
Nuclear Receptor Gene Variants Underlying Disorders/Differences of Sex Development through Abnormal Testicular Development

Abstract
:1. Introduction
2. Testicular Development in Humans
3. Overview of Nuclear Receptors
4. NR5A1 (Nuclear Receptor Subfamily 5 Group A Member 1)
4.1. NR5A1 Variants in 46,XY DSD
4.2. NR5A1 Variants in 46,XX Testicular/Ovotesticular DSD
4.3. Findings Obtained from Recent Studies
4.3.1. Potential Contribution of Digenic/Oligogenic Inheritance to the Broad Phenotypic Spectrum Associated with NR5A1 Variants
4.3.2. Variants in Regulatory Regions of NR5A1 as a Potential Etiology of DSD
5. NR0B1 (Nuclear Receptor Subfamily 0 Group B Member 1)
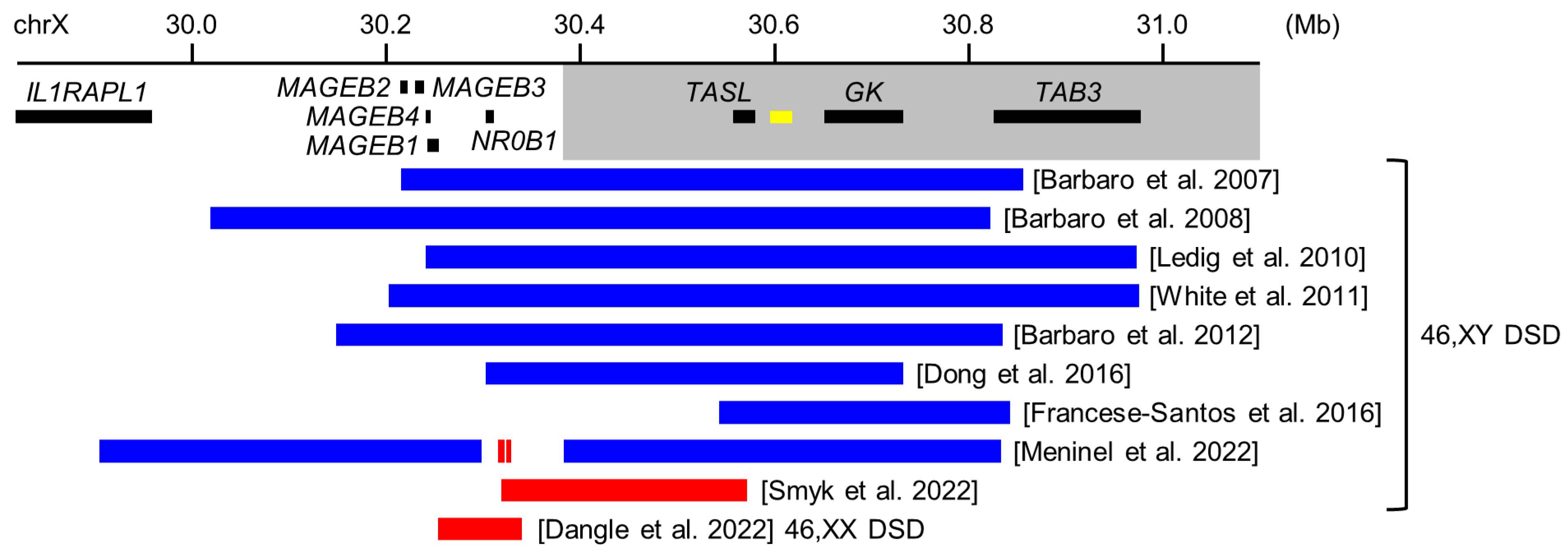
5.1. Copy Number Variants around NR0B1 in 46,XY DSD

5.2. NR0B1 Sequence Variants in 46,XY DSD
5.3. Copy Number Variants around NR0B1 in 46,XX Ovotesticular DSD
5.4. Roles of NR0B1 in Sexual Development: Implications from Studies in Rodents
6. NR2F2 (Nuclear Receptor Subfamily 2 Group F Member 2)
6.1. NR2F2 Variants in 46,XX Testicular/Ovotesticular DSD
6.2. NR2F2 Sequence Variants in 46,XY DSD
6.3. Roles of NR2F2 in Sexual Development: Implications from Studies in Rodents
7. Future Perspective: A New Model to Investigate Molecular Networks in Human Testicular Development
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lundgaard Riis, M.; Jørgensen, A. Deciphering Sex-Specific Differentiation of Human Fetal Gonads: Insight From Experimental Models. Front. Cell. Dev. Biol. 2022, 10, 902082. [Google Scholar] [CrossRef]
- Lee, P.A.; Houk, C.P.; Ahmed, S.F.; Hughes, I.A. International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology Consensus Statement on Management of Intersex Disorders. International Consensus Conference on Intersex. Pediatrics 2006, 118, e488–e500. [Google Scholar] [PubMed]
- Grinspon, R.P.; Bergadá, I.; Rey, R.A. Male Hypogonadism and Disorders of Sex Development. Front. Endocrinol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Délot, E.C.; Vilain, E. Towards Improved Genetic Diagnosis of Human Differences of Sex Development. Nat. Rev. Genet. 2021, 22, 588–602. [Google Scholar] [CrossRef]
- McElreavey, K.; Bashamboo, A. Monogenic Forms of DSD: An Update. Horm. Res. Paediatr. 2023, 96, 182–206. [Google Scholar] [CrossRef] [PubMed]
- Elzaiat, M.; McElreavey, K.; Bashamboo, A. Genetics of 46,XY Gonadal Dysgenesis. Best. Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101633. [Google Scholar] [CrossRef]
- Chiang, H.-S.; Wu, Y.-N.; Wu, C.-C.; Hwang, J.-L. Cytogenic and Molecular Analyses of 46,XX Male Syndrome with Clinical Comparison to Other Groups with Testicular Azoospermia of Genetic Origin. J. Formos. Med. Assoc. 2013, 112, 72–78. [Google Scholar] [CrossRef]
- Dangle, P.; Touzon, M.S.; Reyes-Múgica, M.; Witchel, S.F.; Rajkovic, A.; Schneck, F.X.; Yatsenko, S.A. Female-to-Male Sex Reversal Associated with Unique Xp21.2 Deletion Disrupting Genomic Regulatory Architecture of the Dosage-Sensitive Sex Reversal Region. J. Med. Genet. 2017, 54, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Carvalheira, G.; Malinverni, A.M.; Moysés-Oliveira, M.; Ueta, R.; Cardili, L.; Monteagudo, P.; Mathez, A.L.G.; Verreschi, I.T.; Maluf, M.A.; Shida, M.E.F.; et al. The Natural History of a Man With Ovotesticular 46,XX DSD Caused by a Novel 3-Mb 15q26.2 Deletion Containing NR2F2 Gene. J. Endocr. Soc. 2019, 3, 2107–2113. [Google Scholar] [CrossRef]
- Falah, N.; Posey, J.E.; Thorson, W.; Benke, P.; Tekin, M.; Tarshish, B.; Lupski, J.R.; Harel, T. 22q11.2q13 Duplication Including SOX10 Causes Sex-Reversal and Peripheral Demyelinating Neuropathy, Central Dysmyelinating Leukodystrophy, Waardenburg Syndrome, and Hirschsprung Disease. Am. J. Med. Genet. A 2017, 173, 1066–1070. [Google Scholar] [CrossRef]
- Bashamboo, A.; Eozenou, C.; Jorgensen, A.; Bignon-Topalovic, J.; Siffroi, J.-P.; Hyon, C.; Tar, A.; Nagy, P.; Sólyom, J.; Halász, Z.; et al. Loss of Function of the Nuclear Receptor NR2F2, Encoding COUP-TF2, Causes Testis Development and Cardiac Defects in 46,XX Children. Am. J. Hum. Genet. 2018, 102, 487–493. [Google Scholar] [CrossRef]
- Bashamboo, A.; Donohoue, P.A.; Vilain, E.; Rojo, S.; Calvel, P.; Seneviratne, S.N.; Buonocore, F.; Barseghyan, H.; Bingham, N.; Rosenfeld, J.A.; et al. A Recurrent p.Arg92Trp Variant in Steroidogenic Factor-1 (NR5A1) Can Act as a Molecular Switch in Human Sex Development. Hum. Mol. Genet. 2016, 25, 3446–3453. [Google Scholar] [CrossRef]
- Baetens, D.; Stoop, H.; Peelman, F.; Todeschini, A.-L.; Rosseel, T.; Coppieters, F.; Veitia, R.A.; Looijenga, L.H.J.; De Baere, E.; Cools, M. NR5A1 Is a Novel Disease Gene for 46,XX Testicular and Ovotesticular Disorders of Sex Development. Genet. Med. 2017, 19, 367–376. [Google Scholar] [CrossRef]
