
Functional Proteomic Profiling Analysis in Four Major Types of Gastrointestinal Cancers

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Subsection Acquisition and Preprocessing of TCPA and TCGA Datasets
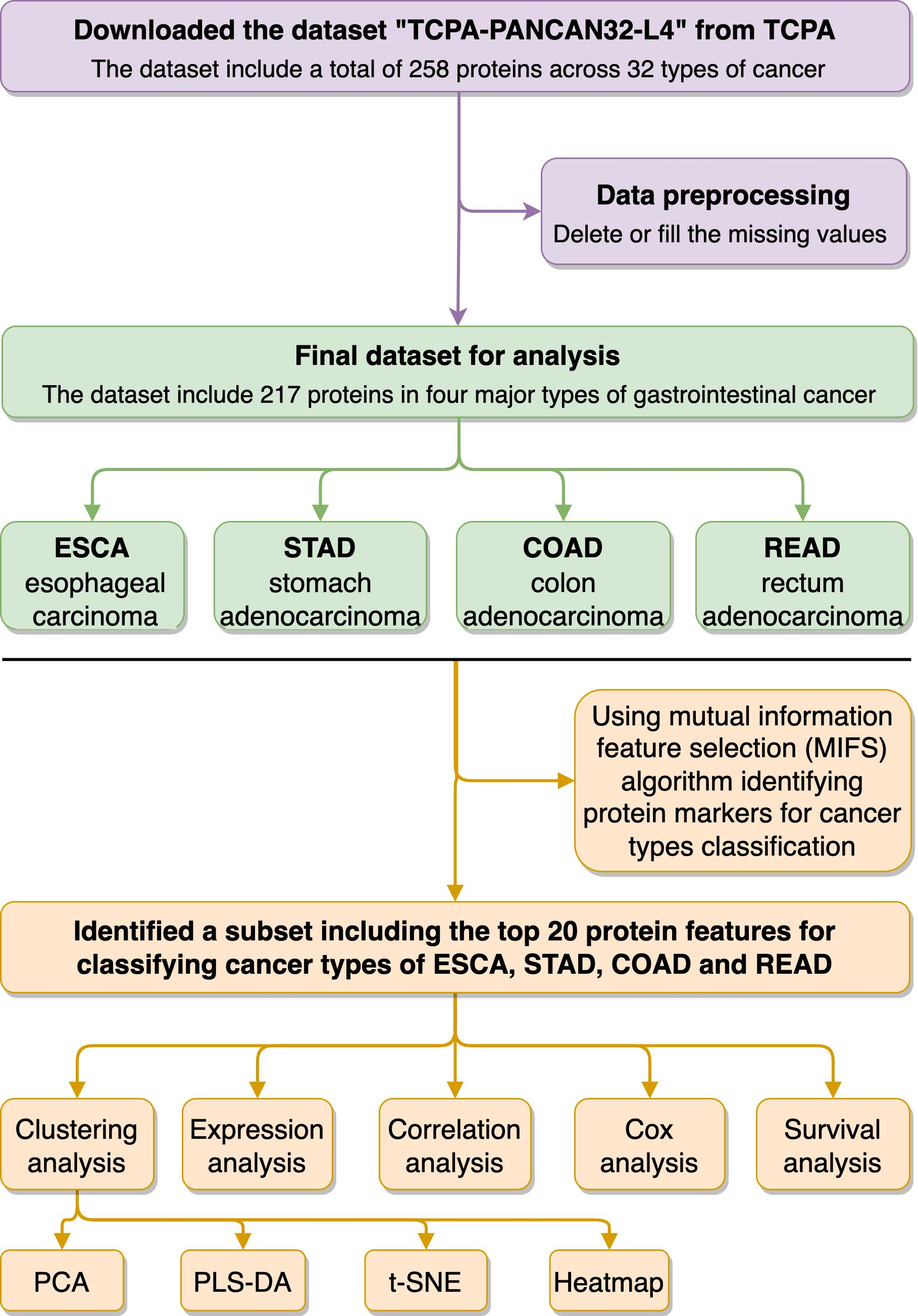
2.2. Functional Proteome Profiling Analysis in the Four Types of Gastrointestinal Cancers
2.3. Using Feature Selection Approaches to Identify Protein Signatures for Classifying Different Cancer Types
2.4. Statistical Analysis
3. Results
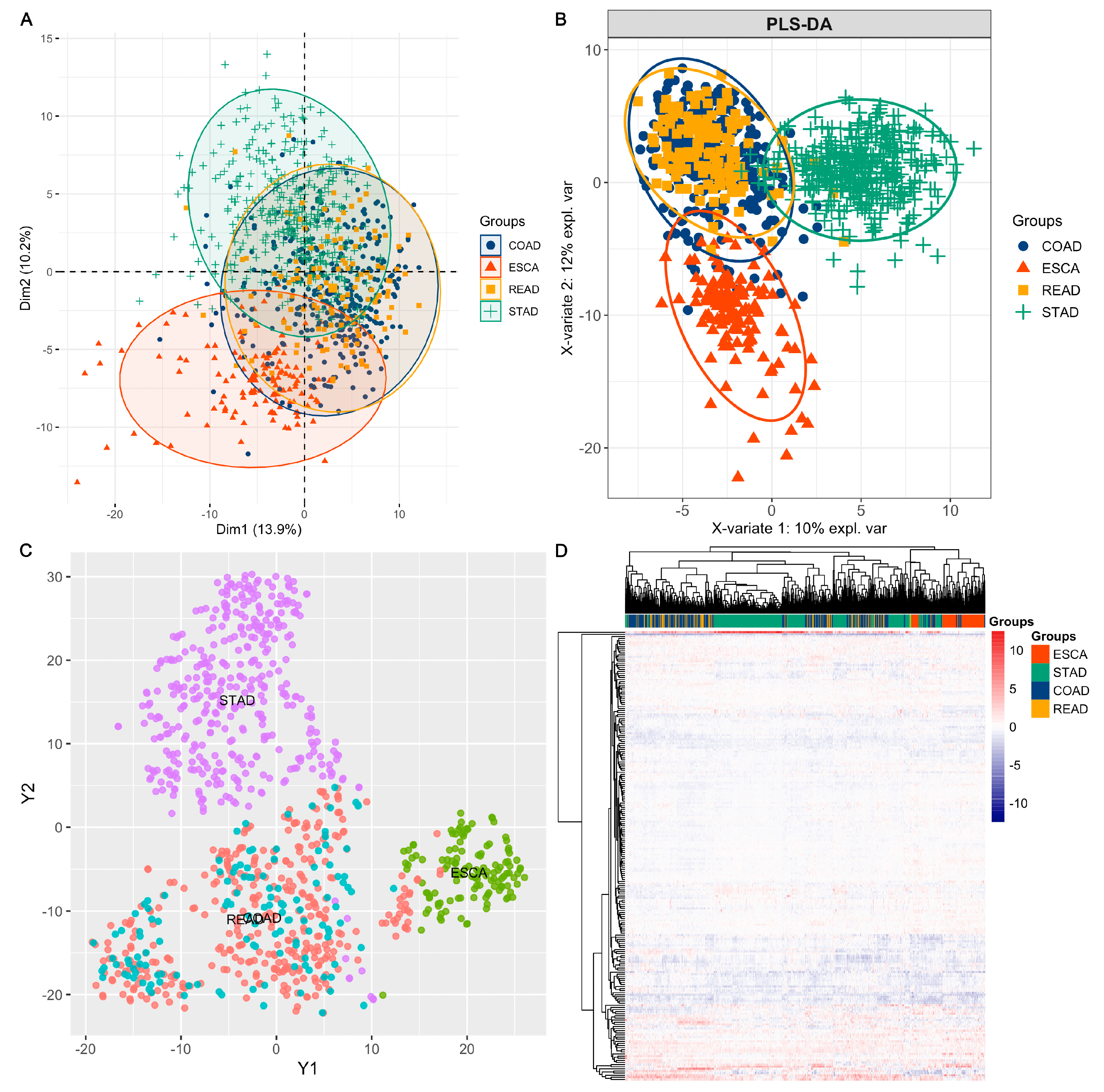
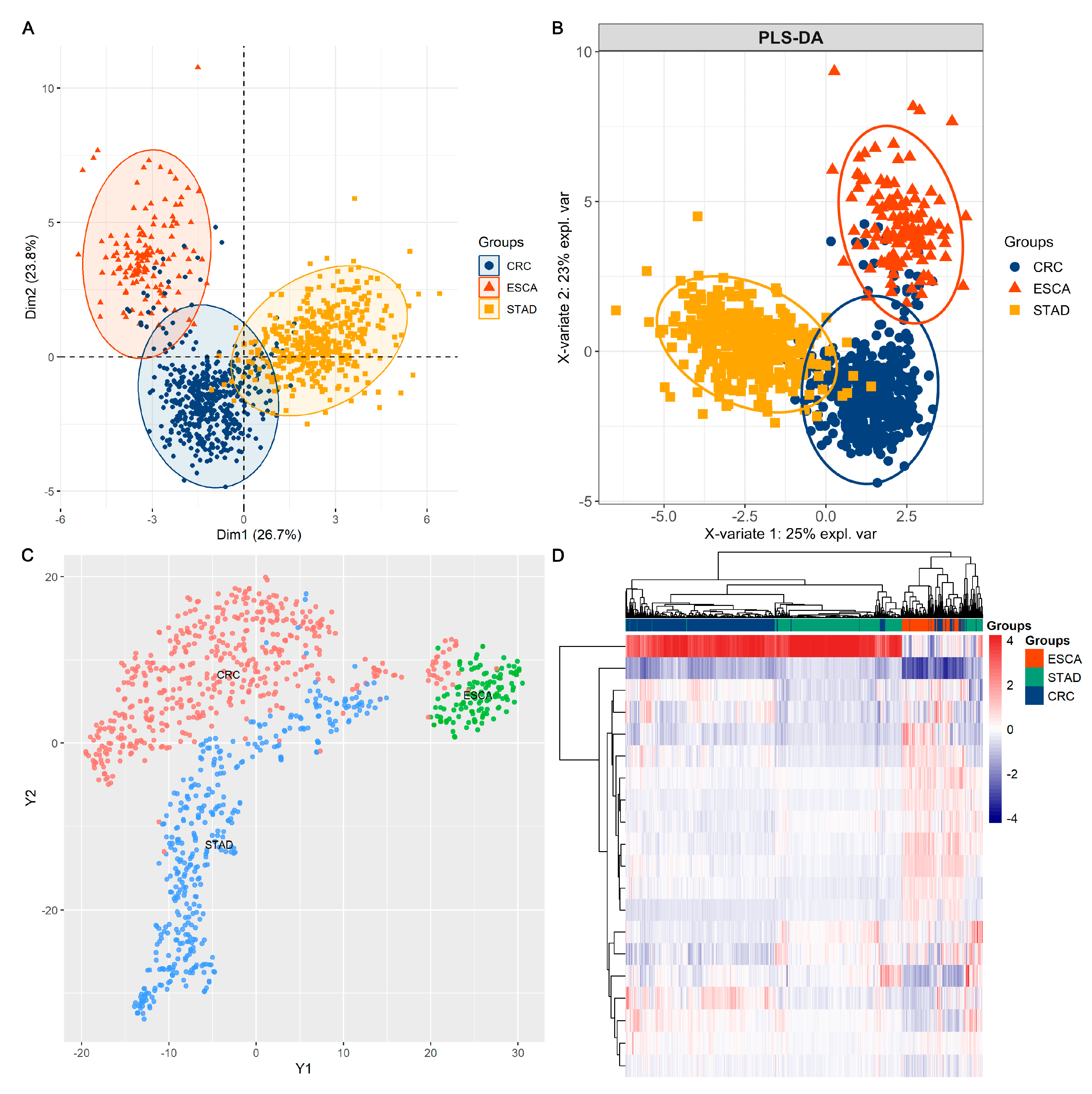
3.1. Overview of the Functional Proteome Profiling across ESCA, STAD, COAD, and READ Tumour Samples
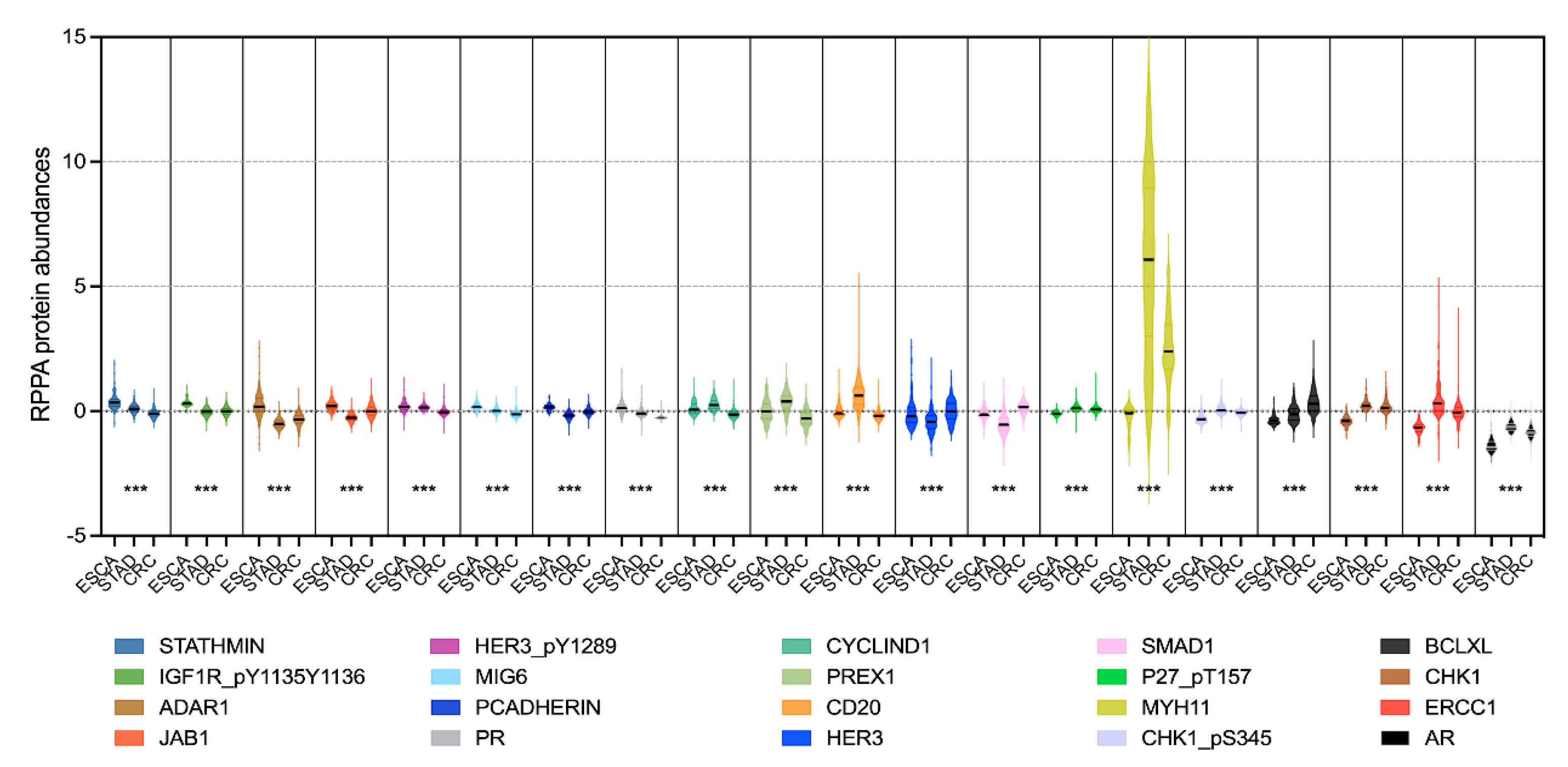
3.2. Using Feature Selection Approaches to Identify Protein Signatures That Help to Classify Different Cancer Types
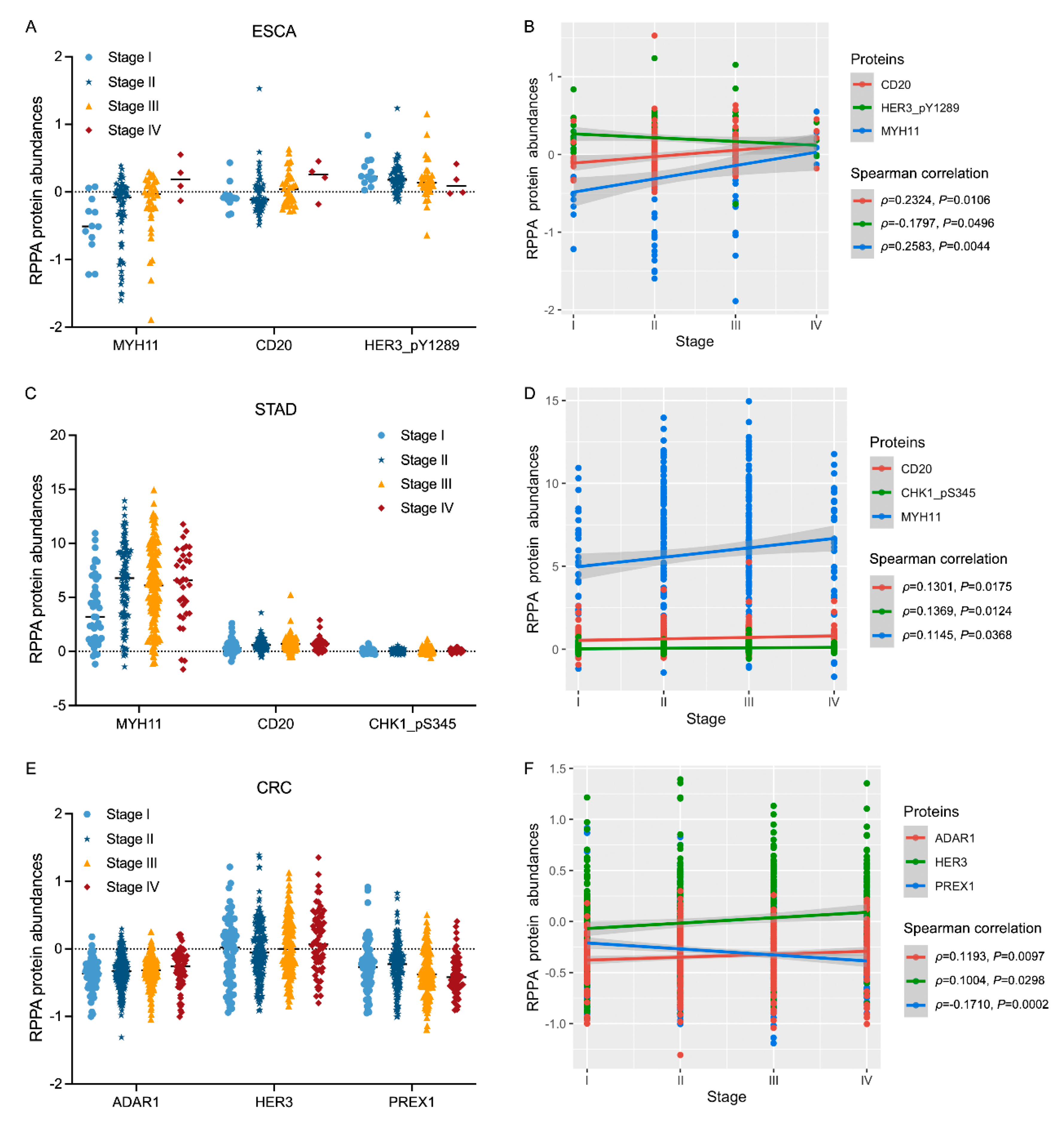
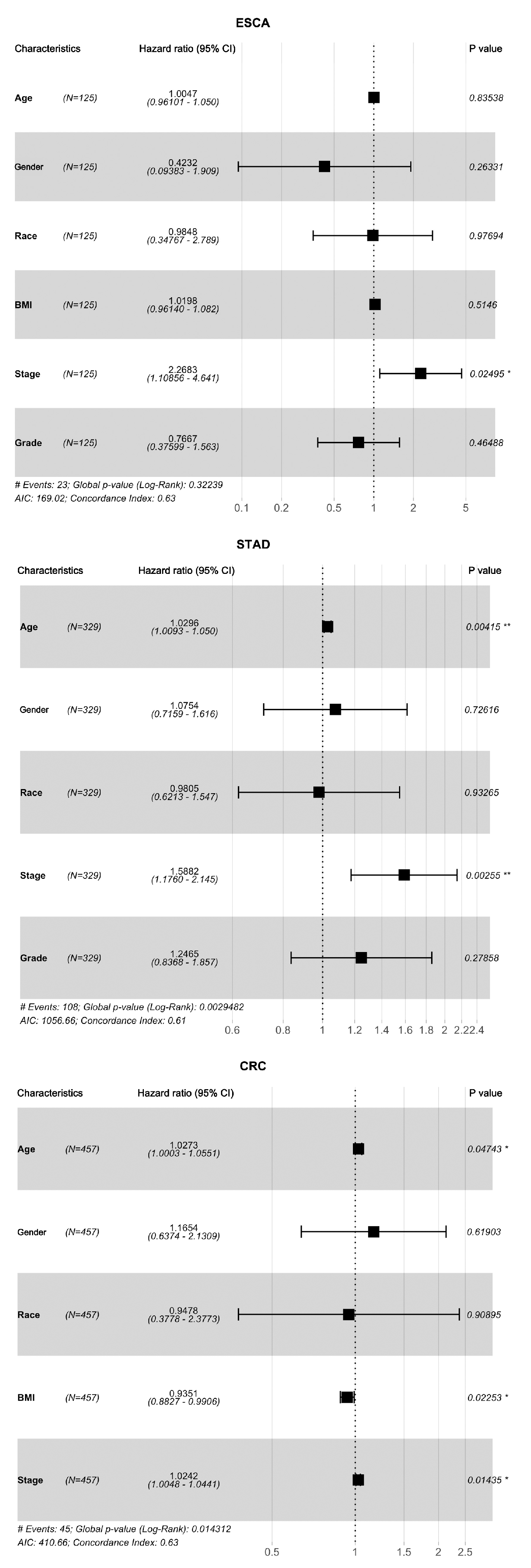
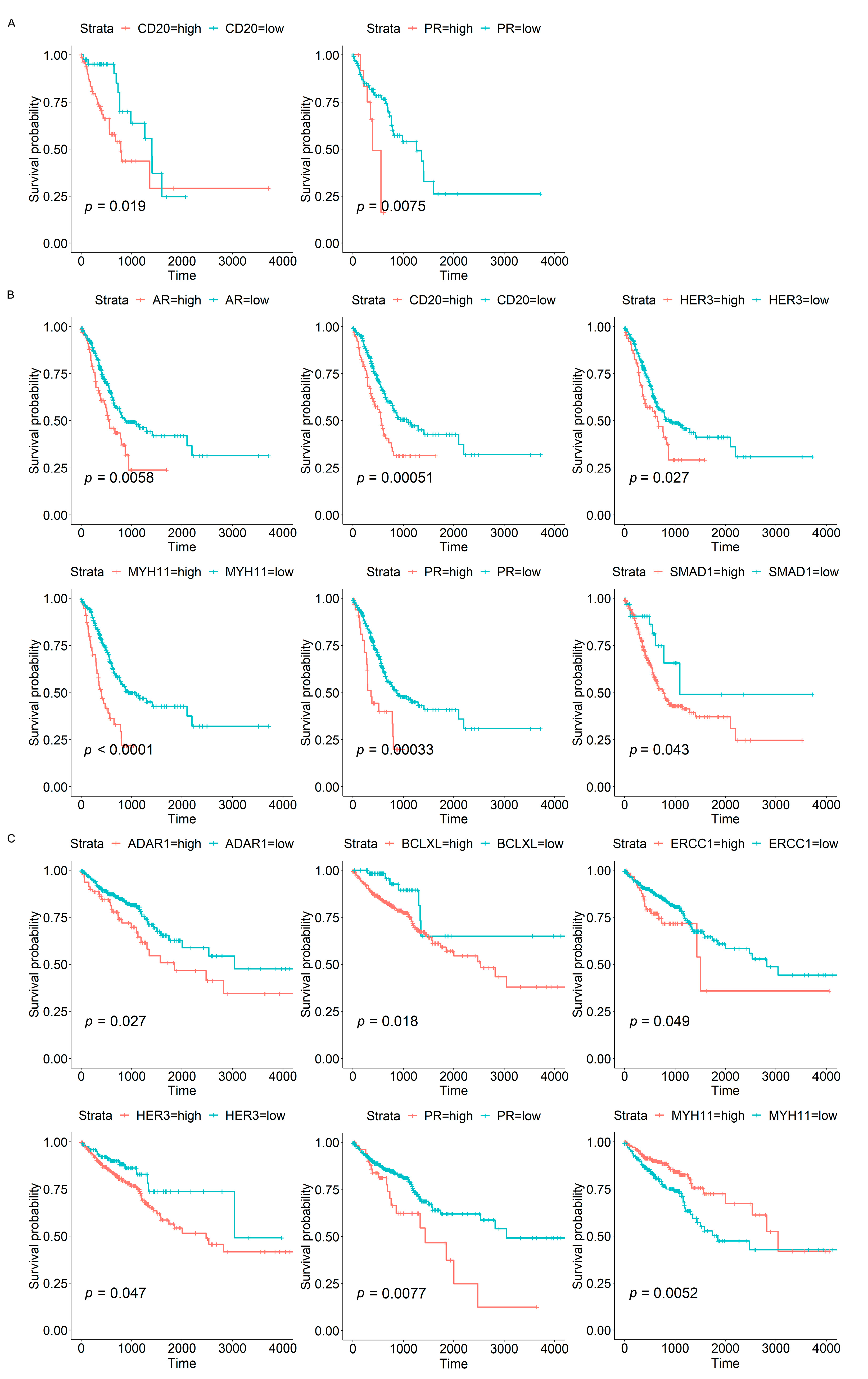
3.3. Associations of Protein Biomarkers with the Clinical Characteristics of Tumours
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e315. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.E.; Kim, N.I.; Lee, J.S.; Park, M.H.; Kang, K. Differentially Expressed Genes in Matched Normal, Cancer, and Lymph Node Metastases Predict Clinical Outcomes in Patients with Breast Cancer. