
Electronic Circular Dichroism Detects Conformational Changes Associated with Proteasome Gating Confirmed Using AFM Imaging

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Secondary Structure Analysis
2.2. Electronic Circular Dichroism (ECD) Experiments
2.3. Atomic Force Microscopy (AFM) Imaging
3. Results and Discussion
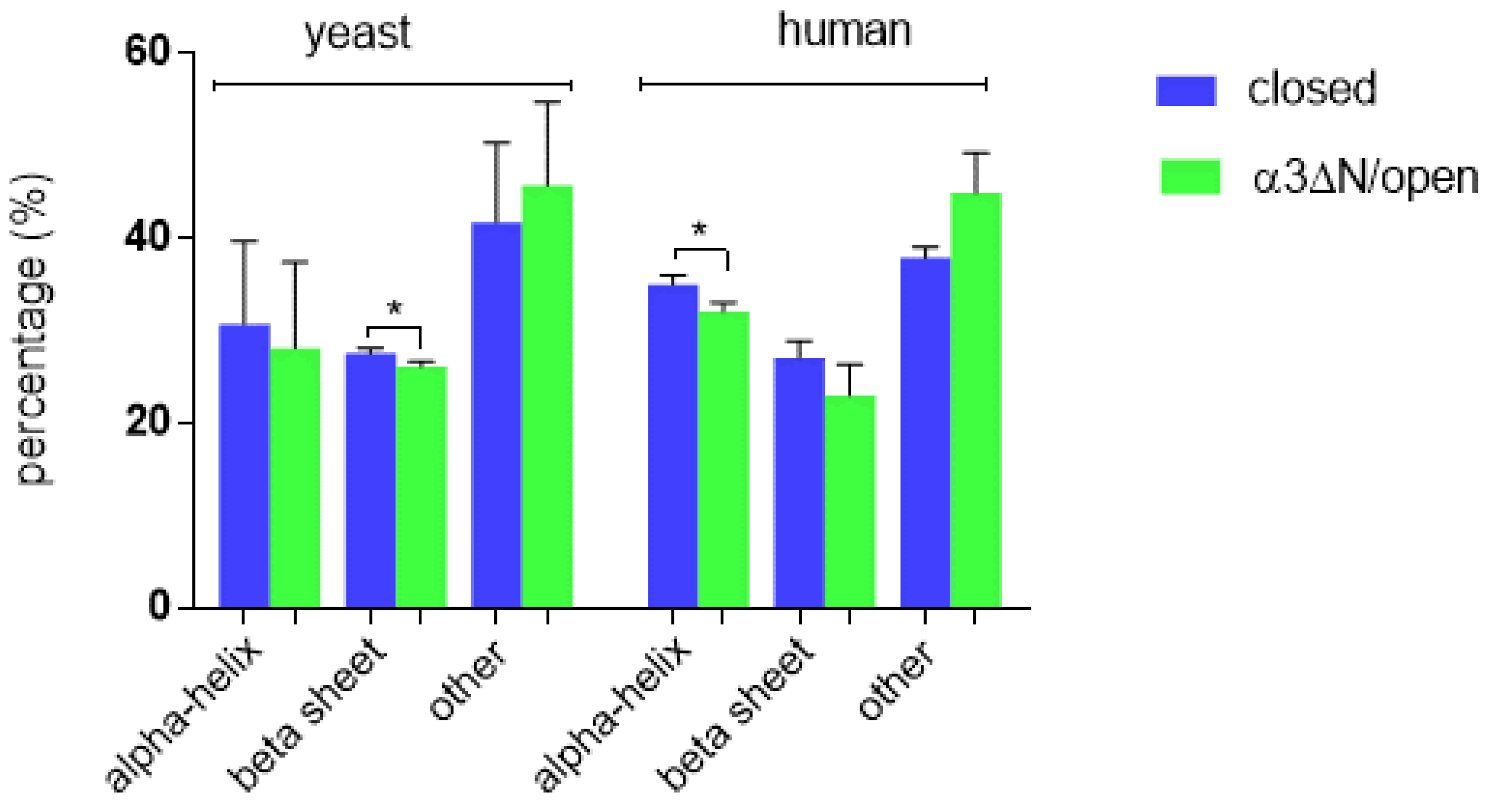
3.1. Structural Analysis
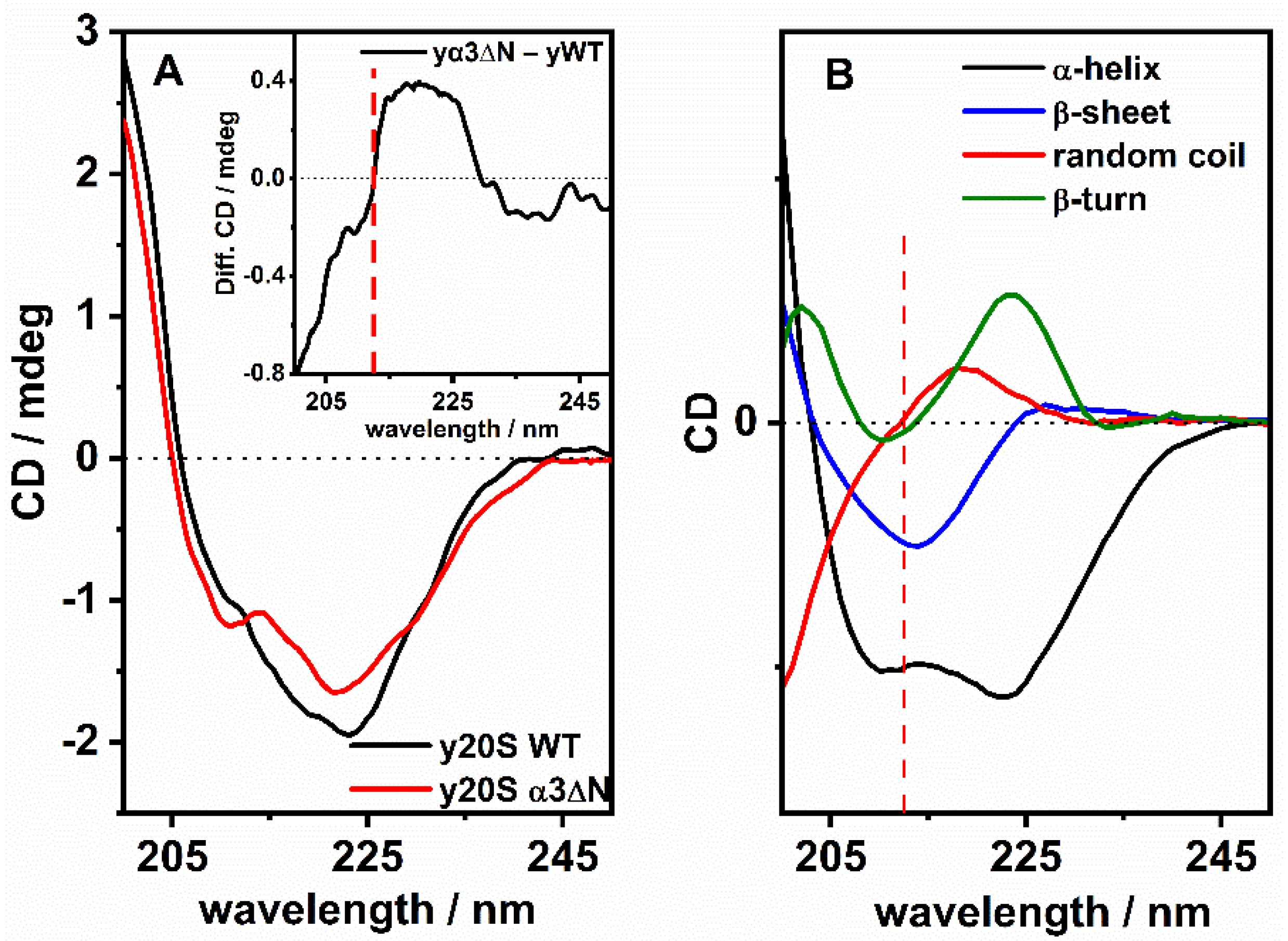
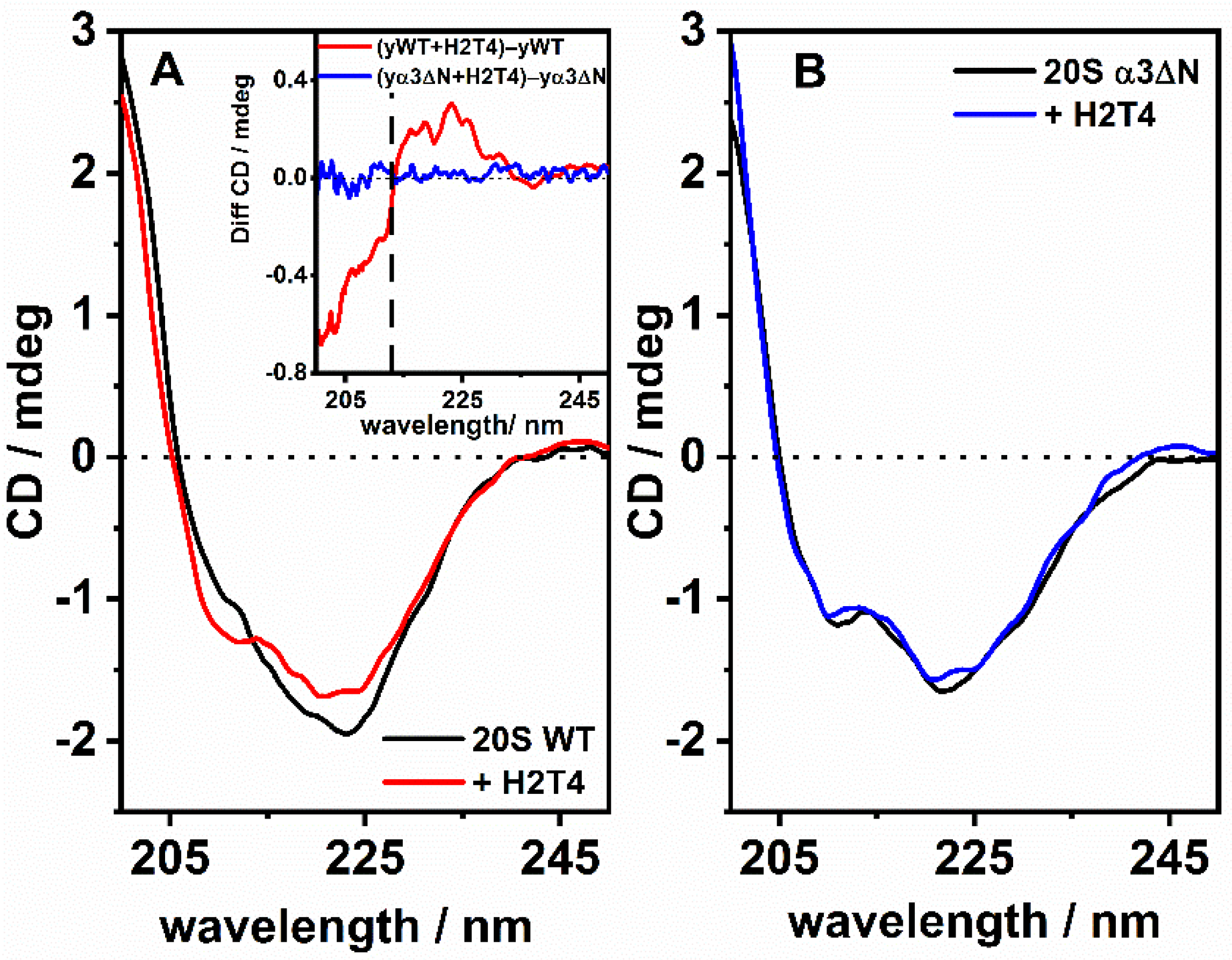
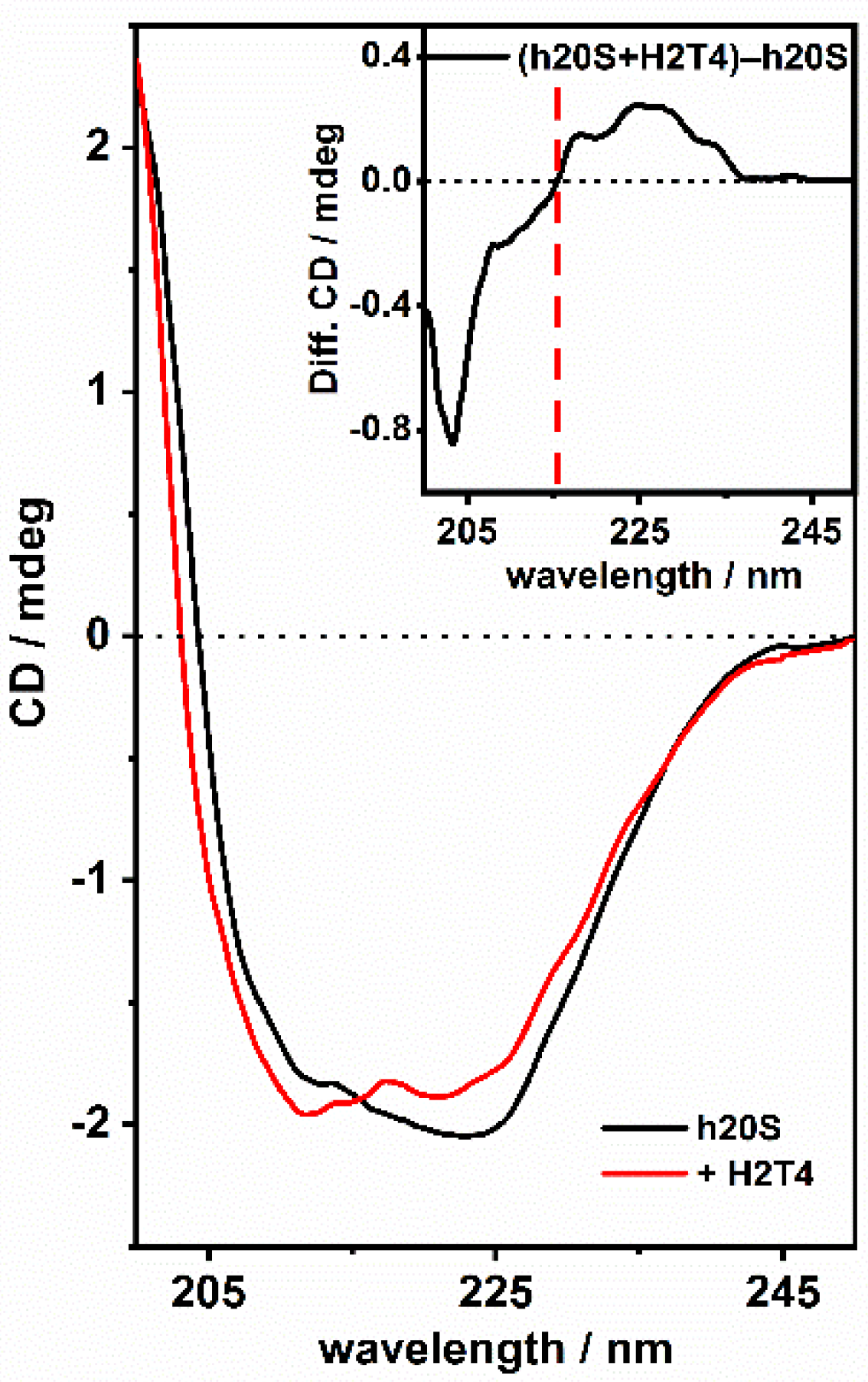
3.2. ECD Studies
3.3. Atomic Force Microscopy (AFM) Imaging
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rousseau, A.; Bertolotti, A. An Evolutionarily Conserved Pathway Controls Proteasome Homeostasis. Nature 2016, 536, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef]
- Deshmukh, F.K.; Yaffe, D.; Olshina, M.A.; Ben-Nissan, G.; Sharon, M. The Contribution of the 20s Proteasome to Proteostasis. Biomolecules 2019, 9, 190. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Tundo, G.R.; Coletta, A.; Marcoux, J.; Koufogeorgou, E.I.; Ciaccio, C.; Santoro, A.M.; Milardi, D.; Grasso, G.; Cozza, P.; et al. The Insulin-Degrading Enzyme Is an Allosteric Modulator of the 20S Proteasome and a Potential Competitor of the 19S. Cell. Mol. Life. Sci. 2018, 75, 3441–3456. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Bianculli, A.; Orlandi, A.; Desimio, M.G.; Arcuri, G.; Coletta, M.; Marini, S. Insulin-Degrading Enzyme (IDE): A Novel Heat Shock-like Protein. J. Biol. Chem. 2013, 288, 2281–2289. [Google Scholar] [CrossRef]
- Gaczynska, M.; Osmulski, P.A. Targeting Protein–Protein Interactions in the Ubiquitin–Proteasome Pathway. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 10, ISBN 978-0-12-814344-5. [Google Scholar]
- Ehlinger, A.; Walters, K.J. Structural Insights into Proteasome Activation by the 19S Regulatory Particle. Biochemistry 2013, 52, 3618–3628. [Google Scholar] [CrossRef]
- Sahu, I.; Glickman, M.H. Proteasome in Action: Substrate Degradation by the 26S Proteasome. Biochem. Soc. Trans. 2021, 49, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Kevei, É.; Kleifeld, O.; Krutauz, D.; Segref, A.; Rinaldi, T.; Erpapazoglou, Z.; Cohen, M.; Reis, N.; Hoppe, T.; et al. Reversible 26S Proteasome Disassembly upon Mitochondrial Stress. Cell Rep. 2014, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Ukmar-Godec, T.; Fang, P.; Ibáñez de Opakua, A.; Henneberg, F.; Godec, A.; Pan, K.-T.; Cima-Omori, M.