
Biological Screening and Crystallographic Studies of Hydroxy γ-Lactone Derivatives to Investigate PPARγ Phosphorylation Inhibition
 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
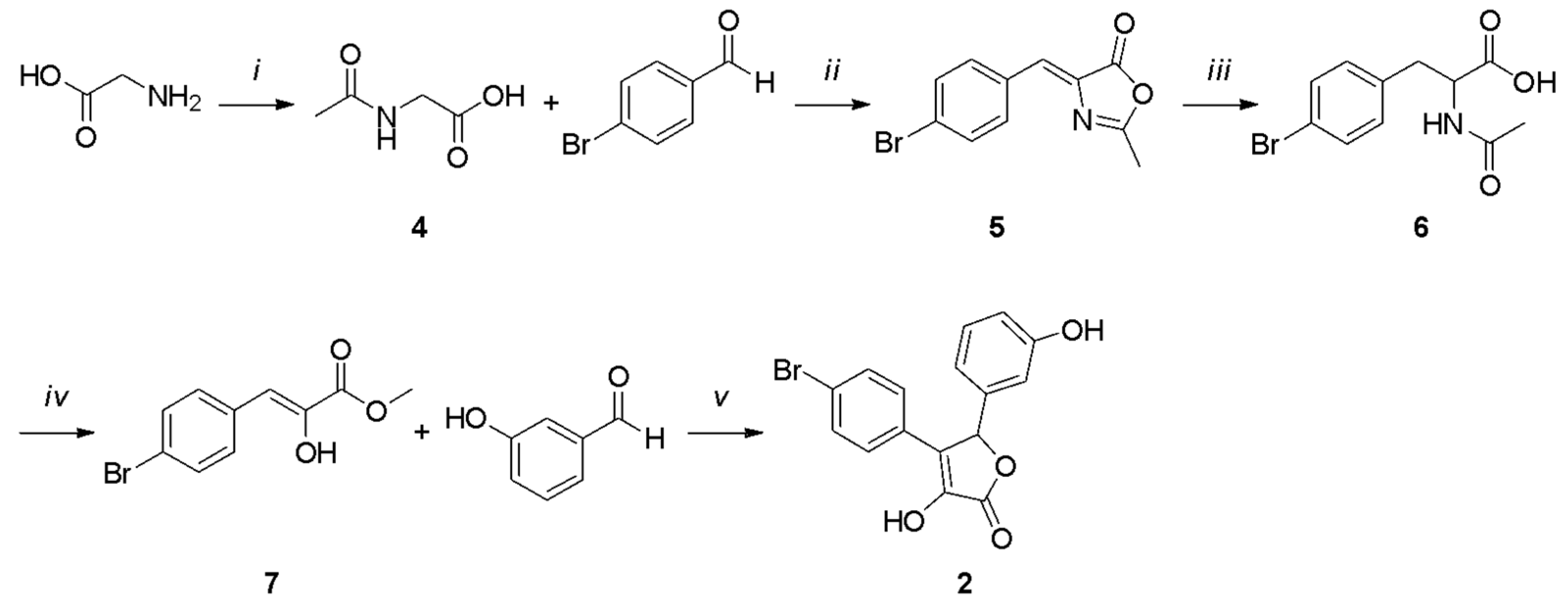
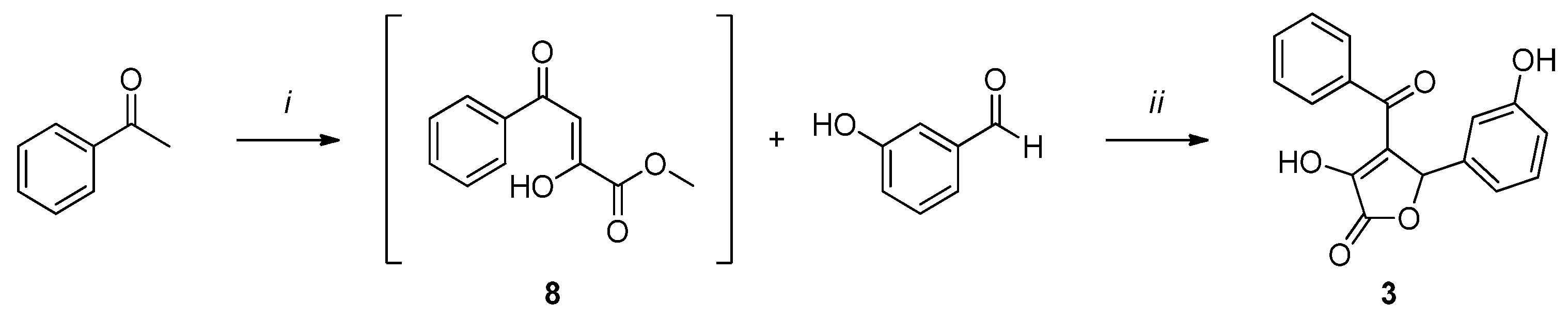
2.1. Chemistry
2.2. Surface Plasmon Resonance
2.3. Transactivation Assay
2.4. Protein Expression and Purification
2.5. Crystallization, Data Collection, and Structure Determination
2.6. In Vitro Kinase Assay
2.7. Pro-Q™ Diamond Phosphoprotein Gel Stain
2.8. Statistical Analysis
2.9. CDK5 Inhibition Test
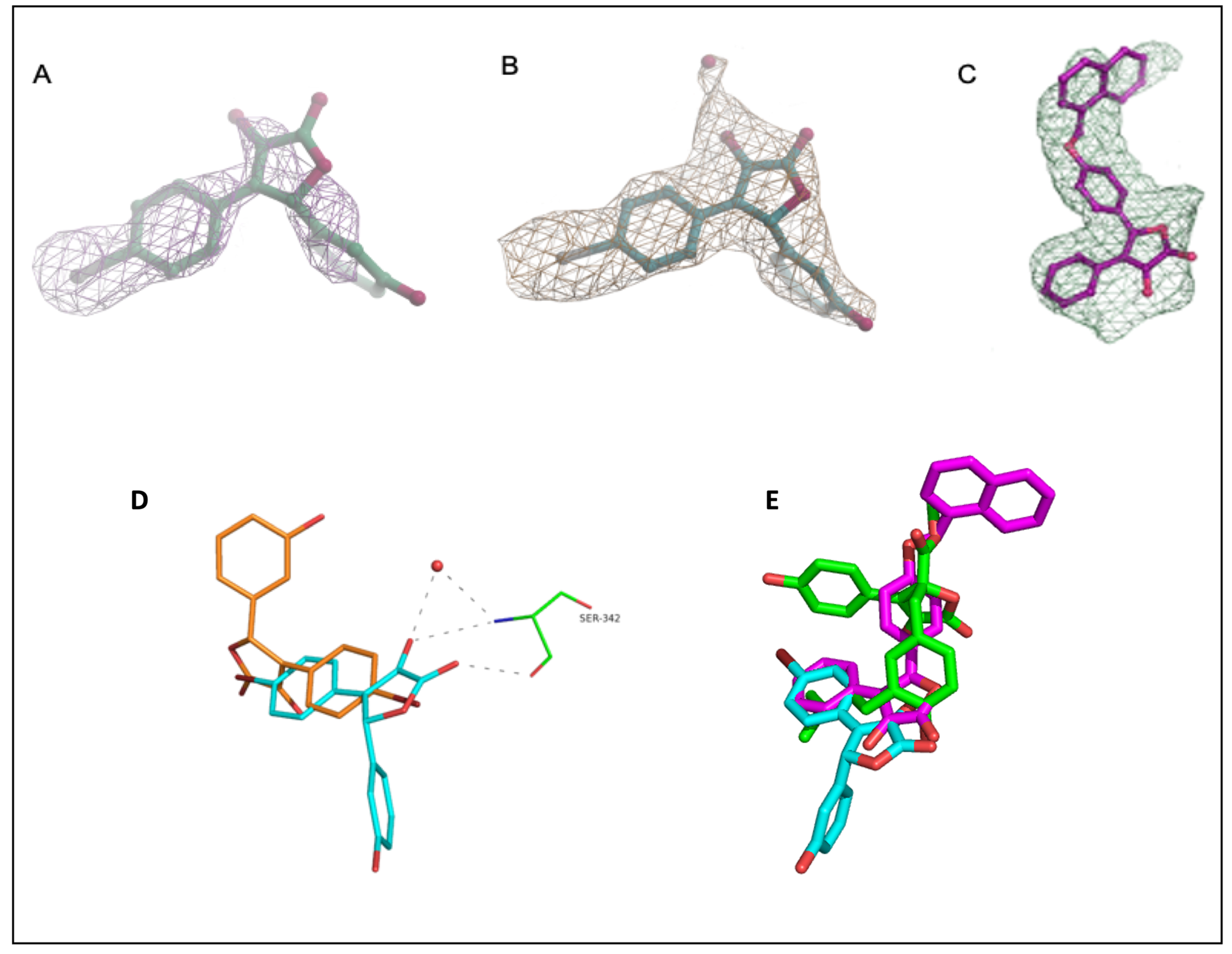
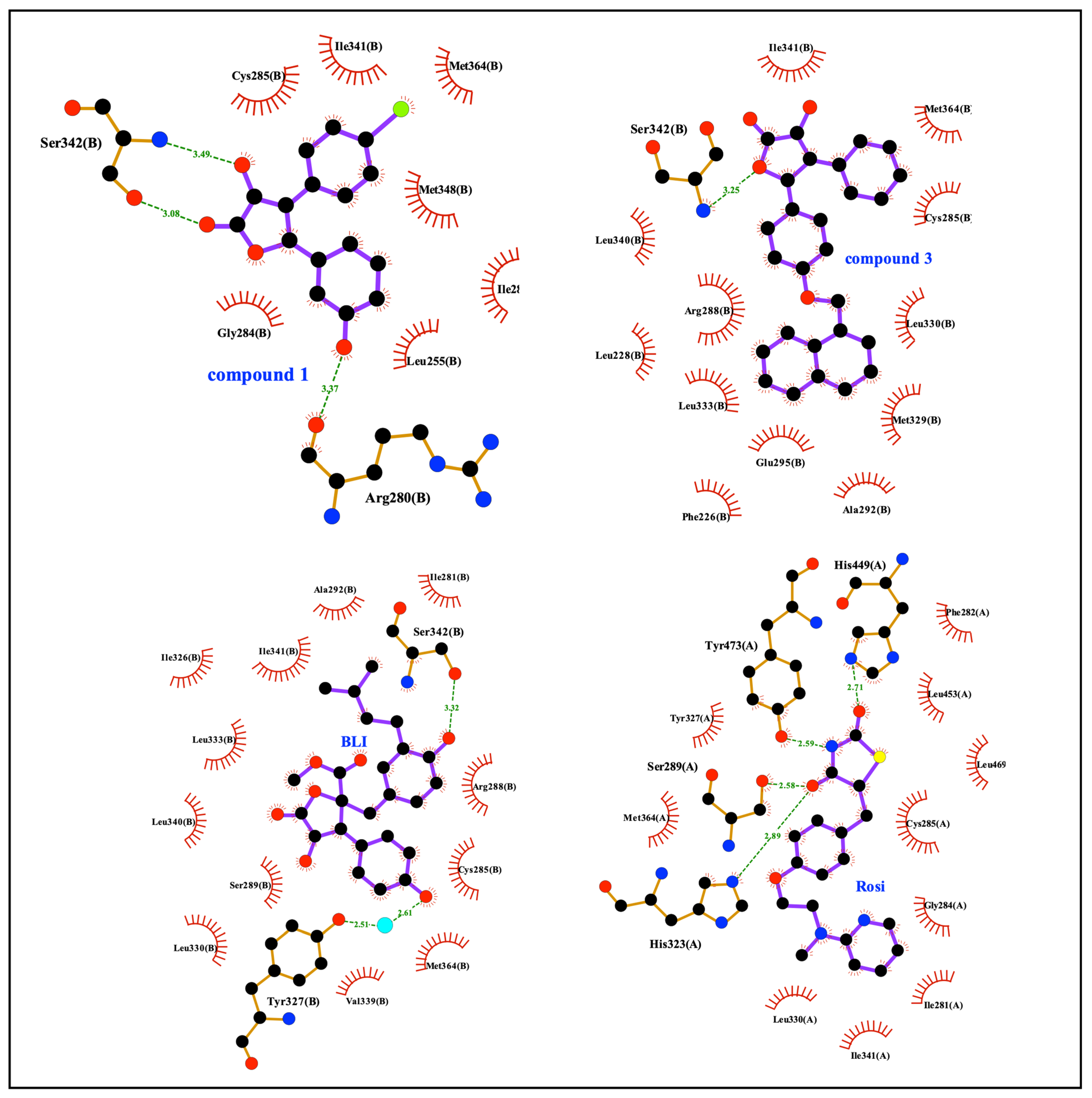
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, J.P.; Akiyama, T.E.; Meinke, P.T. PPARs: Therapeutic Targets for Metabolic Disease. Trends Pharmacol. Sci. 2005, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty Acids and Eicosanoids Regulate Gene Expression through Direct Interactions with Peroxisome Proliferator-Activated Receptors α and γ. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a Member of the Steroid Hormone Receptor Superfamily by Peroxisome Proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the Complex Journey to Obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential Expression of Peroxisome Proliferator-Activated Receptors (PPARs): Tissue Distribution of PPAR-Alpha, -Beta, and -Gamma in the Adult Rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome Proliferator-Activated Receptors and Their Ligands: Nutritional and Clinical Implications—A Review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From Orphan Receptors to Drug Discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand Binding and Co-Activator Assembly of the Peroxisome Proliferator-Activated Receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Ahn, S.; Jang, D.M.; Park, S.C.; An, S.; Shin, J.; Han, B.W.; Noh, M. Cyclin-Dependent Kinase 5 Inhibitor Butyrolactone I Elicits a Partial Agonist Activity of Peroxisome Proliferator-Activated Receptor γ. Biomolecules 2020, 10, 275. [Google Scholar] [CrossRef]