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Rosario, S.R.; Long, M.D.; Affronti, H.C.; Rowsam, A.M.; Eng, K.H.; Smiraglia, D.J. Pan-cancer analysis of transcriptional metabolic dysregulation using The Cancer Genome Atlas. Nat. Commun. 2018, 9, 5330. [Google Scholar] [CrossRef]
- Carino, A.; Graziosi, L.; Marchiano, S.; Biagioli, M.; Marino, E.; Sepe, V.; Zampella, A.; Distrutti, E.; Donini, A.; Fiorucci, S. Analysis of Gastric Cancer Transcriptome Allows the Identification of Histotype Specific Molecular Signatures with Prognostic Potential. Front. Oncol. 2021, 11, 663771. [Google Scholar] [CrossRef]
- Guo, J.N.; Li, M.Q.; Deng, S.H.; Chen, C.; Ni, Y.; Cui, B.B.; Liu, Y.L. Prognostic Immune-Related Analysis Based on Differentially Expressed Genes in Left- and Right-Sided Colon Adenocarcinoma. Front. Oncol. 2021, 11, 640196. [Google Scholar] [CrossRef]
- Suran, M. After the Genome-A Brief History of Proteomics. JAMA 2022, 328, 1168–1169. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef]
- Chen, M.M.; Li, J.; Wang, Y.; Akbani, R.; Lu, Y.; Mills, G.B.; Liang, H. TCPA v3.0: An Integrative Platform to Explore the Pan-Cancer Analysis of Functional Proteomic Data. Mol. Cell Proteom. 2019, 18, S15–S25. [Google Scholar] [CrossRef]
- Li, J.; Akbani, R.; Zhao, W.; Lu, Y.; Weinstein, J.N.; Mills, G.B.; Liang, H. Explore, Visualize, and Analyze Functional Cancer Proteomic Data Using the Cancer Proteome Atlas. Cancer Res. 2017, 77, e51–e54. [Google Scholar] [CrossRef]
