1. Introduction
IDE is a ubiquitous 110 kDa spherical protease with poorly understood functions that is found in both extra- and intracellular compartments, including the cytosol and, also, to a lesser extent, in the plasma membrane, endosomes and peroxisomes. The protein sequence of IDE is strikingly conserved amongst distantly related species, including those that do not produce insulin. Identity scores between sequences from human, chimpanzee, rat, and mouse range between 95% and 99%. In other vertebrates, such as the zebrafish, IDE is still 85% identical to the human sequence. These high identity scores suggest a common evolution, similar 3D structure and functions, and potentially important yet to be identified roles in the biology of the cell. IDE degrades insulin with high efficacy, but also degrades several amyloidogenic peptides, such as amyloid-beta, IGF-II, glucagon, and amylin [
1] Ubiquitin and particularly its less stable forms have also been shown to be degraded by IDE, although ubiquitin can act as an IDE inhibitor, reducing insulin degradation [
2,
3,
4]. Structural analysis by the Tang laboratory has revealed how IDE recognizes and cleaves conformationally metastable amyloidogenic peptides [
5].
IDE is composed of two homologous domains connected by a short hinge, one of which contains the zinc ion binding HXXEH motif [
6]. IDE can adopt an open conformation, giving substrates access to an inner chamber in which the active site is reconstituted upon closure [
5]. Substrates are unfolded inside the inner chamber, which selects substrates of <10 kDa [
7]. IDE has an exosite interacting with the N-terminus of substrates and facilitates their unfolding, as well as an allosteric site that regulates the degradation of short peptides [
8,
9,
10].
In humans and rodents, genetic data and deletion experiments suggest a role of IDE in glucose homeostasis: IDE gene knockout in mice results in glucose intolerance, though the mice remain normoglycemic and are only slightly hyperinsulinemic [
11]. In addition, the IDE gene is linked to T2D and Alzheimer’s disease in humans. Acute treatment with two structurally different inhibitors designed independently by the teams of Deprez-Poulain in collaboration with us and Maianti produced only partially consistent effects on glucose tolerance in mice, underlining the complexity of the interaction between IDE and glucose and insulin homeostasis in vivo [
12,
13].
The reason underlying the high level of sequence conservation across species, and the ubiquitous expression of IDE, including in cells lacking identified IDE substrates, remains unknown. However, over the last decades, experimental findings have established that IDE is also involved in a wide variety of biological processes. Tundo et al. demonstrated that normal and malignant cells exposed to different stresses up-regulate IDE in a heat shock protein-like fashion, and proposed that IDE binds proteins as a “dead end chaperone” [
14]. Consistent with this, IDE was initially reported to function as receptor for the VZV virus but was then shown to bind to an intracellular VZV protein, most likely a misfolded fragment [
15,
16]. Literature reports have also documented that IDE co-immunoprecipitates with the proteasome and ubiquitin [
17,
18]. In line with a possible role in protein scavenging following ER stress, Schmitz et al. reported an IDE-dependent clearance mechanism for ER-localized amyloid-β, suggesting that IDE may be involved in a parallel ERAD pathway that is not dependent on the proteasome [
19]. We have recently found that IDE deficiency triggers the unfolded protein response that is enhanced upon metabolic or pharmacologic stress and is accompanied, at least in pancreatic islet cells, by cell proliferation (Zhu et al., manuscript submitted). Thus, the available data suggest that IDE plays a role in protein homeostasis, particularly in stress situations as well as in glucose metabolism. However, the mechanistic basis for these roles remains unclear.