- Igarashi, M.; Takasawa, K.; Hakoda, A.; Kanno, J.; Takada, S.; Miyado, M.; Baba, T.; Morohashi, K.-I.; Tajima, T.; Hata, K.; et al. Identical NR5A1 Missense Mutations in Two Unrelated 46,XX Individuals with Testicular Tissues. Hum. Mutat. 2017, 38, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, K.; Ogawa, Y.; Terao, M.; Asakura, Y.; Muroya, K.; Hayashi, M.; Ishii, T.; Hasegawa, T.; Sekido, R.; Fukami, M.; et al. Identification of the First Promoter-Specific Gain-of-Function SOX9 Missense Variant (p.E50K) in a Patient with 46,XX Ovotesticular Disorder of Sex Development. Am. J. Med. Genet. A 2021, 185, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Jimenez, L.; Azova, S.; Kremen, J.; Chan, Y.-M.; Elhusseiny, A.M.; Saeed, H.; Goldsmith, J.; Al-Ibraheemi, A.; O’Connell, A.E.; et al. Novel Variants in the Stem Cell Niche Factor WNT2B Define the Disease Phenotype as a Congenital Enteropathy with Ocular Dysgenesis. Eur. J. Hum. Genet. 2021, 29, 998–1007. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; Konrad, D.; Meyer, M.; DeBeaufort, C.; Schoenle, E.J. Ovaries and Female Phenotype in a Girl with 46,XY Karyotype and Mutations in the CBX2 Gene. Am. J. Hum. Genet. 2009, 84, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Cameron, F.J.; Hageman, R.M.; Cooke-Yarborough, C.; Kwok, C.; Goodwin, L.L.; Sillence, D.O.; Sinclair, A.H. A Novel Germ Line Mutation in SOX9 Causes Familial Campomelic Dysplasia and Sex Reversal. Hum. Mol. Genet. 1996, 5, 1625–1630. [Google Scholar] [CrossRef]
- Ledig, S.; Hiort, O.; Wünsch, L.; Wieacker, P. Partial Deletion of DMRT1 Causes 46,XY Ovotesticular Disorder of Sexual Development. Eur. J. Endocrinol. 2012, 167, 119–124. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, C.; Li, Z.; Wu, Z.; Hong, L. Early Gonadal Development and Sex Determination in Mammal. Int. J. Mol. Sci. 2022, 23, 7500. [Google Scholar] [CrossRef]
- Lucas-Herald, A.K.; Bashamboo, A. Gonadal Development. Endocr. Dev. 2014, 27, 1–16. [Google Scholar]
- Culty, M. Gonocytes, the Forgotten Cells of the Germ Cell Lineage. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 1–26. [Google Scholar] [CrossRef]
- Hanley, N.A.; Ball, S.G.; Clement-Jones, M.; Hagan, D.M.; Strachan, T.; Lindsay, S.; Robson, S.; Ostrer, H.; Parker, K.L.; Wilson, D.I. Expression of Steroidogenic Factor 1 and Wilms’ Tumour 1 during Early Human Gonadal Development and Sex Determination. Mech. Dev. 1999, 87, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Overland, M.; Derpinghaus, A.; Aksel, S.; Cao, M.; Ladwig, N.; Cunha, G.R.; Baskin, L.S. Development of the Human Fetal Testis: Morphology and Expression of Cellular Differentiation Markers. Differentiation 2023, 129, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Hanley, N.A.; Hagan, D.M.; Clement-Jones, M.; Ball, S.G.; Strachan, T.; Salas-Cortés, L.; McElreavey, K.; Lindsay, S.; Robson, S.; Bullen, P.; et al. SRY, SOX9, and DAX1 Expression Patterns during Human Sex Determination and Gonadal Development. Mech. Dev. 2000, 91, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Lottrup, G.; Nielsen, J.E.; Maroun, L.L.; Møller, L.M.A.; Yassin, M.; Leffers, H.; Skakkebæk, N.E.; Rajpert-De Meyts, E. Expression Patterns of DLK1 and INSL3 Identify Stages of Leydig Cell Differentiation during Normal Development and in Testicular Pathologies, Including Testicular Cancer and Klinefelter Syndrome. Hum. Reprod. 2014, 29, 1637–1650. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Sosa, E.; Chitiashvili, T.; Nie, X.; Rojas, E.J.; Oliver, E.; Donor, C.; Plath, K.; Hotaling, J.M.; Stukenborg, J.-B.; et al. Single-Cell Analysis of the Developing Human Testis Reveals Somatic Niche Cell Specification and Fetal Germline Stem Cell Establishment. Cell. Stem Cell. 2021, 28, 764–778.e4. [Google Scholar] [CrossRef]
- Pardee, K.; Necakov, A.S.; Krause, H. Nuclear Receptors: Small Molecule Sensors That Coordinate Growth, Metabolism and Reproduction. Subcell. Biochem. 2011, 52, 123–153. [Google Scholar]
- Giguère, V. Orphan Nuclear Receptors: From Gene to Function. Endocr. Rev. 1999, 20, 689–725. [Google Scholar] [CrossRef] [PubMed]
- Meinsohn, M.-C.; Smith, O.E.; Bertolin, K.; Murphy, B.D. The Orphan Nuclear Receptors Steroidogenic Factor-1 and Liver Receptor Homolog-1: Structure, Regulation, and Essential Roles in Mammalian Reproduction. Physiol. Rev. 2019, 99, 1249–1279. [Google Scholar] [CrossRef]