- Wu, H. A Deep Learning-Based Hybrid Feature Selection Approach for Cancer Diagnosis. J. Phys. Conf. Ser. 2021, 1848, 012019. [Google Scholar] [CrossRef]
- Yin, Q.; Chen, W.; Zhang, C.; Wei, Z. A convolutional neural network model for survival prediction based on prognosis-related cascaded Wx feature selection. Lab. Investig. 2022, 102, 1064–1074. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, X.; Tan, Z.; Mei, Y.; Lu, T.; Ji, Y.; Cheng, S.; Xu, Y.; Wang, Z.; Liu, X.; et al. Interferon gamma-related gene signature based on anti-tumor immunity predicts glioma patient prognosis. Front. Genet. 2022, 13, 1053263. [Google Scholar] [CrossRef]
- Qin, X.; Zhang, S.; Yin, D.; Chen, D.; Dong, X. Two-stage feature selection for classification of gene expression data based on an improved Salp Swarm Algorithm. Math. Biosci. Eng. 2022, 19, 13747–13781. [Google Scholar] [CrossRef]
- Shi, Z.; Wen, B.; Gao, Q.; Zhang, B. Feature Selection Methods for Protein Biomarker Discovery from Proteomics or Multiomics Data. Mol. Cell Proteom. 2021, 20, 100083. [Google Scholar] [CrossRef]
- Peng, J.; Xu, H.; Chen, Y.; Wang, W.; Zhu, L.; Shao, Y.; Wang, J. Screening for therapeutic targets of tumor angiogenesis signatures in 31 cancer types and potential insights. Biochem. Biophys. Res. Commun. 2019, 508, 465–471. [Google Scholar] [CrossRef]