-S.; Chari, A.; Mandelkow, E.; Urlaub, H.; et al. Proteasomal Degradation of the Intrinsically Disordered Protein Tau at Single-Residue Resolution. Sci. Adv. 2020, 6, eaba3916. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M.; Ditzel, L.; Groll, M.; Hartmann, C.; Huber, R. The Proteasome. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 295–317. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef]
- de la Peña, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-Engaged 26S Proteasome Structures Reveal Mechanisms for ATP-Hydrolysis–Driven Translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Gaczynska, M. Nanoenzymology of the 20S Proteasome: Proteasomal Actions Are Controlled by the Allosteric Transition. Biochemistry 2002, 41, 7047–7053. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M.; da Cunha, F.M. The Physiological Role of the Free 20S Proteasome in Protein Degradation: A Critical Review. Biochim. Et. Biophys. Acta. Gen. Subj. 2018, 1862, 2948–2954. [Google Scholar] [CrossRef] [PubMed]
- Rabl, J.; Smith, D.M.; Yu, Y.; Chang, S.-C.; Goldberg, A.L.; Cheng, Y. Mechanism of Gate Opening in the 20S Proteasome by the Proteasomal ATPases. Mol. Cell 2008, 30, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Kay, L.E. Proteasome Allostery as a Population Shift between Interchanging Conformers. Proc. Natl. Acad. Sci. USA 2012, 109, E3454–E3462. [Google Scholar] [CrossRef] [PubMed]