- IW, C. The Clinical Significance of PPAR Gamma Agonism. Curr. Mol. Med. 2005, 5, 349–363. [Google Scholar] [CrossRef]
- Guan, Y.F.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones Expand Body Fluid Volume through PPARγ Stimulation of ENaC-Mediated Renal Salt Absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Home, P.D.; Pocock, S.J.; Beck-Nielsen, H.; Curtis, P.S.; Gomis, R.; Hanefeld, M.; Jones, N.P.; Komajda, M.; McMurray, J.J. Rosiglitazone Evaluated for Cardiovascular Outcomes in Oral Agent Combination Therapy for Type 2 Diabetes (RECORD): A Multicentre, Randomised, Open-Label Trial. Lancet 2009, 373, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Shockley, K.R.; Lazarenko, O.P.; Czernik, P.J.; Rosen, C.J.; Churchill, G.A.; Lecka-Czernik, B. PPARγ2 Nuclear Receptor Controls Multiple Regulatory Pathways of Osteoblast Differentiation from Marrow Mesenchymal Stem Cells. J. Cell. Biochem. 2009, 106, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, X.; Yang, M.; Smith, L.C.; Dechow, P.C.; Wan, Y. PGC1beta Mediates PPARgamma Activation of Osteoclastogenesis and Rosiglitazone-Induced Bone Loss. Cell Metab. 2010, 11, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Laghezza, A.; Piemontese, L.; Cerchia, C.; Montanari, R.; Capelli, D.; Giudici, M.; Crestani, M.; Tortorella, P.; Peiretti, F.; Pochetti, G.; et al. Identification of the First PPARα/γ Dual Agonist Able to Bind to Canonical and Alternative Sites of PPARγ and to Inhibit Its Cdk5-Mediated Phosphorylation. J. Med. Chem. 2018, 61, 8282–8298. [Google Scholar] [CrossRef]
- Li, Z.; Ren, Q.; Zhou, Z.; Cai, Z.; Wang, B.; Han, J.; Zhang, L. Discovery of the First-in-Class Dual PPARδ/γ Partial Agonist for the Treatment of Metabolic Syndrome. Eur. J. Med. Chem. 2021, 225, 113807. [Google Scholar] [CrossRef]
- Chandra, A.; Kaur, P.; Sahu, S.K.; Mittal, A. A New Insight into the Treatment of Diabetes by Means of Pan PPAR Agonists. Chem. Biol. Drug Des. 2022, 100, 947–967. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M.; et al. Anti-Diabetic Drugs Inhibit Obesity-Linked Phosphorylation of PPARγ by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic Actions of a Non-Agonist PPARγ Ligand Blocking Cdk5-Mediated Phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef]