- Li, Y.; Kang, K.; Krahn, J.M.; Croutwater, N.; Lee, K.; Umbach, D.M.; Li, L. A comprehensive genomic pan-cancer classification using The Cancer Genome Atlas gene expression data. BMC Genom. 2017, 18, 508. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Li, Z.; Gao, X.; Huang, D. Global Analysis of Microbiota Signatures in Four Major Types of Gastrointestinal Cancer. Front. Oncol. 2021, 11, 685641. [Google Scholar] [CrossRef]
- Zuo, S.; Dai, G.; Ren, X. Identification of a 6-gene signature predicting prognosis for colorectal cancer. Cancer Cell Int. 2019, 19, 6. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Asada, K.; Takasawa, K.; Shimoyama, R.; Sakai, A.; Bolatkan, A.; Shinkai, N.; Kobayashi, K.; Komatsu, M.; Kaneko, S.; et al. Predicting Deep Learning Based Multi-Omics Parallel Integration Survival Subtypes in Lung Cancer Using Reverse Phase Protein Array Data. Biomolecules 2020, 10, 1460. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Liu, H.; Yang, J.; Chen, G. Ensemble feature selection for stable biomarker identification and cancer classification from microarray expression data. Comput. Biol. Med. 2022, 142, 105208. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Hu, Y.; Shangguan, J.; Song, Q.; Xu, J.; Wang, H.; Xue, M.; Wang, L.; Zhang, Y. Identification of key biomarkers for STAD using filter feature selection approaches. Sci. Rep. 2022, 12, 19854. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Ru, X.; Sun, P.; Wang, J. Identification of gene signatures for COAD using feature selection and Bayesian network approaches. Sci. Rep. 2022, 12, 8761. [Google Scholar] [CrossRef]