- Gaczynska, M.; Osmulski, P.A. Harnessing Proteasome Dynamics and Allostery in Drug Design. Antioxid. Redox. Signal. 2014, 21, 2286–2301. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.; Lu, Y.; Ma, Y.-B.; Lee, B.-H.; Yu, Z.; Ouyang, Q.; Finley, D.J.; Kirschner, M.W.; Mao, Y. Structural Basis for Dynamic Regulation of the Human 26S Proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, 12991–12996. [Google Scholar] [CrossRef]
- Santoro, A.M.; D’urso, A.; Cunsolo, A.; Milardi, D.; Purrello, R.; Sbardella, D.; Tundo, G.R.; Diana, D.; Fattorusso, R.; Di Dato, A.; et al. Cooperative Binding of the Cationic Porphyrin Tris-T4 Enhances Catalytic Activity of 20S Proteasome Unveiling a Complex Distribution of Functional States. Int. J. Mol. Sci. 2020, 21, 7190. [Google Scholar] [CrossRef] [PubMed]
- Giżyńska, M.; Witkowska, J.; Karpowicz, P.; Rostankowski, R.; Chocron, E.S.; Pickering, A.M.; Osmulski, P.; Gaczynska, M.; Jankowska, E. Proline- and Arginine-Rich Peptides as Flexible Allosteric Modulators of Human Proteasome Activity. J. Med. Chem. 2019, 62, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM Structures and Dynamics of Substrate-Engaged Human 26S Proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Giletto, M.B.