Overexpression of TaFBA-A10 from Winter Wheat Enhances Freezing Tolerance in Arabidopsis thaliana. J. Plant Growth Regul. 2022, 41, 314–326. [Google Scholar] [CrossRef]
- Sun, Q.; Yamada, T.; Han, Y.; Takano, T. Influence of Salt Stress on C4 Photosynthesis in Miscanthus sinensis Anderss. Plant Biol. 2021, 23, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Cheng, Y.; Zou, X.; Zhang, X. Ethanol Content in Plants of Brassica Napus L. Correlated with Waterlogging Tolerance Index and Regulated by Lactate Dehydrogenase and Citrate Synthase. Acta Physiol. Plant. 2016, 38, 81. [Google Scholar] [CrossRef]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and Metabolomic Data Integration. Brief. Bioinform. 2016, 17, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Gertsman, I.; Barshop, B.A. Promises and Pitfalls of Untargeted Metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef]
- Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An Open-Source MS/MS Sequence Database Search Tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Carvalho, P.C.; Fischer, J.S.G.; Xu, T.; Cociorva, D.; Balbuena, T.S.; Valente, R.H.; Perales, J.; Yates, J.R.; Barbosa, V.C. Search Engine Processor: Filtering and Organizing Peptide Spectrum Matches. Proteomics 2012, 12, 944–949. [Google Scholar] [CrossRef]
- Vargas, L.H.G.; Neto, J.C.R.; de Aquino Ribeiro, J.A.; Ricci-Silva, M.E.; Souza Júnior, M.T.; Rodrigues, C.M.; de Oliveira, A.E.; Abdelnur, P.V. Metabolomics Analysis of Oil Palm (Elaeis guineensis) Leaf: Evaluation of Sample Preparation Steps Using UHPLC–MS/MS. Metabolomics 2016, 12, 153. [Google Scholar] [CrossRef]
- Carvalho da Silva, T.L.; Belo Silva, V.N.; Braga, Í.d.O.; Rodrigues Neto, J.C.; Leão, A.P.; Ribeiro, J.A.d.A.; Valadares, L.F.; Abdelnur, P.V.; Sousa, C.A.F.; Souza Júnior, M.T. Integration of Metabolomics and Transcriptomics Data to Further Characterize Gliricidia sepium (Jacq.) Kunth under High Salinity Stress. Plant Genome 2022, 15, e20182. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Xia, J. Using MetaboAnalyst 4.0 for Metabolomics Data Analysis, Interpretation, and Integration with Other Omics Data. In Computational Methods and Data Analysis for Metabolomics; Li, S., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2104, pp. 337–360. [Google Scholar]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Wold, S. Introduction to Multi-and Megavariate Data Analysis Using Projection Methods (PCA & PLS); Umetrics AB: Umeå, Sweden, 1999. [Google Scholar]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting Network Activity from High Throughput Metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Brink, B.G.; Seidel, A.; Kleinbölting, N.; Nattkemper, T.W.; Albaum, S.P. Omics Fusion—A Platform for Integrative Analysis of Omics Data. J. Integr. Bioinform. 2016, 13, 43–46. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptomics | Number of Features | Up | Down | Non-DE |
| WGS–Proteins | 43,551 | 1138 | 1590 | 40,823 |
| Metabolomics | Number of Peaks | Up | Down | Non-DE |
| Positive Polar | 2843 | 18 | 34 | 2791 |
| Negative Polar | 1855 | 19 | 58 | 1778 |
| Proteomics * | Number of Features | Up | Down | Non-DE |
| LC/MS | 813 | 101 | 70 | 642 |
| Control | Stressed | Total | |