Although wild-type IDE is predominantly located in the cytosol in all cell types studied so far, IDE is also found in the mitochondria. The amino acid sequences of full-length human and mouse IDE features two alternative in-frame initiating methionines at the positions 1 (Met
1) and 42 (Met
42). A bioinformatics analysis of the full-length IDE protein sequence predicts the presence of a mitochondrial targeting sequence (MTS) downstream of Met
1, a prediction confirmed by the mitochondrial localization of eGFP preceded by the N-terminal portion of Met
1-IDE [
20]. Importantly, although the presence of a more efficient Kozak sequence promotes the dominant translation initiation at Met
42 resulting in dominant cytosolic localization, a fraction of IDE can be detected in mitochondria from wild-type CHO and HEK cells, indicating the physiologic relevance of translation initiation at Met
1. Observing highly efficient in vitro cleavage of a synthetic MTS by IDE, possibly related to the frequent presence of arginine residues preferred by IDE in MTS, Leissring and colleagues speculated that IDE might contribute to the physiologic cleavage of MTS assisting other critical enzymes [
21,
22]. However, mitochondrial IDE might also have more important roles. For example, IDE has been suggested to cooperate with the mitochondrial SIRT4 deacetylase to mediate the lysosomal degradation of PTENα and promote survival in response to starvation stress [
23]. Due to its capacity of cleaving amyloid-β, IDE might also help prevent the accumulation of toxic Ab in mitochondria [
24]. Considering the complete lack of information about the biological roles of IDE in mitochondria, we set out to identify interactions of IDE with mitochondrial proteins using a proximity biotinylation approach. We report that both wild-type and protease-dead mitochondrial IDE interact primarily with proteins involved in mitochondrial protein translation and in the citric acid cycle. While the functional impact of these interactions remains to be determined, we speculate that mitochondrial IDE, like the dominant cytosolic form, might be involved in mitochondrial protein homeostasis, particularly in stress situations.
2. Materials and Methods
2.1. Generation of TurboID (Biotin Ligase BirA Mutant) Fusion Constructs, Lentivirus Production and Transduction
Lentiviral constructs were built in the vector pLVX-tdTomato-N1 (Takara, Kyoto, Japan), which was first modified by the insertion between XhoI (5′) and BamHI (3′) in the multiple cloning site of a synthetic gene, containing (5′ to 3′) SmaI and MluI restriction sites, the sequence coding for P2A/T2A self-cleaving peptides (ATNFSLLKQAGDVEENPGP/EGRGSLLTCGDVEENPGP), and td-Tomato, resulting in the vector pLVX-P2A-T2A-tdTomato.
To generate the lentiviral expression constructs IDE-TurboID and IDEMut E111D-TurboID, full-length cDNA (complementary DNA) encoding wt or protease-dead human IDE was amplified from the previously published constructs IDE- or IDEMut E111D-pCRBlunt [
25], with primers adding XhoI at the 5’ end and including an EcoRI site ahead of the stop codon. The PCR product was first cloned as an XhoI-EcoRI fragment into the vector pMA-RQ previously modified by the insertion of a synthetic sequence comprising 79 base pairs at the 3′ end of IDE, including the EcoRI site but not the stop codon, and sequences encoding a GSAGSA linker, the V5 peptide, the TurboID enzyme, and a MluI site at the 3′ end. The sequence encoding TurboID preceded by the V5 peptide was taken from Branon et al. [
26]. Finally, the complete constructs were transferred as XhoI-MluI fragments into pLVX-P2A-T2A-tdTomato.
The construct pLVX-MTS-OVA-V5-TurboID-td-Tomato was produced by amplifying a fragment comprising the GSAGSA linker, V5, and TurboID DNA sequences with primers containing SmaI (5′) and MluI (3′) restriction sites and inserting the PCR product into the plasmid pLVX-MTS-OVA-T2A-P2A-tdTomato previously generated in the laboratory. The latter plasmid carries the mitochondrial targeting sequence (MTS) from human cytochrome c oxidase (MSVLTPLLLRGLTGSARRLPVPRAKIHSL; NCBI, NM_205152.3) and SmaI and MluI sites between the T2A-P2A and tdTomato sequences. Synthetic genes were purchased from GeneArt (Thermo Fisher Scientific, Waltham, MA, USA). The primers for PCR amplification are listed in
Table S1.
Plasmid DNA was extracted and purified with the Nucleobond Xtra Midi EF kit (Macherey-Nagel, Düren, Germany) and used to produce lentiviruses with an average titer of 109 TU/mL. The HEK (human embryonic kidney) 293 cell line was transduced at a multiplicity of infection (MOI) of 5 for 6 h. Cells were selected starting 48 h after infection with puromycin at the concentration of 10 μg/mL. After 2–3 passages in selection media, the percentage of transduced cells was determined by quantifying td-Tomato positive cells by flow cytometry.
