
Transcriptional Regulation Technology for Gene Perturbation in Fission Yeast

Abstract
:1. Introduction
2. Transcriptional Regulation Technologies for S. pombe
2.1. Promoter Replacement
2.2. 3′UTR Disruption
2.3. RNA Interference
2.4. CRISPR–Cas Based Gene Knockdown Technology
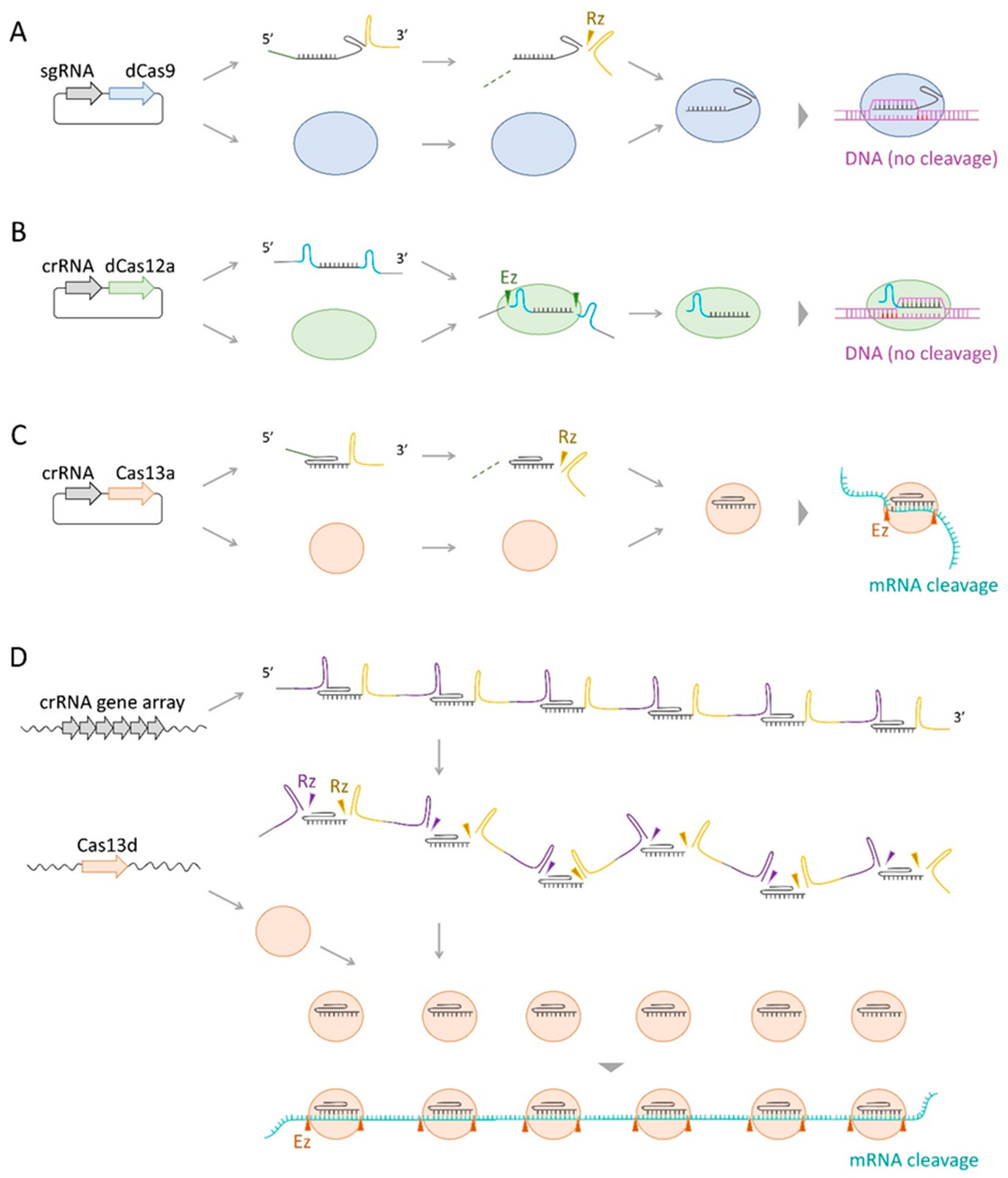
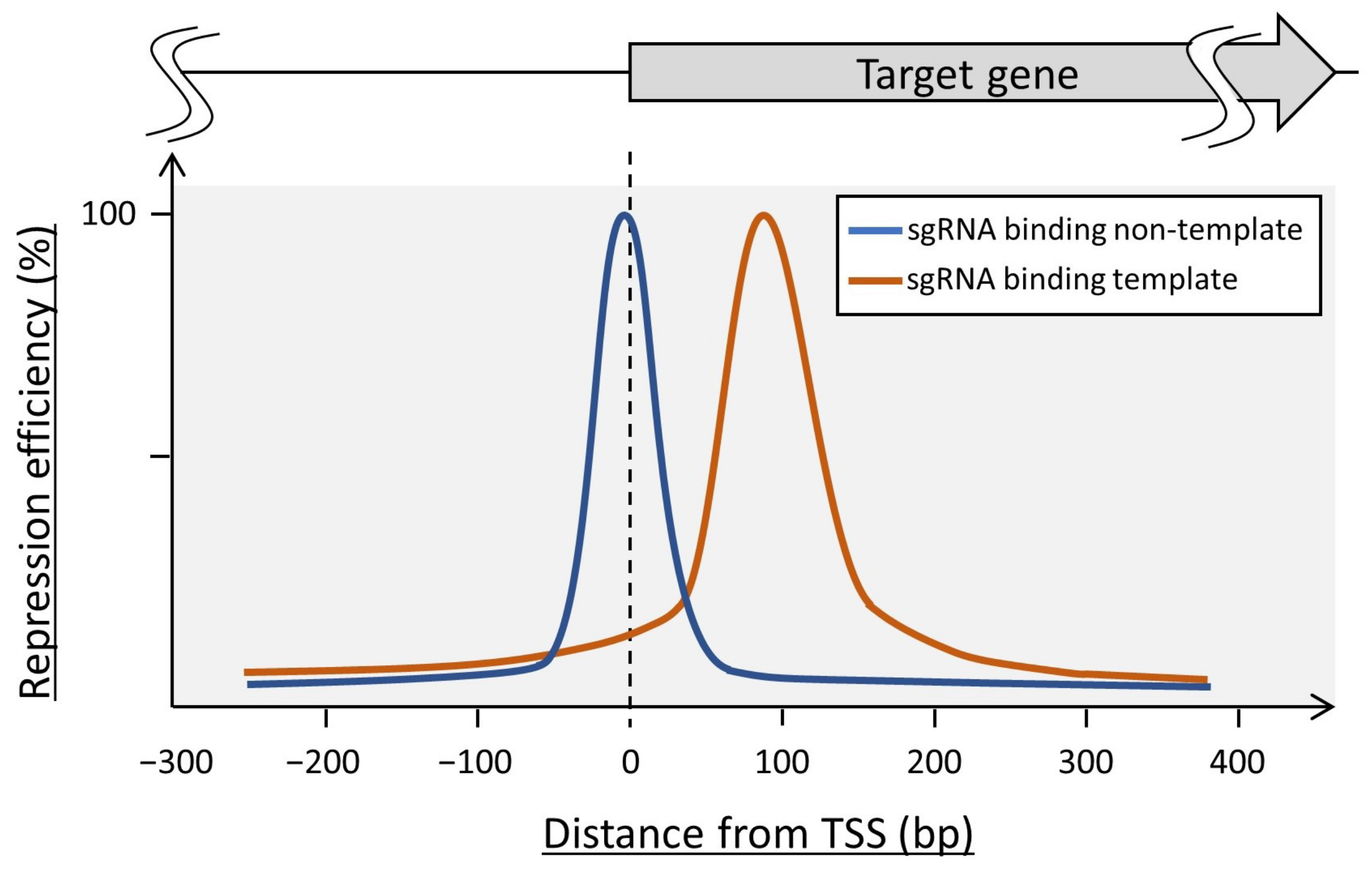
2.4.1. dCas9-Mediated CRISPR Interference
2.4.2. dCas12a-Mediated CRISPR Interference
2.4.3. Cas13-Mediated Gene Knockdown
3. Comparison of Gene Perturbation Technologies in S. pombe
4. Other CRISPR–Cas-Based Technologies on Transcriptional Manipulation
5. Applications of CRISPRi Resources
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3'UTR | 3’-untranslated region |
| AID | auxin-inducible degron |
| Cas | CRISPR associated |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeat |
| CRISPRi | CRISPR interference |
| crRNA | CRISPR RNA |
| DAmP | Decreased Abundance by mRNA Perturbation |
| FKBP12 | FK506 binding protein |
| FRB domain | FKBP12-rapamycin-binding domain (of human mTOR) |
| KRAB | Krüppel associated box |
| Mxi1 | MAX-interacting protein 1 |
| PAM | protospacer adjacent motif |
| RNAi | RNA interference |
| sgRNA | small guide RNA |
| snRNA | small nuclear RNA |
| tracrRNA | transactivating crRNA |
References
- Hoffman, C.S.; Wood, V.; Fantes, P.A. An Ancient Yeast for Young Geneticists: A Primer on the Schizosaccharomyces pombe Model System. Genetics 2015, 201, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Fantes, P.A.; Hoffman, C.S. A Brief History of Schizosaccharomyces pombe Research: A Perspective over the Past 70 Years. Genetics 2016, 203, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.U.; Hayles, J.; Kim, D.; Wood, V.; Park, H.O.; Won, M.; Yoo, H.S.; Duhig, T.; Nam, M.; Palmer, G.; et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2010, 28, 617–623. [Google Scholar] [CrossRef]
- Kanke, M.; Nishimura, K.; Kanemaki, M.; Kakimoto, T.; Takahashi, T.S.; Nakagawa, T.; Masukata, H. Auxin-inducible protein depletion system in fission yeast. BMC Cell Biol. 2011, 12, 8. [Google Scholar] [CrossRef]
- Zhang, X.R.; Zhao, L.; Suo, F.; Gao, Y.; Wu, Q.; Qi, X.; Du, L.L. An improved auxin-inducible degron system for fission yeast. G3 2022, 12, jkab393. [Google Scholar] [CrossRef]