- Amato, A.A.; Rajagopalan, S.; Lin, J.Z.; Carvalho, B.M.; Figueira, A.C.M.; Lu, J.; Ayers, S.D.; Mottin, M.; Silveira, R.L.; Souza, P.C.T.; et al. GQ-16, a Novel Peroxisome Proliferator-Activated Receptor γ (PPARγ) Ligand, Promotes Insulin Sensitization without Weight Gain. J. Biol. Chem. 2012, 287, 28169–28179. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.C.; Weidner, C.; Krausze, J.; Kawamoto, K.; Schroeder, F.C.; Sauer, S.; Büssow, K. Structural Characterization of Amorfrutins Bound to the Peroxisome Proliferator-Activated Receptor γ. J. Med. Chem. 2013, 56, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Pochetti, G.; Mitro, N.; Lavecchia, A.; Gilardi, F.; Besker, N.; Scotti, E.; Aschi, M.; Re, N.; Fracchiolla, G.; Laghezza, A.; et al. Structural Insight into Peroxisome Proliferator-Activated Receptor γ Binding of Two Ureidofibrate-like Enantiomers by Molecular Dynamics, Cofactor Interaction Analysis, and Site-Directed Mutagenesis. J. Med. Chem. 2010, 53, 4354–4366. [Google Scholar] [CrossRef] [PubMed]
- Laghezza, A.; Montanari, R.; Lavecchia, A.; Piemontese, L.; Pochetti, G.; Iacobazzi, V.; Infantino, V.; Capelli, D.; De Bellis, M.; Liantonio, A.; et al. On the Metabolically Active Form of Metaglidasen: Improved Synthesis and Investigation of Its Peculiar Activity on Peroxisome Proliferator-Activated Receptors and Skeletal Muscles. ChemMedChem 2015, 10, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Marciano, D.P.; Kuruvilla, D.S.; Boregowda, S.V.; Asteian, A.; Hughes, T.S.; Garcia-Ordonez, R.; Corzo, C.A.; Khan, T.M.; Novick, S.J.; Park, H.; et al. Pharmacological Repression of PPARγ Promotes Osteogenesis. Nat. Commun. 2015, 6, 7443. [Google Scholar] [CrossRef] [PubMed]
- Marciano, D.P.; Kuruvilla, D.S.; Pascal, B.D.; Griffin, P.R. Identification of Bexarotene as a PPAR γ Antagonist with HDX. PPAR Res. 2015, 2015, 254560. [Google Scholar] [CrossRef]