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.E.; Tepe, J.J. Pipecolic Esters as Minimized Templates for Proteasome Inhibition. Org. Biomol. Chem. 2019, 17, 2734–2746. [Google Scholar] [CrossRef]
- Rennella, E.; Huang, R.; Yu, Z.; Kay, L.E. Exploring Long-Range Cooperativity in the 20S Proteasome Core Particle from Thermoplasma Acidophilum Using Methyl-TROSY-Based NMR. Proc. Natl. Acad. Sci. USA 2020, 117, 5298–5309. [Google Scholar] [CrossRef] [PubMed]
- Di Dato, A.; Cunsolo, A.; Persico, M.; Santoro, A.M.; D’Urso, A.; Milardi, D.; Purrello, R.; Stefanelli, M.; Paolesse, R.; Tundo, G.R.; et al. Electrostatic Map of Proteasome α-Rings Encodes The Design of Allosteric Porphyrin-Based Inhibitors Able to Affect 20S Conformation by Cooperative Binding. Sci. Rep. 2017, 7, 17098. [Google Scholar] [CrossRef]
- Santoro, A.M.; Cunsolo, A.; D’Urso, A.; Sbardella, D.; Tundo, G.R.; Ciaccio, C.; Coletta, M.; Diana, D.; Fattorusso, R.; Persico, M.; et al. Cationic Porphyrins are Tunable Gatekeepers of the 20S Proteasome. Chem. Sci. 2016, 7, 1286. [Google Scholar] [CrossRef]