- Mohamed, S.S.; Salama, M.M.; Kamel, M.; El-Saadany, E.F.; Rizkalla, K.; Chin, J. Prostate cancer multi-feature analysis using trans-rectal ultrasound images. Phys. Med. Biol. 2005, 50, N175–N185. [Google Scholar] [CrossRef]
- Battiti, R. Using mutual information for selecting features in supervised neural net learning. IEEE Trans. Neural Netw. 1994, 5, 537–550. [Google Scholar] [CrossRef]
- Dongrae, C.; Boreom, L. Optimized automatic sleep stage classification using the normalized mutual information feature selection (NMIFS) method. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2017, 2017, 3094–3097. [Google Scholar] [CrossRef]
- Estevez, P.A.; Tesmer, M.; Perez, C.A.; Zurada, J.M. Normalized mutual information feature selection. IEEE Trans. Neural Netw. 2009, 20, 189–201. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Wallace, D.S.; Zent, C.S.; Baran, A.M.; Reagan, P.M.; Casulo, C.; Rice, G.; Friedberg, J.W.; Barr, P.M. Acalabrutinib and High-Frequency Low-Dose Subcutaneous Rituximab for Initial Therapy of Chronic Lymphocytic Leukemia. Blood Adv. 2023, 2022009382. [Google Scholar] [CrossRef]