- Morohashi, K.-I.; Inoue, M.; Baba, T. Coordination of Multiple Cellular Processes by NR5A1/Nr5a1. Endocrinol. Metab. 2020, 35, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ikeda, Y.; Parker, K.L. A Cell-Specific Nuclear Receptor Is Essential for Adrenal and Gonadal Development and Sexual Differentiation. Cell 1994, 77, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Sekido, R.; Lovell-Badge, R. Sex Determination Involves Synergistic Action of SRY and SF1 on a Specific Sox9 Enhancer. Nature 2008, 453, 930–934. [Google Scholar] [CrossRef]
- Achermann, J.C.; Ito, M.; Ito, M.; Hindmarsh, P.C.; Jameson, J.L. A Mutation in the Gene Encoding Steroidogenic Factor-1 Causes XY Sex Reversal and Adrenal Failure in Humans. Nat. Genet. 1999, 22, 125–126. [Google Scholar] [CrossRef]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of Sex Development: Insights from Targeted Gene Sequencing of a Large International Patient Cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef]
- Buonocore, F.; Clifford-Mobley, O.; King, T.F.J.; Striglioni, N.; Man, E.; Suntharalingham, J.P.; Del Valle, I.; Lin, L.; Lagos, C.F.; Rumsby, G.; et al. Next-Generation Sequencing Reveals Novel Genetic Variants (SRY, DMRT1, NR5A1, DHH, DHX37) in Adults With 46,XY DSD. J. Endocr. Soc. 2019, 3, 2341–2360. [Google Scholar] [CrossRef]
- Hughes, L.A.; McKay-Bounford, K.; Webb, E.A.; Dasani, P.; Clokie, S.; Chandran, H.; McCarthy, L.; Mohamed, Z.; Kirk, J.M.W.; Krone, N.P.; et al. Next Generation Sequencing (NGS) to Improve the Diagnosis and Management of Patients with Disorders of Sex Development (DSD). Endocr. Connect. 2019, 8, 100–110. [Google Scholar] [CrossRef]
- Yu, B.-Q.; Liu, Z.-X.; Gao, Y.-J.; Wang, X.; Mao, J.-F.; Nie, M.; Wu, X.-Y. Prevalence of Gene Mutations in a Chinese 46,XY Disorders of Sex Development Cohort Detected by Targeted next-Generation Sequencing. Asian J. Androl. 2021, 23, 69–73. [Google Scholar]
- Ata, A.; Özen, S.; Onay, H.; Uzun, S.; Gökşen, D.; Özkınay, F.; Özbaran, N.B.; Ulman, İ.; Darcan, Ş. A Large Cohort of Disorders of Sex Development and Their Genetic Characteristics: 6 Novel Mutations in Known Genes. Eur. J. Med. Genet. 2021, 64, 104154. [Google Scholar] [CrossRef] [PubMed]
- Globa, E.; Zelinska, N.; Shcherbak, Y.; Bignon-Topalovic, J.; Bashamboo, A.; McElreavey, K. Disorders of Sex Development in a Large Ukrainian Cohort: Clinical Diversity and Genetic Findings. Front. Endocrinol. 2022, 13, 810782. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.L.; Batista, R.L.; Nishi, M.Y.; Lerário, A.M.; Silva, T.E.; de Moraes Narcizo, A.; Benedetti, A.F.F.; de Assis Funari, M.F.; Faria Junior, J.A.; Moraes, D.R.; et al. Contribution of Clinical and Genetic Approaches for Diagnosing 209 Index Cases With 46,XY Differences of Sex Development. J. Clin. Endocrinol. Metab. 2022, 107, e1797–e1806. [Google Scholar] [CrossRef]
- Nagy, O.; Kárteszi, J.; Hartwig, M.; Bertalan, R.; Jávorszky, E.; Erhardt, É.; Patócs, A.; Tornóczky, T.; Balogh, I.; Ujfalusi, A. The Importance of the Multiplex Ligation-Dependent Probe Amplification in the Identification of a Novel Two-Exon Deletion of the NR5A1 Gene in a Patient with 46,XY Differences of Sex Development. Mol. Biol. Rep. 2019, 46, 5595–5601. [Google Scholar] [CrossRef]
- Sreenivasan, R.; Bell, K.; van den Bergen, J.; Robevska, G.; Belluoccio, D.; Dahiya, R.; Leong, G.M.; Dulon, J.; Touraine, P.; Tucker, E.J.; et al. Whole Exome Sequencing Reveals Copy Number Variants in Individuals with Disorders of Sex Development. Mol. Cell. Endocrinol. 2022, 546, 111570. [Google Scholar] [CrossRef]
- Achermann, J.C.; Ozisik, G.; Ito, M.; Orun, U.A.; Harmanci, K.; Gurakan, B.; Jameson, J.L. Gonadal Determination and Adrenal Development Are Regulated by the Orphan Nuclear Receptor Steroidogenic Factor-1, in a Dose-Dependent Manner. J. Clin. Endocrinol. Metab. 2002, 87, 1829–1833. [Google Scholar] [CrossRef]
- Suntharalingham, J.P.; Buonocore, F.; Duncan, A.J.; Achermann, J.C. DAX-1 (NR0B1) and Steroidogenic Factor-1 (SF-1, NR5A1) in Human Disease. Best. Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 607–619. [Google Scholar] [CrossRef]
- Hou, L.; Zhao, M.; Fan, L.; Cao, B.; Chen, J.; Cui, Y.; Polak, M.; Gong, C. One Hundred Twelve Cases of 46, XY DSD Patients after Initial Gender Assignment: A Short-Term Survey of Gender Role and Gender Dysphoria. Orphanet J. Rare Dis. 2021, 16, 416. [Google Scholar] [CrossRef]
- Askari, M.; Rastari, M.; Seresht-Ahmadi, M.; McElreavey, K.; Bashamboo, A.; Razzaghy-Azar, M.; Totonchi, M. A Missense Mutation in NR5A1 Causing Female to Male Sex Reversal: A Case Report. Andrologia 2020, 52, e13585. [Google Scholar] [CrossRef]