- Sprangers, R.; Li, X.; Mao, X.; Rubinstein, J.L.; Schimmer, A.D.; Kay, L.E. TROSY-Based NMR Evidence for a Novel Class of 20S Proteasome Inhibitors. Biochemistry 2008, 47, 6727–6734. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The Inhibition Mechanism of Human 20S Proteasomes Enables Next-Generation Inhibitor Design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S Proteasome from Yeast at 2.4Å Resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A Gated Channel into the Proteasome Core Particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Klose, D.P.; Wallace, B.A.; Janes, R.W. 2Struc: The Secondary Structure Server. Bioinformatics 2010, 26, 2624–2625. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Andersen, C.A.F.; Palmer, A.G.; Brunak, S.; Rost, B. Continuum Secondary Structure Captures Protein Flexibility. Structure 2002, 10, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-Based Protein Secondary Structure Assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Johnson, W.C. Assigning Secondary Structure from Protein Coordinate Data. Proteins 1999, 35, 313–320. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Karpowicz, P.; Jankowska, E.; Bohmann, J.; Pickering, A.M.; Gaczyńska, M. New Peptide-Based Pharmacophore Activates 20S Proteasome. Molecules 2020, 25, 1439. [Google Scholar] [CrossRef] [PubMed]
- Osmulski, P.A.; Hochstrasser, M.; Gaczynska, M. A Tetrahedral Transition State at the Active Sites of the 20S Proteasome is Coupled to Opening of the α-Ring Channel. Structure 2009, 17, 1137–1147. [Google Scholar] [CrossRef]
- Osmulski, P.A.; Gaczynska, M. Rapamycin Allosterically Inhibits the Proteasome. Mol. Pharm. 2013, 84, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Dutaud, D.; Aubry, L.; Sentandreu, M.A.; Ouali, A. Bovine Muscle 20S Proteasome: I. Simple Purification Procedure and Enzymatic Characterization in Relation with Postmortem Conditions. Meat Sci. 2006, 74, 327–336. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
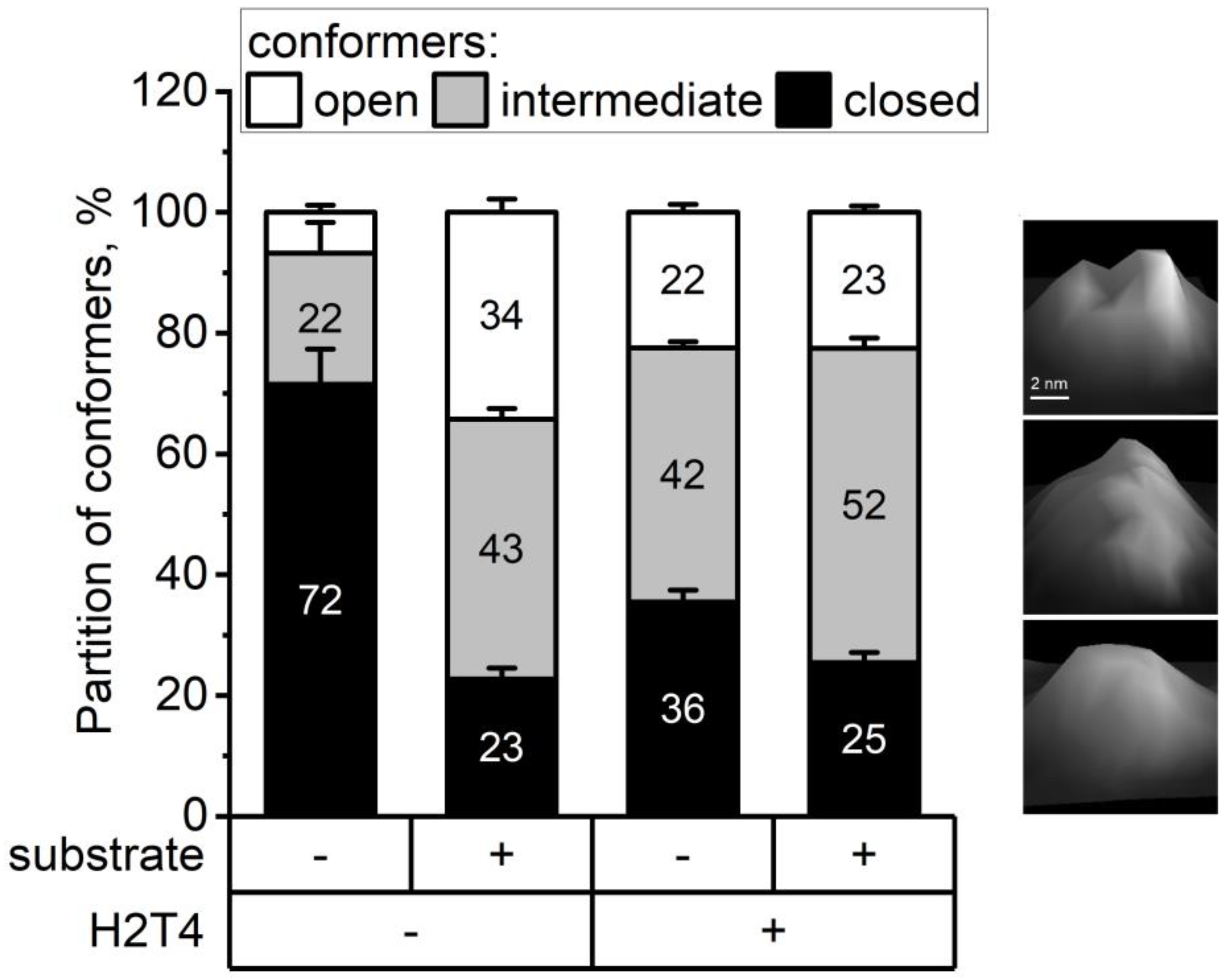{kind=link}
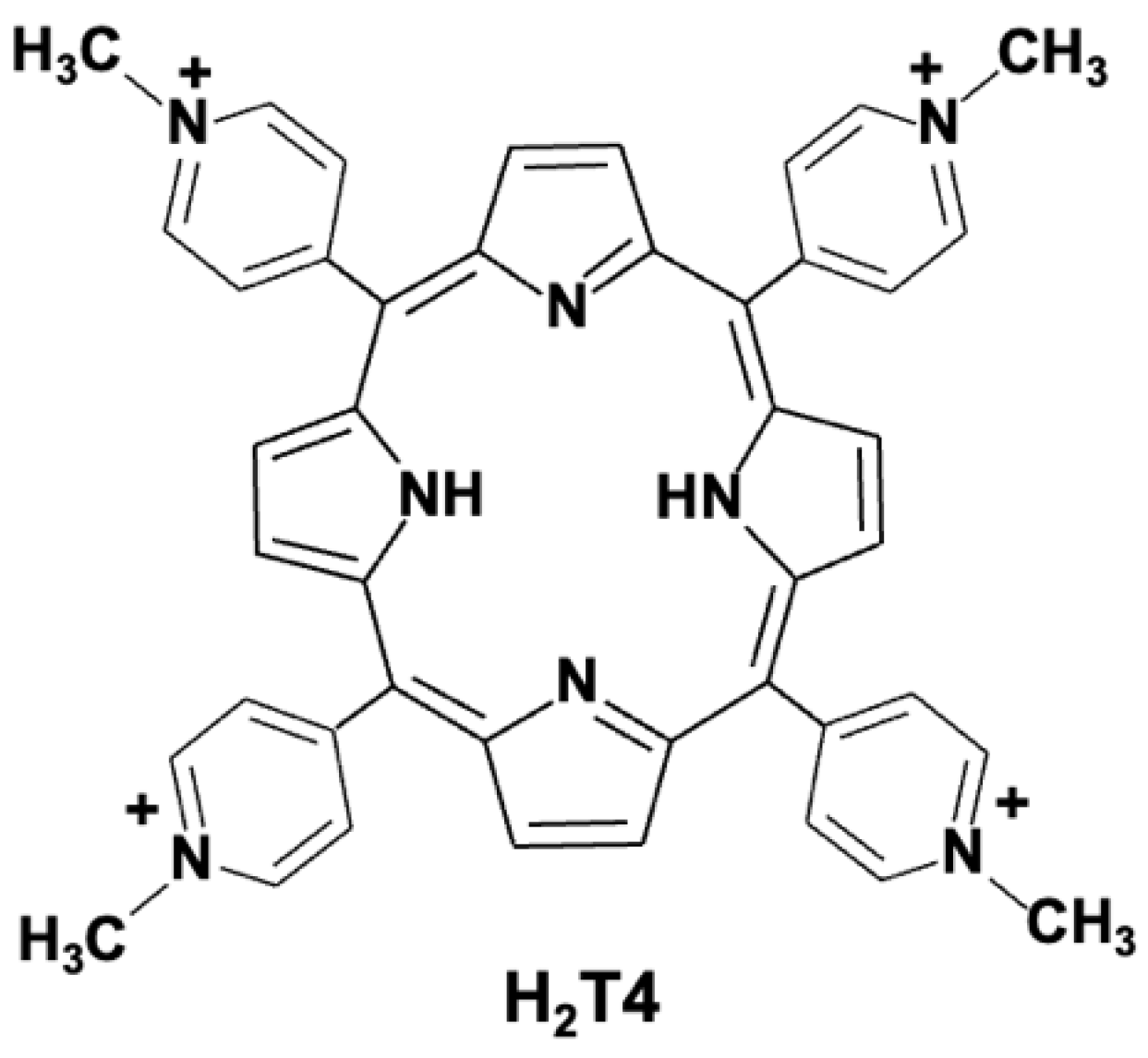{kind=link}
| 20S Conformational State | PDB ID | Organism | Secondary Structure | DSSP a | XTLSSTR | STRIDE |
|---|---|---|---|---|---|---|
| Closed | 5LE5 (1.80 Å) | Homo sapiens | α-Helix | 34.1 | 36.1 | 34.8 |
| β-Sheet | 27.7 | 25.0 | 28.5 | |||
| Other | 38.2 | 38.9 | 36.7 | |||
| Open | 6MSK (3.2 Å) | α-Helix | 32.4 | 30.9 | 32.8 | |
| β-Sheet | 23.7 | 19.4 | 26.0 | |||
| Other | 43.9 | 49.7 | 41.2 | |||
| Closed | 1RYP (1.90 Å) | Saccharomyces cerevisiae | α-Helix | 35.0 | 36.8 | 20.4 |