- Haruki, H.; Nishikawa, J.; Laemmli, U.K. The anchor-away technique: Rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 2008, 31, 925–932. [Google Scholar] [CrossRef]
- Ding, L.; Laor, D.; Weisman, R.; Forsburg, S.L. Rapid regulation of nuclear proteins by rapamycin-induced translocation in fission yeast. Yeast 2014, 31, 253–264. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef]
- Iwaki, T.; Takegawa, K. A set of loxP marker cassettes for Cre-mediated multiple gene disruption in Schizosaccharomyces pombe. Biosci. Biotechnol. Biochem. 2004, 68, 545–550. [Google Scholar] [CrossRef]
- Hirashima, K.; Iwaki, T.; Takegawa, K.; Giga-Hama, Y.; Tohda, H. A simple and effective chromosome modification method for large-scale deletion of genome sequences and identification of essential genes in fission yeast. Nucleic Acids Res. 2006, 34, e11. [Google Scholar] [CrossRef]
- Ishii, K.; Ogiyama, Y.; Chikashige, Y.; Soejima, S.; Masuda, F.; Kakuma, T.; Hiraoka, Y.; Takahashi, K. Heterochromatin integrity affects chromosome reorganization after centromere dysfunction. Science 2008, 321, 1088–1091. [Google Scholar] [CrossRef]
- Bahler, J.; Wu, J.Q.; Longtine, M.S.; Shah, N.G.; McKenzie, A., 3rd; Steever, A.B.; Wach, A.; Philippsen, P.; Pringle, J.R. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 1998, 14, 943–951. [Google Scholar] [CrossRef]
- Schuldiner, M.; Collins, S.R.; Thompson, N.J.; Denic, V.; Bhamidipati, A.; Punna, T.; Ihmels, J.; Andrews, B.; Boone, C.; Greenblatt, J.F.; et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 2005, 123, 507–519. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Housden, B.E.; Muhar, M.; Gemberling, M.; Gersbach, C.A.; Stainier, D.Y.; Seydoux, G.; Mohr, S.E.; Zuber, J.; Perrimon, N. Loss-of-function genetic tools for animal models: Cross-species and cross-platform differences. Nat. Rev. Genet. 2017, 18, 24–40. [Google Scholar] [CrossRef]
- Daga, R.R.; Jimenez, J. Translational control of the cdc25 cell cycle phosphatase: A molecular mechanism coupling mitosis to cell growth. J. Cell Sci. 1999, 112 Pt 18, 3137–3146. [Google Scholar] [CrossRef]