2.2. Confocal Microscopy
4 × 10
4 cells were seeded onto glass coverslips pre-coated with Poly-D-Lysine overnight. The cells were fixed for 12 min with a solution containing 4% paraformaldehyde, 0.2% glutaraldehyde, 60 mM PIPES (piperazine-
N,N′-bis(2-ethanesulfonic acid)), 25 mM HEPES((4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 10 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-
N,N,N′,N′-tetraacetic acid), 2 mM Magnesium acetate, and then permeabilized with 0.2%Triton X-100 in DPBS (Dulbecco’s phosphate-buffered saline) for 10 min. After blocking with 5% donkey serum in PBS-0.05% Tween-20 for 2 h at RT (room temperature), the cells were incubated sequentially with primary antibodies overnight at 4 °C (IDE, V5, TOM20 all at 1:100), and with the appropriate secondary antibodies (1:200) for 2 h at RT (
Table S1). Nuclear counterstaining was carried out using DAPI (4′,6-diamidino-2-phenylindole) at the concentration of 1 μg/mL. The slides were mounted with Vectashield Plus Antifade media. The image acquisitions were performed with a 63x oil immersion objective (NA 1.4) and a laser scanning confocal microscope (TCS SP8-3X STED; Leica Microsystems, Weztlar, Germany). The images were processed with Icy (
https://icy.bioimageanalysis.org, accessed on 9 May 2022).
2.3. Western Blot
Standard denaturing SDS-PAGE analysis was performed using Mini Gel Tank equipment (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were transferred onto PVDF (polyvinylidene difluoride) membrane with the iBlot 2 Dry Blotting System (Thermo Fisher Scientific). The membranes were blocked with BSA (Bovine serum albumin) 5% in TBS (Tris-buffered saline)-Tween 0.1% 1 h at RT and then incubated with the primary antibody overnight at 4 °C and with the secondary antibody 2 h at RT. Antibody binding was visualized by enhanced chemiluminescence with SuperSignalTM West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific), and the images were acquired on the ChemiDoc Imaging System (BioRad, Hercules, CA, USA). Primary and secondary antibodies are listed in the Supplemental Materials (
Table S2).
2.4. TurboID-Based Enzymatic Protein Labeling and Extraction of Biotinylated Proteins for Proteomic Analysis
Proximity labeling by the TurboID enzyme fused to IDE and MutIDE expressed in HEK cells was performed according to the method described by Cho and coworkers [
27]. The TurboID system uses a fast biotin ligase so that incubations of a few minutes are usually sufficient for the biotinylation of neighboring proteins. Briefly, 20 × 10
6 HEK293T cells expressing the IDE-TurboID proteins were cultured at 37 °C 5% CO
2 in Dulbecco’s Modified Eagle Medium (DMEM) containing 10 U/mL penicillin-streptomycin, 2 mM Glutamine (Sigma-Aldrich, St. Louis, MO, USA), and 10% dialyzed FCS (Fetal calf serum, Eurobio Scientific, Saclay, France) for 24 h. The cells were then incubated with 50 μM biotin (Sigma-Aldrich) for 15 min. The labeling reaction was stopped by placing the cells on ice and washing out the biotin excess with ice-cold DPBS (Thermo Fischer Scientific). The cells were detached via pipetting with 10 mL of ice-cold DPBS and centrifuged at 300×
g. The cell pellet was lysed in 2 mL of a radio immunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific) supplemented by 1x protease inhibitor cocktail (Complete EDTA (Ethylenediaminetetraacetic acid)-free protease inhibitors, Roche Diagnostic, Basel, Switzerland) and incubated on ice for 30 min. The cell lysate was clarified by centrifugation at 13,000×
g at 4 °C for 10 min and transferred to a fresh tube. The protein concentration was measured by a BCA (bicinchoninic acid) protein assay (Bio-Rad, Hercules, CA, USA). For the enrichment of the biotinylated proteins, 2.5 mg proteins in a 1.8 mL RIPA buffer were incubated with 200 μL Streptavidin magnetic beads (Thermo Fisher Scientific) overnight at 4 °C. Afterward, the supernatant was removed using a magnetic rack to pellet the beads, which were washed twice with the RIPA buffer (1 mL, 2 min), once with KCl 1 M (1 mL, 2 min), once with Na
2CO
3 0.1 M (1 mL, 10 s), and once with urea 2 M in 10 mM Tris-HCl (pH 8.0) (1 mL, 10 s). After the final wash, the beads were transferred in a 1 mL RIPA buffer to a fresh tube and washed again with a RIPA buffer (1 mL, 2 min). The enriched biotinylated proteins were eluted from the beads in 80 μL of 4× Laemmli buffer (Biorad) supplemented with 2 mM biotin and 20 mM DDT (dichloro-diphenyl-trichloroethane) at 95 °C for 10 min. The input, the flow-through, and the enriched material from each sample were analyzed by western blot before performing a proteomic analysis.