- Domenice, S.; Machado, A.Z.; Ferreira, F.M.; Ferraz-de-Souza, B.; Lerario, A.M.; Lin, L.; Nishi, M.Y.; Gomes, N.L.; da Silva, T.E.; Silva, R.B.; et al. Wide Spectrum of NR5A1-Related Phenotypes in 46,XY and 46,XX Individuals. Birth Defects Res. Part C Embryo Today Rev. 2016, 108, 309–320. [Google Scholar]
- Knarston, I.M.; Robevska, G.; van den Bergen, J.A.; Eggers, S.; Croft, B.; Yates, J.; Hersmus, R.; Looijenga, L.H.J.; Cameron, F.J.; Monhike, K.; et al. NR5A1 Gene Variants Repress the Ovarian-Specific WNT Signaling Pathway in 46,XX Disorders of Sex Development Patients. Hum. Mutat. 2019, 40, 207–216. [Google Scholar] [CrossRef]
- Takasawa, K.; Igarashi, M.; Ono, M.; Takemoto, A.; Takada, S.; Yamataka, A.; Ogata, T.; Morio, T.; Fukami, M.; Kashimada, K. Phenotypic Variation in 46,XX Disorders of Sex Development Due to the NR5A1 p.R92W Variant: A Sibling Case Report and Literature Review. Sex. Dev. 2017, 11, 284–288. [Google Scholar] [CrossRef]
- Swartz, J.M.; Ciarlo, R.; Guo, M.H.; Abrha, A.; Weaver, B.; Diamond, D.A.; Chan, Y.-M.; Hirschhorn, J.N. A 46,XX Ovotesticular Disorder of Sex Development Likely Caused by a Steroidogenic Factor-1 (NR5A1) Variant. Horm. Res. Paediatr. 2017, 87, 191–195. [Google Scholar] [CrossRef]
- Song, Y.; Fan, L.; Gong, C. Phenotype and Molecular Characterizations of 30 Children From China With NR5A1 Mutations. Front. Pharmacol. 2018, 9, 1224. [Google Scholar] [CrossRef] [PubMed]
- Miyado, M.; Inui, M.; Igarashi, M.; Katoh-Fukui, Y.; Takasawa, K.; Hakoda, A.; Kanno, J.; Kashimada, K.; Miyado, K.; Tamano, M.; et al. The p.R92W Variant of NR5A1/Nr5a1 Induces Testicular Development of 46,XX Gonads in Humans, but Not in Mice: Phenotypic Comparison of Human Patients and Mutation-Induced Mice. Biol. Sex. Differ. 2016, 7, 56. [Google Scholar] [CrossRef]
- Hatano, O.; Takayama, K.; Imai, T.; Waterman, M.R.; Takakusu, A.; Omura, T.; Morohashi, K. Sex-Dependent Expression of a Transcription Factor, Ad4BP, Regulating Steroidogenic P-450 Genes in the Gonads during Prenatal and Postnatal Rat Development. Development 1994, 120, 2787–2797. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, K.; Kashimada, K.; Pelosi, E.; Takagi, M.; Morio, T.; Asahara, H.; Schlessinger, D.; Mizutani, S.; Koopman, P. FOXL2 Transcriptionally Represses Sf1 Expression by Antagonizing WT1 during Ovarian Development in Mice. FASEB J. 2014, 28, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Fabbri-Scallet, H.; de Sousa, L.M.; Maciel-Guerra, A.T.; Guerra-Júnior, G.; de Mello, M.P. Mutation Update for the NR5A1 Gene Involved in DSD and Infertility. Hum. Mutat. 2020, 41, 58–68. [Google Scholar] [CrossRef]
- Hattori, A.; Zukeran, H.; Igarashi, M.; Toguchi, S.; Toubaru, Y.; Inoue, T.; Katoh-Fukui, Y.; Fukami, M. A Novel C-Terminal Truncating NR5A1 Mutation in Dizygotic Twins. Hum. Genome Var. 2017, 4, 17008. [Google Scholar] [CrossRef] [PubMed]
- Camats, N.; Flück, C.E.; Audí, L. Oligogenic Origin of Differences of Sex Development in Humans. Int. J. Mol. Sci. 2020, 21, 1809. [Google Scholar] [CrossRef]
- Kouri, C.; Sommer, G.; Flück, C.E. Oligogenic Causes of Human Differences of Sex Development: Facing the Challenge of Genetic Complexity. Horm. Res. Paediatr. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Mazen, I.; Mekkawy, M.; Kamel, A.; Essawi, M.; Hassan, H.; Abdel-Hamid, M.; Amr, K.; Soliman, H.; El-Ruby, M.; Torky, A.; et al. Advances in Genomic Diagnosis of a Large Cohort of Egyptian Patients with Disorders of Sex Development. Am. J. Med. Genet. A 2021, 185, 1666–1677. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, J.; Zhou, X.; Yang, J.; Ji, Y.; Xu, C. Characteristics and Possible Mechanisms of 46, XY Differences in Sex Development Caused by Novel Compound Variants in NR5A1 and MAP3K1. Orphanet J. Rare Dis. 2021, 16, 268. [Google Scholar] [CrossRef] [PubMed]
- Zidoune, H.; Ladjouze, A.; Chellat-Rezgoune, D.; Boukri, A.; Dib, S.A.; Nouri, N.; Tebibel, M.; Sifi, K.; Abadi, N.; Satta, D.; et al. Novel Genomic Variants, Atypical Phenotypes and Evidence of a Digenic/Oligogenic Contribution to Disorders/Differences of Sex Development in a Large North African Cohort. Front. Genet. 2022, 13, 900574. [Google Scholar] [CrossRef]