- Choi, S.S.; Kim, E.S.; Jung, J.E.; Marciano, D.P.; Jo, A.; Koo, J.Y.; Choi, S.Y.; Yang, Y.R.; Jang, H.J.; Kim, E.K.; et al. PPARγ Antagonist Gleevec Improves Insulin Sensitivity and Promotes the Browning of White Adipose Tissue. Diabetes 2016, 65, 829–839. [Google Scholar] [CrossRef]
- Asteian, A.; Blayo, A.L.; He, Y.; Koenig, M.; Shin, Y.; Kuruvilla, D.S.; Corzo, C.A.; Cameron, M.D.; Lin, L.; Ruiz, C.; et al. Design, Synthesis, and Biological Evaluation of Indole Biphenylcarboxylic Acids as PPARγ Antagonists. ACS Med. Chem. Lett. 2015, 6, 998–1003. [Google Scholar] [CrossRef]
- Brusotti, G.; Montanari, R.; Capelli, D.; Cattaneo, G.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Peiretti, F.; Bonardo, B.; Paiardini, A.; et al. Betulinic Acid Is a PPARγ Antagonist That Improves Glucose Uptake, Promotes Osteogenesis and Inhibits Adipogenesis. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef]
- Montanari, R.; Capelli, D.; Yamamoto, K.; Awaishima, H.; Nishikata, K.; Barendregt, A.; Heck, A.J.R.; Loiodice, F.; Altieri, F.; Paiardini, A.; et al. Insights into Pparγphosphorylation and Its Inhibition Mechanism. J. Med. Chem. 2020, 63, 4811–4823. [Google Scholar] [CrossRef] [PubMed]
- Weber, V.; Coudert, P.; Rubat, C.; Duroux, E.; Vallée-Goyet, D.; Gardette, D.; Bria, M.; Albuisson, E.; Leal, F.; Gramain, J.C.; et al. Novel 4,5-Diaryl-3-Hydroxy-2(5H)-Furanones as Anti-Oxidants and Anti-Inflammatory Agents. Bioorganic Med. Chem. 2002, 10, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, C.; Li, J. Comparison of Vitamin c and Its Derivative Antioxidant Activity: Evaluated by Using Density Functional Theory. ACS Omega 2020, 5, 25467–25475. [Google Scholar] [CrossRef]
- Macan, A.M.; Kraljević, T.G.; Raić-Malić, S. Therapeutic Perspective of Vitamin C and Its Derivatives. Antioxidants 2019, 8, 247. [Google Scholar] [CrossRef] [PubMed]
- Naturale, G.; Lamblin, M.; Commandeur, C.; Felpin, F.X.; Dessolin, J. Direct C–H Alkylation of Naphthoquinones with Amino Acids Through a Revisited Kochi–Anderson Radical Decarboxylation: Trends in Reactivity and Applications. Eur. J. Org. Chem. 2012, 2012, 5774–5788. [Google Scholar] [CrossRef]