|---|---|---|---|
| Peptide Spectrum Match (PSM) | 5419 | 5391 | 10,808 |
| Total number of peptides | 3234 | 2872 | 4254 |
| Number of unique peptides | 1805 | 1606 | 2426 |
| Total number of proteins entries | 1497 | 1436 | 1809 |
| Total number of proteins using the maximum parsimony criterion | 826 | 831 | 1019 |
| Entry | Class | Fold Change | p-Value | Signal in Control | Signal in Stressed | Gene ID at NCBI | Description |
|---|---|---|---|---|---|---|---|
| A0A6I9RY35 | UP | 3.50631 | 0.00860 | 0.00027 | 0.00094 | LOC105054572 | probable inactive purple acid phosphatase 29 |
| A0A6I9QVF6 | UP | 3.25426 | 0.02982 | 0.00104 | 0.00340 | LOC105040203 | GTP-binding nuclear protein |
| A0A6I9R375 | UP | 3.25426 | 0.02982 | 0.00085 | 0.00275 | LOC105043116 | GTP-binding nuclear protein |
| A0A6I9RFH3 | UP | 3.25426 | 0.02982 | 0.00104 | 0.00340 | LOC105047773 | GTP-binding nuclear protein |
| A0A6I9QCS1 | UP | 2.87620 | 0.00511 | 0.00059 | 0.00169 | LOC105033701 | Proteasome subunit alpha type |
| A0A6I9QQJ4 | UP | 2.43453 | 0.01697 | 0.00103 | 0.00251 | LOC105039272 | 60S ribosomal protein L35a-1 |
| B3TLX9 | UP | 2.43453 | 0.01697 | 0.00103 | 0.00251 | LOC105037063 | 60S ribosomal protein L35a-1 |
| A0A6I9QWA8 | UP | 2.33349 | 0.01927 | 0.00071 | 0.00165 | LOC105039716 | Succinate-semialdehyde dehydrogenase |
| A0A6I9RG83 | UP | 2.14817 | 0.02330 | 0.00062 | 0.00133 | LOC105045986 | uncharacterized protein LOC105045986 |
| B3TLY5 | UP | 1.83395 | 0.00630 | 0.00105 | 0.00193 | CAT2 | Catalase |
| A0A6I9QQQ6 | UP | 1.76320 | 0.00012 | 0.00068 | 0.00119 | LOC105039332 | V-ATPase 69 kDa subunit |
| A0A6I9R4U7 | UP | 1.63284 | 0.00286 | 0.00276 | 0.00450 | LOC105044322 | Malate dehydrogenase |
| A0A6I9S1Z5 | DOWN | −1.63290 | 0.00151 | 0.00166 | 0.00101 | LOC105055575 | ruBisCO large subunit-binding protein subunit alpha |
| A0A6I9QJN4 | DOWN | −1.84374 | 0.00267 | 0.00177 | 0.00096 | LOC105036569 | CBBY-like protein |
| A0A6I9RPV6 | DOWN | −1.84374 | 0.00267 | 0.00177 | 0.00096 | LOC105051320 | CBBY-like protein |
| A0A6J0PH47 | DOWN | −1.88477 | 0.00179 | 0.00395 | 0.00210 | LOC105044080 | Ferredoxin—NADP reductase, chloroplastic |
| A0A6I9S9I9 | DOWN | −2.00037 | 0.01375 | 0.00091 | 0.00045 | LOC105058225 | uncharacterized protein LOC105058225 |
| A0A6I9RWU5 | DOWN | −2.19127 | 0.02157 | 0.00284 | 0.00129 | LOC105054048 | actin-101 |
| A0A6I9RC26 | DOWN | −2.21145 | 0.01945 | 0.00172 | 0.00078 | LOC105047077 | sorbitol dehydrogenase isoform X2 |
| A0A6I9RDE7 | DOWN | −2.21145 | 0.01945 | 0.00172 | 0.00078 | LOC105047077 | sorbitol dehydrogenase isoform X1 |
| Query Mass | Matched Compound | Matched Form | Mass Difference | Compound Name | FDR | Log2(FC) |
|---|---|---|---|---|---|---|
| 145.01452 | C00026 | M-H[–] | 2.69 × 10−4 | Oxoglutaric acid | 0.0106 | –0.4146 |
| 616.17640 | C00032 | M[1+] | 8.96 × 10−4 | Heme | 0.0039 | 2.8661 |
| 106.04953 | C00049 | M-CO+H[1+] | 2.53 × 10−4 | L-Aspartic acid | 0.0292 | 0.9617 |
| 306.07651 | C00051 | M-H[–] | 2.27 × 10−5 | Glutathione | 0.0204 | 1.5265 |
| 289.03241 | C00117 | M+CH3COO[–] | 3.46 × 10−5 | D-Ribose 5-phosphate | 0.0475 | –0.9714 |
| 427.01748 | C00224 | M(C13)-H[–] | 1.46 × 10−3 | Adenosine phosphosulfate | 0.0172 | –0.6544 |