- Phan, T.D.A.; Duong, T.T.; Thi Nhu Pham, D.; Hoang Dang, M.; Thanh Ly, T.; Thi Tuyet Ngo, H.; Ngo, D.Q.; Trinh, N.D.T.; Le Ly, U.; Anh Thai, T.; et al. A Multicenter Study of Clinicopathology and Immunohistochemical Distinction between Adult and Pediatric Large B-Cell Lymphoma. Fetal Pediatr. Pathol. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Ribatti, D.; Vacca, A.; Einsele, H. Targeting B-cell non Hodgkin lymphoma: New and old tricks. Leuk. Res. 2016, 42, 93–104. [Google Scholar] [CrossRef]
- Jo, Y.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Somatic Mutations and Intratumoral Heterogeneity of MYH11 Gene in Gastric and Colorectal Cancers. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 562–566. [Google Scholar] [CrossRef]
- Chen, W.; Hu, J.; He, Y.; Yu, L.; Liu, Y.; Cheng, Y.; Jia, B.; Li, X.; Yu, G.; Wang, Y. The Interaction Between SMAD1 and YAP1 Is Correlated with Increased Resistance of Gastric Cancer Cells to Cisplatin. Appl. Biochem. Biotechnol. 2022, 1–18. [Google Scholar] [CrossRef]
- Zheng, G.L.; Zhang, G.J.; Zhao, Y.; Zheng, Z.C. The Interplay between RNA Editing Regulator ADAR1 and Immune Environment in Colorectal Cancer. J. Oncol. 2023, 2023, 9315027. [Google Scholar] [CrossRef]
- Li, M.; Zhou, C. Progesterone receptor gene serves as a prognostic biomarker associated with immune infiltration in gastric cancer: A bioinformatics analysis. Transl. Cancer Res. 2021, 10, 2663–2677. [Google Scholar] [CrossRef]