- Yamashita, A.; Norton, E.B.; Kaplan, J.A.; Niu, C.; Loganzo, F.; Hernandez, R.; Beyer, C.F.; Annable, T.; Musto, S.; Discafani, C.; et al. Synthesis and Activity of Novel Analogs of Hemiasterlin as Inhibitors of Tubulin Polymerization: Modification of the A Segment. Bioorganic Med. Chem. Lett. 2004, 14, 5317–5322. [Google Scholar] [CrossRef]
- Shymanska, N.V.; Pierce, J.G. Stereoselective Synthesis of Quaternary Pyrrolidine-2,3-Diones and β-Amino Acids. Org. Lett. 2017, 19, 2961–2964. [Google Scholar] [CrossRef]
- Ji, R.J.; Shi, W.M.; Tian, D.Y.; Zhang, G.P.; Wang, H. Facile Synthesis of Novel Dithioacetal–Naphthalene Derivatives as Potential Activators for Plant Resistance Induction. RSC Adv. 2019, 9, 32375–32381. [Google Scholar] [CrossRef]
- Fischer, M.J.E. Amine Coupling through EDC/NHS: A Practical Approach. Methods Mol. Biol. 2010, 627, 55–73. [Google Scholar]
- Holdgate, G.A.; Rawlins, P.; Bista, M.; Stubbs, C.J. Using Biophysical Methods to Optimize Compound Residence Time. In Applied Biophysics for Drug Discovery; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 197–216. [Google Scholar]
- Fracchiolla, G.; Laghezza, A.; Piemontese, L.; Parente, M.; Lavecchia, A.; Pochetti, G.; Montanari, R.; Giovanni, C.D.; Carbonara, G.; Tortorella, P.; et al. Synthesis, Biological Evaluation and Molecular Investigation of Fluorinated Peroxisome Proliferator-Activated Receptors α/γ Dual Agonists. Bioorganic Med. Chem. 2012, 20, 2141–2151. [Google Scholar] [CrossRef]
- Hollon, T.; Yoshimura, F.K. Variation in Enzymatic Transient Gene Expression Assays. Anal. Biochem. 1989, 182, 411–418. [Google Scholar] [CrossRef]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural Basis for PPAR Partial or Full Activation Revealed by a Novel Ligand Binding Mode. Sci. Rep. 2016, 6, 34792. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How Good Are My Data and What Is the Resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Winter, G. Xia2: An Expert System for Macromolecular Crystallography Data Reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar] [CrossRef]