- Hou, M.C.; Wiley, D.J.; Verde, F.; McCollum, D. Mob2p interacts with the protein kinase Orb6p to promote coordination of cell polarity with cell cycle progression. J. Cell Sci. 2003, 116, 125–135. [Google Scholar] [CrossRef]
- Shams-Eldin, H.; Azzouz, N.; Eckert, V.; Blaschke, T.; Kedees, M.H.; Hubel, A.; Schwarz, R.T. The Schizosaccharomyces pombe GPI8 gene complements a Saccharomyces cerevisiae GPI8 anchoring mutant. Yeast 2001, 18, 33–39. [Google Scholar] [CrossRef]
- Forsburg, S.L. Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res. 1993, 21, 2955–2956. [Google Scholar] [CrossRef] [PubMed]
- Roguev, A.; Bandyopadhyay, S.; Zofall, M.; Zhang, K.; Fischer, T.; Collins, S.R.; Qu, H.; Shales, M.; Park, H.O.; Hayles, J.; et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science 2008, 322, 405–410. [Google Scholar] [CrossRef]
- Muhlrad, D.; Parker, R. Aberrant mRNAs with extended 3’ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 1999, 5, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Cameron, D.M.; Collins, S.R.; Schuldiner, M.; Stewart-Ornstein, J.; Newman, H.W.; Braun, S.; Madhani, H.D.; Krogan, N.J.; Weissman, J.S. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 2008, 5, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef]
- Hall, I.M.; Shankaranarayana, G.D.; Noma, K.; Ayoub, N.; Cohen, A.; Grewal, S.I. Establishment and maintenance of a heterochromatin domain. Science 2002, 297, 2232–2237. [Google Scholar] [CrossRef]
- Raponi, M.; Arndt, G.M. Double-stranded RNA-mediated gene silencing in fission yeast. Nucleic Acids Res. 2003, 31, 4481–4489. [Google Scholar] [CrossRef]
- Sigova, A.; Rhind, N.; Zamore, P.D. A single Argonaute protein mediates both transcriptional and posttranscriptional silencing in Schizosaccharomyces pombe. Genes Dev. 2004, 18, 2359–2367. [Google Scholar] [CrossRef]
- Iida, T.; Nakayama, J.; Moazed, D. siRNA-mediated heterochromatin establishment requires HP1 and is associated with antisense transcription. Mol. Cell 2008, 31, 178–189. [Google Scholar] [CrossRef]