- Oliveira, L.L.; Bergmann, A.; Melo, A.C.; Thuler, L.C. Prognostic factors associated with overall survival in patients with oral cavity squamous cell carcinoma. Med. Oral Patol. Oral Cir. Bucal 2020, 25, e523–e531. [Google Scholar] [CrossRef]
- Alonso, J.E.; Han, A.Y.; Kuan, E.C.; Strohl, M.; Clair, J.M.; St John, M.A.; Ryan, W.R.; Heaton, C.M. The survival impact of surgical therapy in squamous cell carcinoma of the hard palate. Laryngoscope 2018, 128, 2050–2055. [Google Scholar] [CrossRef]
- Lu, C.H.; Lee, S.H.; Liu, K.H.; Hung, Y.S.; Wang, C.H.; Lin, Y.C.; Yeh, T.S.; Chou, W.C. Older age impacts on survival outcome in patients receiving curative surgery for solid cancer. Asian J. Surg. 2018, 41, 333–340. [Google Scholar] [CrossRef]
- Petrelli, F.; Cortellini, A.; Indini, A.; Tomasello, G.; Ghidini, M.; Nigro, O.; Salati, M.; Dottorini, L.; Iaculli, A.; Varricchio, A.; et al. Association of Obesity with Survival Outcomes in Patients with Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e213520. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Number of Cases | |||
|---|---|---|---|---|
| ESCA | STAD | CRC | ||
| Age at initial pathologic diagnosis (year) | <65 | 73 | 151 | 188 |
| ≥65 | 53 | 201 | 293 | |
| Not reported | 0 | 5 | 3 | |
| Gender demographic | Male | 108 | 236 | 251 |
| Female | 18 | 121 | 230 | |
| Not reported | 0 | 0 | 3 | |
| Race demographic | Asian | 43 | 66 | 12 |
| Black | 2 | 5 | 52 | |
| White | 73 | 232 | 240 | |