- Mazen, I.; Abdel-Hamid, M.; Mekkawy, M.; Bignon-Topalovic, J.; Boudjenah, R.; El Gammal, M.; Essawi, M.; Bashamboo, A.; McElreavey, K. Identification of NR5A1 Mutations and Possible Digenic Inheritance in 46,XY Gonadal Dysgenesis. Sex. Dev. 2016, 10, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Camats, N.; Fernández-Cancio, M.; Audí, L.; Schaller, A.; Flück, C.E. Broad Phenotypes in Heterozygous NR5A1 46,XY Patients with a Disorder of Sex Development: An Oligogenic Origin? Eur. J. Hum. Genet. 2018, 26, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, L.; Wang, N.; Zhu, H.; Han, B.; Sun, F.; Yao, H.; Zhang, Q.; Zhu, W.; Cheng, T.; et al. Next-Generation Sequencing Reveals Genetic Landscape in 46, XY Disorders of Sexual Development Patients with Variable Phenotypes. Hum. Genet. 2018, 137, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Martínez de LaPiscina, I.; Mahmoud, R.A.; Sauter, K.-S.; Esteva, I.; Alonso, M.; Costa, I.; Rial-Rodriguez, J.M.; Rodríguez-Estévez, A.; Vela, A.; Castano, L.; et al. Variants of STAR, AMH and ZFPM2/FOG2 May Contribute towards the Broad Phenotype Observed in 46,XY DSD Patients with Heterozygous Variants of NR5A1. Int. J. Mol. Sci. 2020, 21, 8554. [Google Scholar] [CrossRef]
- Okazaki, A.; Ott, J. Machine Learning Approaches to Explore Digenic Inheritance. Trends Genet. 2022, 38, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Schäffer, A.A. Digenic Inheritance in Medical Genetics. J. Med. Genet. 2013, 50, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Bashamboo, A.; Ferraz-de-Souza, B.; Lourenço, D.; Lin, L.; Sebire, N.J.; Montjean, D.; Bignon-Topalovic, J.; Mandelbaum, J.; Siffroi, J.-P.; Christin-Maitre, S.; et al. Human Male Infertility Associated with Mutations in NR5A1 Encoding Steroidogenic Factor 1. Am. J. Hum. Genet. 2010, 87, 505–512. [Google Scholar] [CrossRef]
- Jorgensen, J.S.; Nilson, J.H. AR Suppresses Transcription of the LHbeta Subunit by Interacting with Steroidogenic Factor-1. Mol. Endocrinol. 2001, 15, 1505–1516. [Google Scholar]
- Cunha, G.R.; Cao, M.; Aksel, S.; Derpinghaus, A.; Baskin, L.S. Mouse-Human Species Differences in Early Testicular Development and Its Implications. Differentiation 2023, 129, 79–95. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, P.J.; Mitchell, R.T.; Monteiro, A.; O’Hara, L.; Cruickshanks, L.; der Grinten, H.C.; Brown, P.; Abel, M.; Smith, L.B. Androgen Receptor Expression Is Required to Ensure Development of Adult Leydig Cells and to Prevent Development of Steroidogenic Cells with Adrenal Characteristics in the Mouse Testis. BMC Dev. Biol. 2019, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Lottrup, G.; Jørgensen, A.; Nielsen, J.E.; Jørgensen, N.; Duno, M.; Vinggaard, A.M.; Skakkebæk, N.E.; Rajpert-De Meyts, E. Identification of a Novel Androgen Receptor Mutation in a Family with Multiple Components Compatible with the Testicular Dysgenesis Syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, L.; Cofini, M.; Leonardi, A.; Bertozzi, M.; Penta, L.; Esposito, S. Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS). Int. J. Environ. Res. Public. Health 2019, 16, 1268. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Willatt, L.; Homfray, T.; Woods, C.G. A SOX9 Duplication and Familial 46,XX Developmental Testicular Disorder. N. Engl. J. Med. 2011, 364, 91–93. [Google Scholar] [CrossRef]
- Kim, G.-J.; Sock, E.; Buchberger, A.; Just, W.; Denzer, F.; Hoepffner, W.; German, J.; Cole, T.; Mann, J.; Seguin, J.H.; et al. Copy Number Variation of Two Separate Regulatory Regions Upstream of SOX9 Causes Isolated 46,XY or 46,XX Disorder of Sex Development. J. Med. Genet. 2015, 52, 240–247. [Google Scholar] [CrossRef]
- Fabbri-Scallet, H.; Werner, R.; Guaragna, M.S.; de Andrade, J.G.R.; Maciel-Guerra, A.T.; Hornig, N.C.; Hiort, O.; Guerra-Júnior, G.; de Mello, M.P. Can Non-Coding NR5A1 Gene Variants Explain Phenotypes of Disorders of Sex Development? Sex. Dev. 2022, 16, 242–250. [Google Scholar] [CrossRef]
- Shima, Y.; Miyabayashi, K.; Baba, T.; Otake, H.; Katsura, Y.; Oka, S.; Zubair, M.; Morohashi, K. Identification of an Enhancer in the Ad4BP/SF-1 Gene Specific for Fetal Leydig Cells. Endocrinology 2012, 153, 417–425. [Google Scholar] [CrossRef]
- Ludbrook, L.M.; Harley, V.R. Sex Determination: A “window” of DAX1 Activity. Trends Endocrinol. Metab. 2004, 15, 116–121. [Google Scholar] [CrossRef]
- Lalli, E.; Sassone-Corsi, P. DAX-1, an Unusual Orphan Receptor at the Crossroads of Steroidogenic Function and Sexual Differentiation. Mol. Endocrinol. 2003, 17, 1445–1453. [Google Scholar] [CrossRef]
- Ludbrook, L.M.; Bernard, P.; Bagheri-Fam, S.; Ryan, J.; Sekido, R.; Wilhelm, D.; Lovell-Badge, R.; Harley, V.R. Excess DAX1 Leads to XY Ovotesticular Disorder of Sex Development (DSD) in Mice by Inhibiting Steroidogenic Factor-1 (SF1) Activation of the Testis Enhancer of SRY-Box-9 (Sox9). Endocrinology 2012, 153, 1948–1958. [Google Scholar] [CrossRef]
- Barbaro, M.; Oscarson, M.; Schoumans, J.; Staaf, J.; Ivarsson, S.A.; Wedell, A. Isolated 46,XY Gonadal Dysgenesis in Two Sisters Caused by a Xp21.2 Interstitial Duplication Containing the DAX1 Gene. J. Clin. Endocrinol. Metab. 2007, 92, 3305–3313. [Google Scholar] [CrossRef]