- Simmer, F.; Buscaino, A.; Kos-Braun, I.C.; Kagansky, A.; Boukaba, A.; Urano, T.; Kerr, A.R.; Allshire, R.C. Hairpin RNA induces secondary small interfering RNA synthesis and silencing in trans in fission yeast. EMBO Rep. 2010, 11, 112–118. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Zhao, Y.; Boeke, J.D. CRISPR-Cas12a system in fission yeast for multiplex genomic editing and CRISPR interference. Nucleic Acids Res. 2020, 48, 5788–5798. [Google Scholar] [CrossRef]
- Ishikawa, K.; Soejima, S.; Masuda, F.; Saitoh, S. Implementation of dCas9-mediated CRISPRi in the fission yeast Schizosaccharomyces pombe. G3 2021, 11, jkab051. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, S.; Fu, C. Shotgun knockdown of RNA by CRISPR-Cas13d in fission yeast. J. Cell Sci. 2023, 136, jcs260769. [Google Scholar] [CrossRef]
- Jing, X.; Xie, B.; Chen, L.; Zhang, N.; Jiang, Y.; Qin, H.; Wang, H.; Hao, P.; Yang, S.; Li, X. Implementation of the CRISPR-Cas13a system in fission yeast and its repurposing for precise RNA editing. Nucleic Acids Res. 2018, 46, e90. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef]
- Hayashi, A.; Tanaka, K. Short-Homology-Mediated CRISPR/Cas9-Based Method for Genome Editing in Fission Yeast. G3 2019, 9, 1153–1163. [Google Scholar] [CrossRef]
- Jacobs, J.Z.; Ciccaglione, K.M.; Tournier, V.; Zaratiegui, M. Implementation of the CRISPR-Cas9 system in fission yeast. Nat. Commun. 2014, 5, 5344. [Google Scholar] [CrossRef]
- Rodriguez-Lopez, M.; Cotobal, C.; Fernandez-Sanchez, O.; Borbaran Bravo, N.; Oktriani, R.; Abendroth, H.; Uka, D.; Hoti, M.; Wang, J.; Zaratiegui, M.; et al. A CRISPR/Cas9-based method and primer design tool for seamless genome editing in fission yeast. Wellcome Open Res. 2016, 1, 19. [Google Scholar] [CrossRef]
- Zhang, X.R.; He, J.B.; Wang, Y.Z.; Du, L.L. A Cloning-Free Method for CRISPR/Cas9-Mediated Genome Editing in Fission Yeast. G3 2018, 8, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Soejima, S.; Saitoh, S. Genetic knockdown of obscure, conserved, essential genes using CRISPR interference methods in the fission yeast S. pombe. J. Cell Sci. 2023, in press. [Google Scholar]
- Horlbeck, M.A.; Witkowsky, L.B.; Guglielmi, B.; Replogle, J.M.; Gilbert, L.A.; Villalta, J.E.; Torigoe, S.E.; Tjian, R.; Weissman, J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 2016, 5, e12677. [Google Scholar] [CrossRef]