| Not reported | 8 | 54 | 180 | |
| BMI | ≤18.4 | 4 | No information | 4 |
| 18.5–23.9 | 60 | 65 | ||
| 24.0–27.9 | 29 | 74 | ||
| ≥28 | 28 | 117 | ||
| Not reported | 5 | 224 | ||
| Tumour stage | Stage I | 12 | 45 | 74 |
| Stage II | 66 | 105 | 185 | |
| Stage III | 38 | 150 | 144 | |
| Stage IV | 4 | 33 | 66 | |
| Not reported | 6 | 24 | 15 | |
| Neoplasm histologic grade | Grade 1 | 15 | 9 | No information |
| Grade 2 | 55 | 120 | ||
| Grade 3 | 32 | 219 | ||
| Not reported | 24 | 9 | ||
| OS_status | Alive | 82 | 189 | 361 |
| Dead | 43 | 140 | 96 | |
| Not reported | 1 | 28 | 27 | |
| OS_time (day) | Alive | 576.56 ± 527.05 | 711.61 ± 573.43 | 878.75 ± 740.52 |
| Dead | 479.56 ± 427.18 | 421.28 ± 341.58 | 677.34 ± 644.99 | |
| Total number of cases | 126 | 357 | 484 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Gao, X.; Wang, J. Functional Proteomic Profiling Analysis in Four Major Types of Gastrointestinal Cancers. Biomolecules 2023, 13, 701. https://doi.org/10.3390/biom13040701
Wang Y, Gao X, Wang J. Functional Proteomic Profiling Analysis in Four Major Types of Gastrointestinal Cancers. Biomolecules. 2023; 13(4):701. https://doi.org/10.3390/biom13040701
Chicago/Turabian StyleWang, Yangyang, Xiaoguang Gao, and Jihan Wang. 2023. "Functional Proteomic Profiling Analysis in Four Major Types of Gastrointestinal Cancers" Biomolecules 13, no. 4: 701. https://doi.org/10.3390/biom13040701