2.5. Identification and Quantification of Proteins by NanoLC-MS/MS
Sample digestion was performed on S-TrapTM microcolumns (Protifi, Hutington, CA, USA) according to the manufacturer’s instructions. Samples (60 µL, IP) were supplemented with 20% SDS to a final concentration of 5%, reduced with 20 mM TCEP, and alkylated with 50 mM CAA (chloracetamide) for 15 min at room temperature. Aqueous 27.5% phosphoric acid was then added to a final concentration of 2.5%, followed by the addition of a binding buffer (90% methanol, 100 mM TEAB, pH 8). The mixtures were then loaded onto S-Trap columns and centrifuged for 30 s at 3000× g. The columns were washed 6 times (3000× g, 30 s) to ensure the complete removal of SDS. The samples were digested with 1 µg trypsin (Promega, Madison, WI, USA) at 47 °C for 1.5 h. After elution, the peptides were dried under a vacuum and resuspended in 20 µL of 2% ACN and 0.1% formic acid in HPLC grade water prior to MS analysis.
An amount of 2 µL of the tryptic peptides were injected onto a nanoElute HPLC system (Bruker Daltonics, Leipzig, Germany) coupled to a timsTOF Pro mass spectrometer (Bruker Daltonics). HPLC separation (Solvent A: 0.1% formic acid in water, 2% acetonitrile; Solvent B: 0.1% formic acid in acetonitrile) was performed at 250 nL/min using a packed emitter column (C18, 25 cm × 75 μm 1.6 μm) (Ion Optics, Fitzroy, Australia) using gradient elution (2–11% solvent B for 19 min; 11–16% for 7 min; 16–25% for 4 min; 25–80% for 3 min and, finally, 80% for 7 min to wash the column). The data were acquired using the PASEF (parallel accumulation serial fragmentation) acquisition method. Measurements were performed over the m/z range from 100 to 1700 Th. The range of ion mobility values was from 0.85 to 1.3 V s/cm2 (1/K0). The total cycle time was set to 1.2 s, and the number of PASEF MS/MS scans was set to 6.
2.6. Data Processing after LC-MS/MS Acquisition
The raw file folders (.d) were processed with MaxQuant 2.0.1 software and searched with the Andromeda search engine in the UniProtKB/Swiss-Prot Homo Sapiens database (version 01-02-2021, 20,396 entries). To search for parent and fragment mass ions, we set an initial mass deviation of 4.5 ppm and 20 ppm, respectively. The minimum peptide length was set at seven amino acids, and strict specificity for the trypsin cleavage was required, allowing up to two missed cleavage sites. Carbamidomethylation (Cys) was defined as a fixed modification, while oxidation (Met) and N-term acetylation were defined as variable modifications. Matching between the series was not allowed. The minimum number of LFQ (Label Free quantification) ratios was set at 2. False discovery rates (FDR) at the protein and peptide level were set at 1%. Scores were calculated in MaxQuant as described previously [
28]. The reverse and common contaminants were removed from MaxQuant output. Proteins were quantified according to the MaxQuant label-free algorithm using LFQ intensities. Finally, a match between runs was allowed during the analysis.
Four independent immunoprecipitations per group were analyzed with Perseus software (version 1.6.15.0), which is freely available at
www.perseus-framework.org, accessed on 16 March 2022 [
29]. Protein intensities were log2 transformed, and proteins identified in at least four replicates in at least one group were statistically tested (volcano plot, FDR = 0.05 and S0 = 0.1) after the imputation of the missing value by a Gaussian random number distribution with a standard deviation of 30% from the standard deviation of the measured values and a downward shift of 1.8 standard deviations from the mean. To increase the selectivity of the candidate, a double comparison was performed between the IDE/mutIDE (with biotin) vs the negative control (with biotin), and IDE/mutIDE (with biotin) vs. IDE/mutIDE (without biotin).