- Smith, J.D.; Suresh, S.; Schlecht, U.; Wu, M.; Wagih, O.; Peltz, G.; Davis, R.W.; Steinmetz, L.M.; Parts, L.; St Onge, R.P. Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol. 2016, 17, 45. [Google Scholar] [CrossRef]
- Givens, R.M.; Lai, W.K.; Rizzo, J.M.; Bard, J.E.; Mieczkowski, P.A.; Leatherwood, J.; Huberman, J.A.; Buck, M.J. Chromatin architectures at fission yeast transcriptional promoters and replication origins. Nucleic Acids Res. 2012, 40, 7176–7189. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Fonfara, I.; Richter, H.; Bratovic, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Cheng, Q.; Zheng, X.; Zhao, G.; Wang, J. Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov. 2017, 3, 17018. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e614. [Google Scholar] [CrossRef]
- Yan, W.X.; Chong, S.; Zhang, H.; Makarova, K.S.; Koonin, E.V.; Cheng, D.R.; Scott, D.A. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 2018, 70, 327–339.e325. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Guo, X.; Li, Z.; Cao, L.; Liu, S.; Guo, Y.; Wang, G.; Luo, Y.; Zhang, Z.; et al. The collateral activity of RfxCas13d can induce lethality in a RfxCas13d knock-in mouse model. Genome Biol. 2023, 24, 20. [Google Scholar] [CrossRef]
- Guo, L.Y.; Bian, J.; Davis, A.E.; Liu, P.; Kempton, H.R.; Zhang, X.; Chemparathy, A.; Gu, B.; Lin, X.; Rane, D.A.; et al. Multiplexed genome regulation in vivo with hyper-efficient Cas12a. Nat. Cell Biol. 2022, 24, 590–600. [Google Scholar] [CrossRef]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Greenside, P.G.; Natoli, T.; Lahr, D.L.; Wadden, D.; Tirosh, I.; Narayan, R.; Root, D.E.; Golub, T.R.; Subramanian, A.; et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017, 15, e2003213. [Google Scholar] [CrossRef] [PubMed]
- Stojic, L.; Lun, A.T.L.; Mangei, J.; Mascalchi, P.; Quarantotti, V.; Barr, A.R.; Bakal, C.; Marioni, J.C.; Gergely, F.; Odom, D.T. Specificity of RNAi, LNA and CRISPRi as loss-of-function methods in transcriptional analysis. Nucleic Acids Res. 2018, 46, 5950–5966. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Nishimasu, H.; Shi, X.; Ishiguro, S.; Gao, L.; Hirano, S.; Okazaki, S.; Noda, T.; Abudayyeh, O.O.; Gootenberg, J.S.; Mori, H.; et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 2018, 361, 1259–1262. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Momen-Roknabadi, A.; Oikonomou, P.; Zegans, M.; Tavazoie, S. An inducible CRISPR interference library for genetic interrogation of Saccharomyces cerevisiae biology. Commun. Biol. 2020, 3, 723. [Google Scholar] [CrossRef]