| 172.98600 | C00262 | M+K-2H[–] | 7.55 × 10−4 | Hypoxanthine | 0.0004 | –1.5351 |
| 203.22237 | C00750 | M+H[1+] | 6.58 × 10−4 | Spermine | 0.0036 | 2.4559 |
| 163.04033 | C00811 | M-H[–] | 2.65 × 10−4 | 4-Hydroxycinnamic acid | 0.0065 | –0.3818 |
| 162.02134 | C01419 | M-NH3+H[1+] | 6.49 × 10−4 | Cysteinylglycine | 0.0263 | 1.2145 |
| 260.02535 | C05345 | M(C13)-H[–] | 4.92 × 10−4 | Beta-D-Fructose 6-phosphate | 0.0489 | –1.0337 |
| 359.11946 | C05399 | M-H+O[–] | 3.09 × 10−5 | Melibiitol | 0.0103 | –1.6922 |
| 254.09610 | C05401 | M(C13)-H[–] | 1.95 × 10−4 | Galactosylglycerol | 0.0410 | –0.7515 |
| 326.09623 | C05839 | M(C13)-H[–] | 6.61 × 10−5 | cis-beta-D-Glucosyl-2-hydroxycinnamate | 0.0472 | –1.4286 |
| 277.06946 | C05911 | M-CO+H[1+] | 1.11 × 10−3 | Pentahydroxyflavanone | 0.0143 | –1.0759 |
| 337.05555 | C10107 | M+H2O+H[1+] | 1.09 × 10−4 | Myricetin | 0.0313 | –2.4012 |
| 337.00976 | C11453 | M+CH3COO[–] | 8.02 × 10−4 | 2-C-Methyl-D-erythritol 2,4-cyclodiphosphate | 0.0272 | 0.8232 |
| 259.02223 | C17214 | M+Cl37[–] | 1.45 × 10−4 | 2-(3′-Methylthio)propylmalic acid | 0.0222 | –0.9440 |
| 447.91027 | G00005 | M(C13)+2H [2+] | 1.30 × 10−3 | (GlcNAc)2 (Man)3 (PP-Dol)1 | 0.0263 | 0.4044 |
| Pathway | Pathway ID | Occurrence of Transcripts | Occurrence of Proteins | Occurrence of Metabolites | Occurrence of Unique Molecule |
|---|---|---|---|---|---|
| Cysteine and methionine metabolism | 270 | 15 | 5 | 2 | 20 |
| Glycolysis/Gluconeogenesis | 10 | 17 | 3 | 1 | 20 |
| Glyoxylate and dicarboxylate metabolism | 630 | 14 | 4 | 1 | 16 |
| Carbon fixation in photosynthetic organisms | 710 | 12 | 2 | 2 | 15 |
| Glycine, serine and threonine metabolism | 260 | 11 | 2 | 1 | 14 |
| Pentose phosphate pathway | 30 | 10 | 4 | 2 | 14 |
| Glutathione metabolism | 480 | 9 | 3 | 3 | 13 |
| Amino sugar and nucleotide sugar metabolism | 520 | 10 | 2 | 1 | 12 |
| Carbon fixation pathways in prokaryotes | 720 | 7 | 6 | 1 | 11 |
| Citrate cycle (TCA cycle) | 20 | 5 | 4 | 1 | 8 |
| Butanoate metabolism | 650 | 4 | 2 | 1 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bittencourt, C.B.; Carvalho da Silva, T.L.; Rodrigues Neto, J.C.; Vieira, L.R.; Leão, A.P.; de Aquino Ribeiro, J.A.; Abdelnur, P.V.; de Sousa, C.A.F.; Souza, M.T., Jr. Insights from a Multi-Omics Integration (MOI) Study in Oil Palm (Elaeis guineensis Jacq.) Response to Abiotic Stresses: Part One—Salinity. Plants 2022, 11, 1755. https://doi.org/10.3390/plants11131755
Bittencourt CB, Carvalho da Silva TL, Rodrigues Neto JC, Vieira LR, Leão AP, de Aquino Ribeiro JA, Abdelnur PV, de Sousa CAF, Souza MT Jr. Insights from a Multi-Omics Integration (MOI) Study in Oil Palm (Elaeis guineensis Jacq.) Response to Abiotic Stresses: Part One—Salinity. Plants. 2022; 11(13):1755. https://doi.org/10.3390/plants11131755
Chicago/Turabian StyleBittencourt, Cleiton Barroso, Thalliton Luiz Carvalho da Silva, Jorge Cândido Rodrigues Neto, Letícia Rios Vieira, André Pereira Leão, José Antônio de Aquino Ribeiro, Patrícia Verardi Abdelnur, Carlos Antônio Ferreira de Sousa, and Manoel Teixeira Souza, Jr. 2022. "Insights from a Multi-Omics Integration (MOI) Study in Oil Palm (Elaeis guineensis Jacq.) Response to Abiotic Stresses: Part One—Salinity" Plants 11, no. 13: 1755. https://doi.org/10.3390/plants11131755