2.7. IDE Interactome Data Analysis
Functional protein networks of the IDE and MutIDE interactome data were annotated with STRING Software (version 11.5) [
30]. Lists of (i) all proteins significantly enriched in IDE or MutIDE samples incubated with biotin relative to both control samples (samples without biotin and OVA samples), and of proteins; (ii) enriched in biotin samples relative to OVA controls only with fold change > 3.5; or (iii) enriched in biotin samples relative to samples without biotin only with fold change > 5.5, were inputted into the String database in the section “multiple protein analysis”. The organism was set as Homo Sapiens. The parameter chosen for network analysis was “full string network” with a minimum required interaction score of 0.04. Interactions were limited to protein query only. The PPI (Protein Protein interaction) enrichment
p-value was ≤10
−16 and the average local clustering coefficient varied between 0.5–0.6 for the data sets analyzed.
The pathway enrichment analysis was analyzed with Metascape (
https://metascape.org, accessed on 10 March 2023) [
31]. The list of selected genes was submitted to Metascape (Version v3.5.20230101). The process enrichment analysis was carried out with the following ontology sources: KEGG Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways, CORUM, and WikiPathways. The complete human genome was used as the enrichment background. Terms with a
p-value < 0.01, a minimum count of 3, and an enrichment factor > 1.5 (i.e., the ratio between the counts observed and the counts expected by chance) were collected and grouped into clusters based on their membership similarities.
p-values are calculated based on the cumulative hypergeometric distribution, and q-values are calculated using the Benjamini-Hochberg procedure to account for multiple testing. The most statistically significant term within a term cluster is chosen to represent the cluster. Top clusters with their representative enriched terms (one per cluster) are depicted as bar graphs with a discrete color scale indicating statistical significance expressed as −log10.
4. Discussion