- Navaza, J. AMoRe: An Automated Package for Molecular Replacement. Acta Crystallogr. Sect. A 1994, 50, 157–163. [Google Scholar] [CrossRef]
- Montanari, R.; Saccoccia, F.; Scotti, E.; Crestani, M.; Godio, C.; Gilardi, F.; Loiodice, F.; Fracchiolla, G.; Laghezza, A.; Tortorella, P.; et al. Crystal Structure of the Peroxisome Proliferator-Activated Receptor γ (PPARγ) Ligand Binding Domain Complexed with a Novel Partial Agonist: A New Region of the Hydrophobic Pocket Could Be Exploited for Drug Design. J. Med. Chem. 2008, 51, 7768–7776. [Google Scholar] [CrossRef]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; Delano, W.L.; Gros, P.; Grossekunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [CrossRef]
- Brunger, A.T. Version 1.2 of the Crystallography and NMR System. Nat. Protoc. 2007, 2, 2728–2733. [Google Scholar] [CrossRef]
- Torices, R.; Muñoz-Pajares, A.J. PHENIX: An R Package to Estimate a Size-Controlled Phenotypic Integration Index. Appl. Plant Sci. 2015, 3, 1400104. [Google Scholar] [CrossRef]
- Montanari, R.; Capelli, D.; Tava, A.; Galli, A.; Laghezza, A.; Tortorella, P.; Loiodice, F.; Pochetti, G. Screening of Saponins and Sapogenins from Medicago Species as Potential PPARγ Agonists and X-Ray Structure of the Complex PPARγ/Caulophyllogenin. Sci. Rep. 2016, 6, 27658. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.G. Modeling Taylor Dispersion Injections: Determination of Kinetic/Affinity Interaction Constants and Diffusion Coefficients in Label-Free Biosensing. Anal. Biochem. 2012, 421, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Calleri, E.; Fracchiolla, G.; Montanari, R.; Pochetti, G.; Lavecchia, A.; Loiodice, F.; Laghezza, A.; Piemontese, L.; Massolini, G.; Temporini, C. Frontal Affinity Chromatography with MS Detection of the Ligand Binding Domain of PPARγ Receptor: Ligand Affinity Screening and Stereoselective Ligand–Macromolecule Interaction. J. Chromatogr. A 2012, 1232, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Peiretti, F.; Montanari, R.; Capelli, D.; Bonardo, B.; Colson, C.; Amri, E.Z.; Grimaldi, M.; Balaguer, P.; Ito, K.; Roeder, R.G.; et al. A Novel N-Substituted Valine Derivative with Unique Peroxisome Proliferator-Activated Receptor Γbinding Properties and Biological Activities. J. Med. Chem. 2020, 63, 13124–13139. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial Agonists Activate PPARgamma Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder Maps: Improving OMIT Maps by Excluding Bulk Solvent. Acta Crystallogr. D Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Hughes, T.S.; Chalmers, M.J.; Novick, S.; Kuruvilla, D.S.; Chang, M.R.; Kamenecka, T.M.; Rance, M.; Johnson, B.A.; Burris, T.P.; Griffin, P.R.; et al. Ligand and Receptor Dynamics Contribute to the Mechanism of Graded PPARγ Agonism. Structure 2012, 20, 139–150. [Google Scholar] [CrossRef]
- Bae, H.; Jang, J.Y.; Choi, S.S.; Lee, J.J.; Kim, H.; Jo, A.; Lee, K.J.; Choi, J.H.; Suh, S.W.; Park, S.B. Mechanistic Elucidation Guided by Covalent Inhibitors for the Development of Anti-Diabetic PPARγ Ligands. Chem. Sci. 2016, 7, 5523–5529. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capelli, D.; Cazzaniga, G.; Mori, M.; Laghezza, A.; Loiodice, F.; Quaglia, M.; Negro, E.; Meneghetti, F.; Villa, S.; Montanari, R. Biological Screening and Crystallographic Studies of Hydroxy γ-Lactone Derivatives to Investigate PPARγ Phosphorylation Inhibition. Biomolecules 2023, 13, 694. https://doi.org/10.3390/biom13040694
Capelli D, Cazzaniga G, Mori M, Laghezza A, Loiodice F, Quaglia M, Negro E, Meneghetti F, Villa S, Montanari R. Biological Screening and Crystallographic Studies of Hydroxy γ-Lactone Derivatives to Investigate PPARγ Phosphorylation Inhibition. Biomolecules. 2023; 13(4):694. https://doi.org/10.3390/biom13040694
Chicago/Turabian StyleCapelli, Davide, Giulia Cazzaniga, Matteo Mori, Antonio Laghezza, Fulvio Loiodice, Martina Quaglia, Elisa Negro, Fiorella Meneghetti, Stefania Villa, and Roberta Montanari. 2023. "Biological Screening and Crystallographic Studies of Hydroxy γ-Lactone Derivatives to Investigate PPARγ Phosphorylation Inhibition" Biomolecules 13, no. 4: 694. https://doi.org/10.3390/biom13040694