| β-Sheet | 27.8 | 26.8 | 27.9 | |||
| Other | 37.2 | 36.4 | 51.7 | |||
| Mutant/Open | 1G0U (2.40 Å) | α-Helix | 33.4 | 33.6 | 17.4 | |
| β-Sheet | 26.4 | 25.5 | 26.5 | |||
| Other | 40.2 | 40.9 | 56.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Urso, A.; Purrello, R.; Cunsolo, A.; Milardi, D.; Fattorusso, C.; Persico, M.; Gaczynska, M.; Osmulski, P.A.; Santoro, A.M. Electronic Circular Dichroism Detects Conformational Changes Associated with Proteasome Gating Confirmed Using AFM Imaging. Biomolecules 2023, 13, 704. https://doi.org/10.3390/biom13040704
D’Urso A, Purrello R, Cunsolo A, Milardi D, Fattorusso C, Persico M, Gaczynska M, Osmulski PA, Santoro AM. Electronic Circular Dichroism Detects Conformational Changes Associated with Proteasome Gating Confirmed Using AFM Imaging. Biomolecules. 2023; 13(4):704. https://doi.org/10.3390/biom13040704
Chicago/Turabian StyleD’Urso, Alessandro, Roberto Purrello, Alessandra Cunsolo, Danilo Milardi, Caterina Fattorusso, Marco Persico, Maria Gaczynska, Pawel A. Osmulski, and Anna Maria Santoro. 2023. "Electronic Circular Dichroism Detects Conformational Changes Associated with Proteasome Gating Confirmed Using AFM Imaging" Biomolecules 13, no. 4: 704. https://doi.org/10.3390/biom13040704