In this study, we had set out to obtain initial leads on the role of the fraction of IDE localizing to mitochondria by using a fast proximity biotinylation approach and HEK cells. The expression of a full-length human IDE cDNA starting at Met1 resulted in strongly dominant mitochondrial localization, which was consistent with previous reports. The short-term incubation of fusion protein-expressing cells with biotin resulted in the readily detectable biotinylation of client proteins, allowing for the proteomic identification of candidate proteins interacting with IDE. Following standard recommendations, potentially interacting proteins were identified through comparison with a non-biotin and with a compartmentalization control.
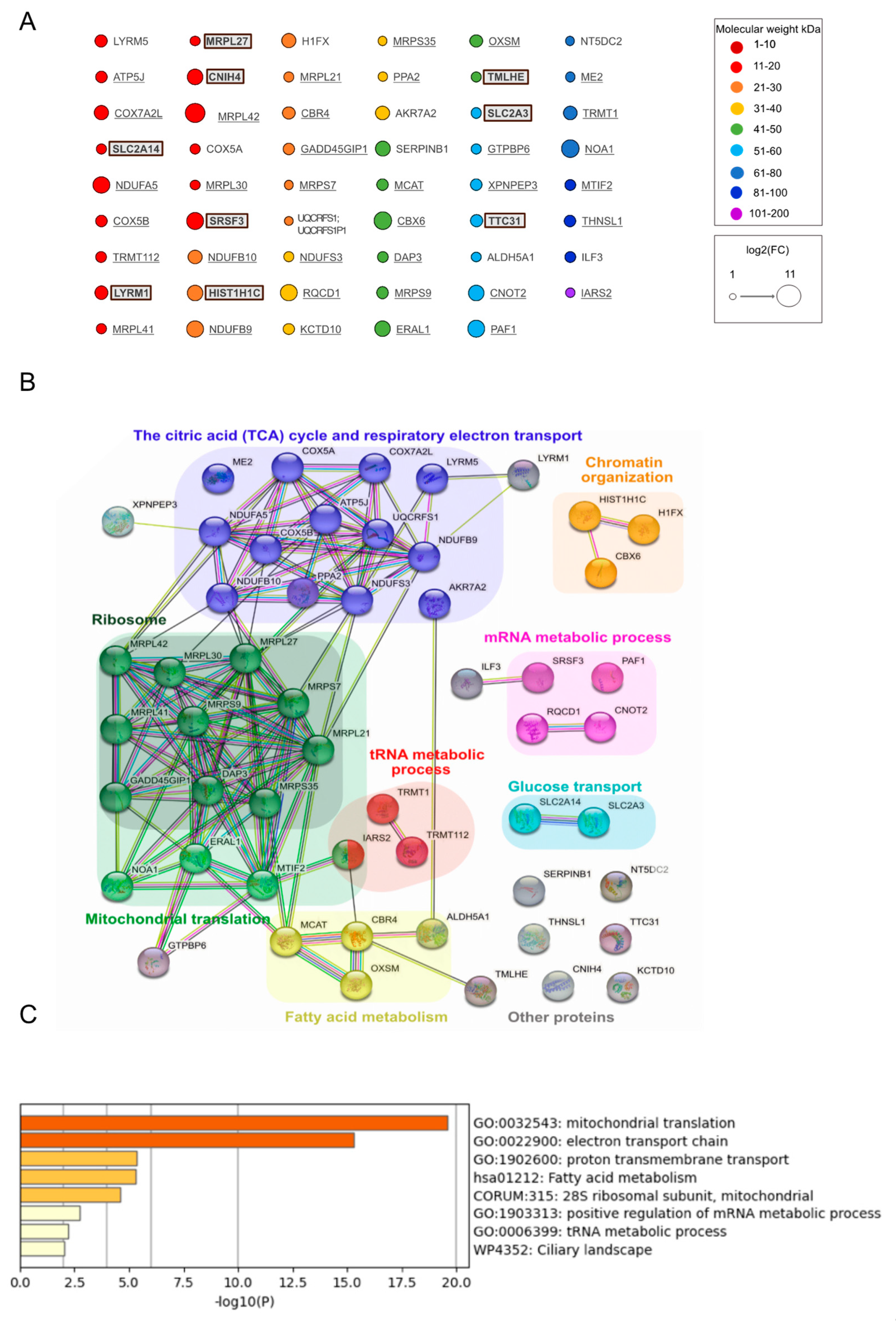
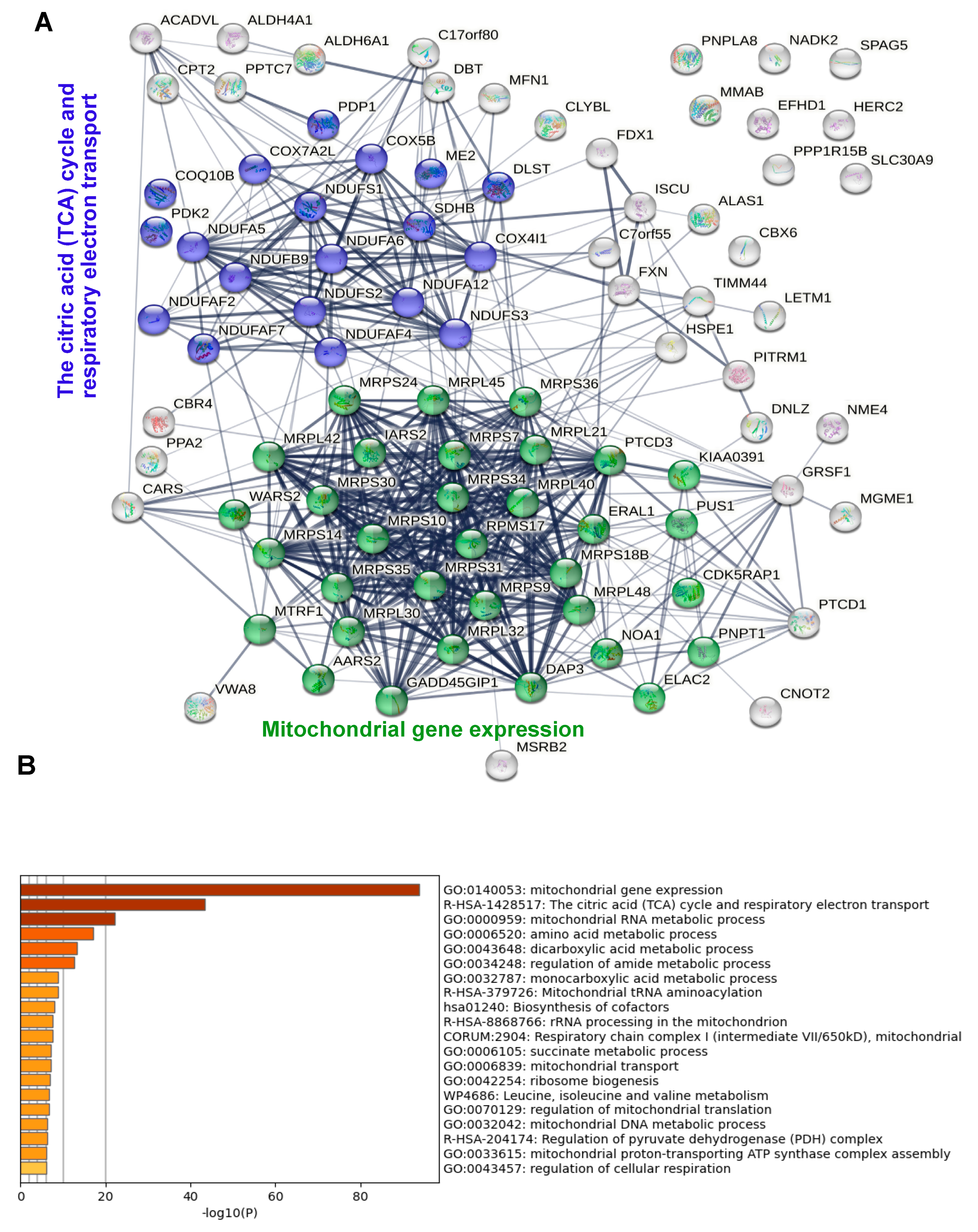
Both the confocal microscopy experiments and the results of the proteomics indicate that IDE localizes to the mitochondrial matrix, a conclusion also supported by the analysis of the IDE MTS using the DeepMito algorithm [
34]. Given the physiologic localization of cytochrome C oxidase, the same conclusion applied (not surprisingly) to the OVA control. A manual as well as a bioinformatics analysis of the proteomics results strongly suggested that mitochondrial IDE interacts with the mitochondrial protein translation machinery, and with the proteins of the respiratory chain. This was true when both controls were considered but was even more evident when only the no-biotin control was considered. In the latter case, six (wt IDE) and seven (MutIde) of the top ten enriched proteins were ribosomal subunits. Our data also suggest that wt and mutant IDE interact with the same mitochondrial proteins.
We were surprised to find a significant number of non-mitochondrial proteins, some of them linked to protein networks related to RNA processing and stress responses, in the IDE interactome. Given the absence of these proteins when only the no-biotin control was considered, this finding almost certainly resulted from the partial localization of IDE but not OVA fusion proteins in the cytosol due to distinct efficiencies of the IDE and OVA MTS. The relevance