- Barbaro, M.; Cicognani, A.; Balsamo, A.; Löfgren, A.; Baldazzi, L.; Wedell, A.; Oscarson, M. Gene Dosage Imbalances in Patients with 46,XY Gonadal DSD Detected by an in-House-Designed Synthetic Probe Set for Multiplex Ligation-Dependent Probe Amplification Analysis. Clin. Genet. 2008, 73, 453–464. [Google Scholar] [CrossRef]
- Barbaro, M.; Cook, J.; Lagerstedt-Robinson, K.; Wedell, A. Multigeneration Inheritance through Fertile XX Carriers of an NR0B1 (DAX1) Locus Duplication in a Kindred of Females with Isolated XY Gonadal Dysgenesis. Int. J. Endocrinol. 2012, 2012, 504904. [Google Scholar] [CrossRef]
- Bardoni, B.; Zanaria, E.; Guioli, S.; Floridia, G.; Worley, K.C.; Tonini, G.; Ferrante, E.; Chiumello, G.; McCabe, E.R.; Fraccaro, M. A Dosage Sensitive Locus at Chromosome Xp21 Is Involved in Male to Female Sex Reversal. Nat. Genet. 1994, 7, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yi, Y.; Yao, H.; Yang, Z.; Hu, H.; Liu, J.; Gao, C.; Zhang, M.; Zhou, L.; Yi, X.; et al. Targeted Next-Generation Sequencing Identification of Mutations in Patients with Disorders of Sex Development. BMC Med. Genet. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Gimelli, G.; Giglio, S.; Zuffardi, O.; Alhonen, L.; Suppola, S.; Cusano, R.; Lo Nigro, C.; Gatti, R.; Ravazzolo, R.; Seri, M. Gene Dosage of the Spermidine/Spermine N(1)-Acetyltransferase (SSAT) Gene with Putrescine Accumulation in a Patient with a Xp21.1p22.12 Duplication and Keratosis Follicularis Spinulosa Decalvans (KFSD). Hum. Genet. 2002, 111, 235–241. [Google Scholar] [CrossRef]
- Ledig, S.; Hiort, O.; Scherer, G.; Hoffmann, M.; Wolff, G.; Morlot, S.; Kuechler, A.; Wieacker, P. Array-CGH Analysis in Patients with Syndromic and Non-Syndromic XY Gonadal Dysgenesis: Evaluation of Array CGH as Diagnostic Tool and Search for New Candidate Loci. Hum. Reprod. 2010, 25, 2637–2646. [Google Scholar] [CrossRef] [PubMed]
- Rjiba, K.; Slimani, W.; Gaddas, M.; Hassine, I.H.; Jelloul, A.; Khelifa, H.B.; El Amri, F.; Zaouali, M.; Mcelreavey, K.; Saad, A.; et al. Anomalies in Human Sex Determination: Usefulness of a Combined Cytogenetic Approach to Characterize an Additional Case with Xp Functional Disomy Associated to 46,XY Gonadal Dysgenesis. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 25–34. [Google Scholar]
- Sukumaran, A.; Desmangles, J.-C.; Gartner, L.A.; Buchlis, J. Duplication of Dosage Sensitive Sex Reversal Area in a 46, XY Patient with Normal Sex Determining Region of Y Causing Complete Sex Reversal. J. Pediatr. Endocrinol. Metab. 2013, 26, 775–779. [Google Scholar] [CrossRef] [PubMed]
- White, S.; Ohnesorg, T.; Notini, A.; Roeszler, K.; Hewitt, J.; Daggag, H.; Smith, C.; Turbitt, E.; Gustin, S.; van den Bergen, J.; et al. Copy Number Variation in Patients with Disorders of Sex Development Due to 46,XY Gonadal Dysgenesis. PLoS ONE 2011, 6, e17793. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.Y.; Chu, G.M.; Li, P.P.; He, R. Phenotype and Genetic Characteristics in 20 Chinese Patients with 46,XY Disorders of Sex Development. J. Endocrinol. Investig. 2023. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dabovic, B.; Zanaria, E.; Bardoni, B.; Lisa, A.; Bordignon, C.; Russo, V.; Matessi, C.; Traversari, C.; Camerino, G. A Family of Rapidly Evolving Genes from the Sex Reversal Critical Region in Xp21. Mamm. Genome 1995, 6, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Smyk, M.; Berg, J.S.; Pursley, A.; Curtis, F.K.; Fernandez, B.A.; Bien-Willner, G.A.; Lupski, J.R.; Cheung, S.W.; Stankiewicz, P. Male-to-Female Sex Reversal Associated with an Approximately 250 Kb Deletion Upstream of NR0B1 (DAX1). Hum. Genet. 2007, 122, 63–70. [Google Scholar] [CrossRef]
- Meinel, J.A.; Yumiceba, V.; Künstner, A.; Schultz, K.; Kruse, N.; Kaiser, F.J.; Holterhus, P.-M.; Claviez, A.; Hiort, O.; Busch, H.; et al. Disruption of the Topologically Associated Domain at Xp21.2 Is Related to 46,XY Gonadal Dysgenesis. J. Med. Genet. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Francese-Santos, A.P.; Meinel, J.A.; Piveta, C.S.C.; Andrade, J.G.R.; Barros, B.A.; Fabbri-Scallet, H.; Gil-da-Silva-Lopes, V.L.; Guerra-Junior, G.; Künstner, A.; Busch, H.; et al. A Novel Look at Dosage-Sensitive Sex Locus Xp21.2 in a Case of 46,XY Partial Gonadal Dysgenesis without NR0B1 Duplication. Int. J. Mol. Sci. 2022, 24, 494. [Google Scholar] [CrossRef]
- Miclea, D.; Alkhzouz, C.; Bucerzan, S.; Grigorescu-Sido, P.; Popp, R.A.; Pascanu, I.M.; Cret, V.; Ghervan, C.; Blaga, L.; Zaharie, G. Molecular and Cytogenetic Analysis of Romanian Patients with Differences in Sex Development. Diagnostics 2021, 11, 2107. [Google Scholar] [CrossRef]