- McGlincy, N.J.; Meacham, Z.A.; Reynaud, K.K.; Muller, R.; Baum, R.; Ingolia, N.T. A genome-scale CRISPR interference guide library enables comprehensive phenotypic profiling in yeast. BMC Genom. 2021, 22, 205. [Google Scholar] [CrossRef]
- Muller, R.; Meacham, Z.A.; Ferguson, L.; Ingolia, N.T. CiBER-seq dissects genetic networks by quantitative CRISPRi profiling of expression phenotypes. Science 2020, 370, eabb9662. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Method | Material Preparation | Conditional Induction | Systematic Design | Relative Throughput | Relative Specificity |
|---|---|---|---|---|---|
| genetic mutation | genetic screening | Yes a | No | + | +++ |
| AID | degron tag insertion | Yes | Yes | ++ | +++ |
| anchor-away | FRB tag insertion | Yes | Yes | ++ | +++ |
| Cre–loxP | loxP sites insertion | Yes | Yes | ++ | +++ |
| promoter replacement | promoter replacement | Yes | Yes | ++ | +++ |
| DAmP | marker gene insertion | No | Yes | ++ | +++ |
| RNAi | long hairpin RNA (chromosome or plasmid) | Yes | No | ++ | + |
| CRISPRi (dCas9) | short oligo DNA cloning (plasmid) | Yes | Yes | +++ | ++ |
| CRISPRi (dCas12a) | short oligo DNA cloning (plasmid) | No b | No c | ++ c | ++ |
| Cas13a | short oligo DNA cloning (plasmid) | Yes | No c | ++ c | ++ d |
| Cas13d | multiplex crRNA gene cloning (chromosome) | Yes | Yes | ++ | ++ d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishikawa, K.; Saitoh, S. Transcriptional Regulation Technology for Gene Perturbation in Fission Yeast. Biomolecules 2023, 13, 716. https://doi.org/10.3390/biom13040716
Ishikawa K, Saitoh S. Transcriptional Regulation Technology for Gene Perturbation in Fission Yeast. Biomolecules. 2023; 13(4):716. https://doi.org/10.3390/biom13040716
Chicago/Turabian StyleIshikawa, Ken, and Shigeaki Saitoh. 2023. "Transcriptional Regulation Technology for Gene Perturbation in Fission Yeast" Biomolecules 13, no. 4: 716. https://doi.org/10.3390/biom13040716