of the non-mitochondrial proteins and protein networks as IDE interactants will only become clear upon appropriately controlled investigations of Met42-encoded cytosolic IDE. However, proteomic and genomic studies of tissues from IDE-deficient mice in our laboratory are consistent with a role for cytosolic IDE in stress responses and RNA processing (Zhu et al., manuscript submitted).
Non-mitochondrial proteins were not only present in significant numbers in the IDE interactome when both controls were considered, but represented the vast majority of strongly enriched proteins when only the OVA control was considered, a finding requiring explanation. First, due to its strong MTS, OVA very likely was entirely absent from the cytosol, resulting in the extremely low background biotinylation of cytosolic proteins in the OVA sample, with the consequence of relatively strong enrichment factors for proteins biotinylated by cytosolic TurboId-IDE. Second, and because of this, the inspection of the complete list of enriched proteins relative to the OVA control (
Table S2) shows that many ribosomal and other mitochondrial proteins are present, but with lower enrichment factors. Third, the globally lower enrichment factors in the comparison of IDE to OVA controls only suggests that samples incubated with exogenous biotin may display a higher background and therefore a lower enrichment, a factor biasing the detection to cellular compartments devoid of an exogenous biotin ligase (the cytosol in OVA samples).
What might be the role of mitochondrial IDE and its interactions? The fact that almost all proteins biotinylated by both wt and mutant IDE have molecular weights inconsistent with the selectivity of IDE suggests that IDE may have few or no proteolytic substrates in mitochondria. We speculate that IDE may play a role in mitochondrial protein homeostasis and stress responses, possibly as a chaperone-like protein, as has been suggested for the cytosolic protein [
14,
35]. The impact of IDE on mitochondrial function at the steady state and in stress situations remains to be evaluated.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