- Bertalan, R.; Bencsik, Z.; Mezei, P.; Vajda, Z.; Butz, H.; Patócs, A. Novel Frameshift Mutation of the NR0B1(DAX1) in Two Tall Adult Brothers. Mol. Biol. Rep. 2019, 46, 4599–4604. [Google Scholar] [CrossRef]
- Yu, T.; Wang, J.; Yu, Y.; Huang, X.; Fu, Q.; Shen, Y.; Chen, F. X-Linked Adrenal Hypoplasia Congenita and Hypogonadotropic Hypogonadism: Identification and in Vitro Study of a Novel Small Indel in the NR0B1 Gene. Mol. Med. Rep. 2016, 13, 4039–4045. [Google Scholar] [CrossRef]
- Hodžić, A.; Maver, A.; Plaseska-Karanfilska, D.; Ristanović, M.; Noveski, P.; Zorn, B.; Terzic, M.; Kunej, T.; Peterlin, B. De Novo Mutations in Idiopathic Male Infertility-A Pilot Study. Andrology 2021, 9, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Raffin-Sanson, M.-L.; Oudet, B.; Salenave, S.; Brailly-Tabard, S.; Pehuet, M.; Christin-Maitre, S.; Morel, Y.; Young, J. A Man with a DAX1/NR0B1 Mutation, Normal Puberty, and an Intact Hypothalamic-Pituitary-Gonadal Axis but Deteriorating Oligospermia during Long-Term Follow-Up. Eur. J. Endocrinol. 2013, 168, K45–K50. [Google Scholar] [CrossRef]
- Seminara, S.B.; Achermann, J.C.; Genel, M.; Jameson, J.L.; Crowley, W.F. X-Linked Adrenal Hypoplasia Congenita: A Mutation in DAX1 Expands the Phenotypic Spectrum in Males and Females. J. Clin. Endocrinol. Metab. 1999, 84, 4501–4509. [Google Scholar] [CrossRef]
- Mou, L.; Xie, N.; Yang, L.; Liu, Y.; Diao, R.; Cai, Z.; Li, H.; Gui, Y. A Novel Mutation of DAX-1 Associated with Secretory Azoospermia. PLoS ONE 2015, 10, e0133997. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.N.; Ito, M.; Saunders, T.L.; Camper, S.A.; Jameson, J.L. Role of Ahch in Gonadal Development and Gametogenesis. Nat. Genet. 1998, 20, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Meeks, J.J.; Weiss, J.; Jameson, J.L. Dax1 Is Required for Testis Determination. Nat. Genet. 2003, 34, 32–33. [Google Scholar] [CrossRef]
- Bouma, G.J.; Albrecht, K.H.; Washburn, L.L.; Recknagel, A.K.; Churchill, G.A.; Eicher, E.M. Gonadal Sex Reversal in Mutant Dax1 XY Mice: A Failure to Upregulate Sox9 in Pre-Sertoli Cells. Development 2005, 132, 3045–3054. [Google Scholar] [CrossRef]
- Kumar, S.; Kim, H.J.; Lee, C.-H.; Choi, H.-S.; Lee, K. Leydig Cell-Specific DAX1-Deleted Mice Has Higher Testosterone Level in the Testis During Pubertal Development. Reprod. Sci. 2022, 29, 955–962. [Google Scholar] [CrossRef]
- Shima, H.; Yatsuga, S.; Nakamura, A.; Sano, S.; Sasaki, T.; Katsumata, N.; Suzuki, E.; Hata, K.; Nakabayashi, K.; Momozawa, Y.; et al. NR0B1 Frameshift Mutation in a Boy with Idiopathic Central Precocious Puberty. Sex. Dev. 2016, 10, 205–209. [Google Scholar] [CrossRef]
- Guzzetti, C.; Bizzarri, C.; Pisaneschi, E.; Mucciolo, M.; Bellacchio, E.; Ibba, A.; Casula, L.; Novelli, A.; Loche, S.; Cappa, M. Next-Generation Sequencing Identifies Different Genetic Defects in 2 Patients with Primary Adrenal Insufficiency and Gonadotropin-Independent Precocious Puberty. Horm. Res. Paediatr. 2018, 90, 203–211. [Google Scholar] [CrossRef]
- Durmaz, E.; Turkkahraman, D.; Berdeli, A.; Atan, M.; Karaguzel, G.; Akcurin, S.; Bircan, I. A Novel DAX-1 Mutation Presented with Precocious Puberty and Hypogonadotropic Hypogonadism in Different Members of a Large Pedigree. J. Pediatr. Endocrinol. Metab. 2013, 26, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.W.; Kang, S.Y.; Kim, G.H.; Yoo, H.W.; Yu, J. Central Precocious Puberty in a Patient with X-Linked Adrenal Hypoplasia Congenita and Xp21 Contiguous Gene Deletion Syndrome. Ann. Pediatr. Endocrinol. Metab. 2013, 18, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wei, H.; Shen, L.; Kumar C, S.; Chen, Q.; Chen, Y.; Kumar, S.A. A Novel Stop-Loss DAX1 Variant Affecting Its Protein-Interaction with SF1 Precedes the Adrenal Hypoplasia Congenital with Rare Spontaneous Precocious Puberty and Elevated Hypothalamic-Pituitary-Gonadal/Adrenal Axis Responses. Eur. J. Med. Genet. 2021, 64, 104192. [Google Scholar] [CrossRef]
- Polvani, S.; Pepe, S.; Milani, S.; Galli, A. COUP-TFII in Health and Disease. Cells 2019, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Y.; Ge, R.; Zirkin, B.R. Leydig Cell Stem Cells: Identification, Proliferation and Differentiation. Mol. Cell. Endocrinol. 2017, 445, 65–73. [Google Scholar] [CrossRef]
- van den Driesche, S.; Walker, M.; McKinnell, C.; Scott, H.M.; Eddie, S.L.; Mitchell, R.T.; Seckl, J.R.; Drake, A.J.; Smith, L.B.; Anderson, R.A.; et al. Proposed Role for COUP-TFII in Regulating Fetal Leydig Cell Steroidogenesis, Perturbation of Which Leads to Masculinization Disorders in Rodents. PLoS ONE 2012, 7, e37064. [Google Scholar] [CrossRef]
- Eliveld, J.; van Daalen, S.K.M.; de Winter-Korver, C.M.; van der Veen, F.; Repping, S.; Teerds, K.; van Pelt, A.M.M. A Comparative Analysis of Human Adult Testicular Cells Expressing Stem Leydig Cell Markers in the Interstitium, Vasculature, and Peritubular Layer. Andrology 2020, 8, 1265–1276. [Google Scholar] [CrossRef]
- Di-Luoffo, M.; Brousseau, C.; Tremblay, J.J. MEF2 and NR2F2 Cooperate to Regulate Akr1c14 Gene Expression in Mouse MA-10 Leydig Cells. Andrology 2016, 4, 335–344. [Google Scholar] [CrossRef]
- Mehanovic, S.; Mendoza-Villarroel, R.E.; Viger, R.S.; Tremblay, J.J. The Nuclear Receptor COUP-TFII Regulates Amhr2 Gene Transcription via a GC-Rich Promoter Element in Mouse Leydig Cells. J. Endocr. Soc. 2019, 3, 2236–2257. [Google Scholar] [CrossRef]
- Mendoza-Villarroel, R.E.; Di-Luoffo, M.; Camiré, E.; Giner, X.C.; Brousseau, C.; Tremblay, J.J. The INSL3 Gene Is a Direct Target for the Orphan Nuclear Receptor, COUP-TFII, in Leydig Cells. J. Mol. Endocrinol. 2014, 53, 43–55. [Google Scholar] [CrossRef]
- Mendoza-Villarroel, R.E.; Robert, N.M.; Martin, L.J.; Brousseau, C.; Tremblay, J.J. The Nuclear Receptor NR2F2 Activates Star Expression and Steroidogenesis in Mouse MA-10 and MLTC-1 Leydig Cells. Biol. Reprod. 2014, 91, 26. [Google Scholar] [CrossRef] [PubMed]
- Kilcoyne, K.R.; Smith, L.B.; Atanassova, N.; Macpherson, S.; McKinnell, C.; van den Driesche, S.; Jobling, M.S.; Chambers, T.J.G.; De Gendt, K.; Verhoeven, G.; et al. Fetal Programming of Adult Leydig Cell Function by Androgenic Effects on Stem/Progenitor Cells. Proc. Natl. Acad. Sci. USA 2014, 111, E1924–E1932. [Google Scholar] [CrossRef]
- Qin, J.; Tsai, M.-J.; Tsai, S.Y. Essential Roles of COUP-TFII in Leydig Cell Differentiation and Male Fertility. PLoS ONE 2008, 3, e3285. [Google Scholar] [CrossRef]
- Bhattacharya, I.; Dey, S. Emerging Concepts on Leydig Cell Development in Fetal and Adult Testis. Front. Endocrinol. 2022, 13, 1086276. [Google Scholar] [CrossRef]
- Teerds, K.J.; Huhtaniemi, I.T. Morphological and Functional Maturation of Leydig Cells: From Rodent Models to Primates. Hum. Reprod. Update 2015, 21, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Di-Luoffo, M.; Pierre, K.J.; Robert, N.M.; Girard, M.-J.; Tremblay, J.J. The Nuclear Receptors SF1 and COUP-TFII Cooperate on the Insl3 Promoter in Leydig Cells. Reproduction 2022, 164, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Giner, X.C.; Pierre, K.J.; Robert, N.M.; Tremblay, J.J. A 35-Bp Conserved Region Is Crucial for Insl3 Promoter Activity in Mouse MA-10 Leydig Cells. Int. J. Mol. Sci. 2022, 23, 15060. [Google Scholar] [CrossRef]
- Gonen, N.; Eozenou, C.; Mitter, R.; Elzaiat, M.; Stévant, I.; Aviram, R.; Bernardo, A.S.; Chervova, A.; Wankanit, S.; Frachon, E.; et al. In Vitro Cellular Reprogramming to Model Gonad Development and Its Disorders. Sci. Adv. 2023, 9, eabn9793. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| HGNC-Approved Gene Symbol | Unified Nomenclature | Class | Phenotype | OMIM | HGMD | Numbers of Variants 1 |
|---|---|---|---|---|---|---|
| RARA | NR1B1 | II | 46,XX DSD (Mayer-Rokitansky-Küster-Hauser syndrome) | unknown | Yes | 1 |
| NR2F2 | NR2F2 | III | 46,XX testicular/ovotesticular DSD (syndromic) | Yes | Yes | 3 |
| ESR1 | NR3A1 | I | 46,XX DSD (Mayer-Rokitansky-Küster-Hauser syndrome) | unknown | Yes | 3 |
| 46,XY DSD | unknown | Yes | 1 | |||
| ESR2 | NR3A2 | I | 46,XY DSD | unknown | Yes | 4 |
| AR | NR3C4 | I | 46,XY DSD (androgen insensitivity syndrome) | Yes | Yes | 647 |
| 46,XY DSD (gonadal dysgenesis) | unknown | Yes | 1 | |||
| NR5A1 | NR5A1 | IV | 46,XX testicular/ovotesticular DSD (nonsyndromic) | Yes | Yes | 1 |
| 46,XY DSD (gonadal dysgenesis, nonsyndromic) | Yes | Yes | 232 | |||
| NR0B1 | NR0B1 | unclassifiable | 46,XX ovotesticular DSD (nonsyndromic) | unknown | Yes | 1 |
| 46,XY DSD (gonadal dysgenesis, syndromic and nonsyndromic) | Yes | Yes | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hattori, A.; Fukami, M. Nuclear Receptor Gene Variants Underlying Disorders/Differences of Sex Development through Abnormal Testicular Development. Biomolecules 2023, 13, 691. https://doi.org/10.3390/biom13040691
Hattori A, Fukami M. Nuclear Receptor Gene Variants Underlying Disorders/Differences of Sex Development through Abnormal Testicular Development. Biomolecules. 2023; 13(4):691. https://doi.org/10.3390/biom13040691
Chicago/Turabian StyleHattori, Atsushi, and Maki Fukami. 2023. "Nuclear Receptor Gene Variants Underlying Disorders/Differences of Sex Development through Abnormal Testicular Development" Biomolecules 13, no. 4: 691. https://doi.org/10.3390/biom13040691