
Alpha-Synuclein mRNA Level Found Dependent on L444P Variant in Carriers and Gaucher Disease Patients on Enzyme Replacement Therapy
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Blood Samples
2.2. Biobanking
2.3. α-Synuclein mRNA Analysis
2.4. α-Synuclein Total and Oligomer Protein Concentration
2.5. Statistical Analysis
3. Results
3.1. Patients’ Characteristics
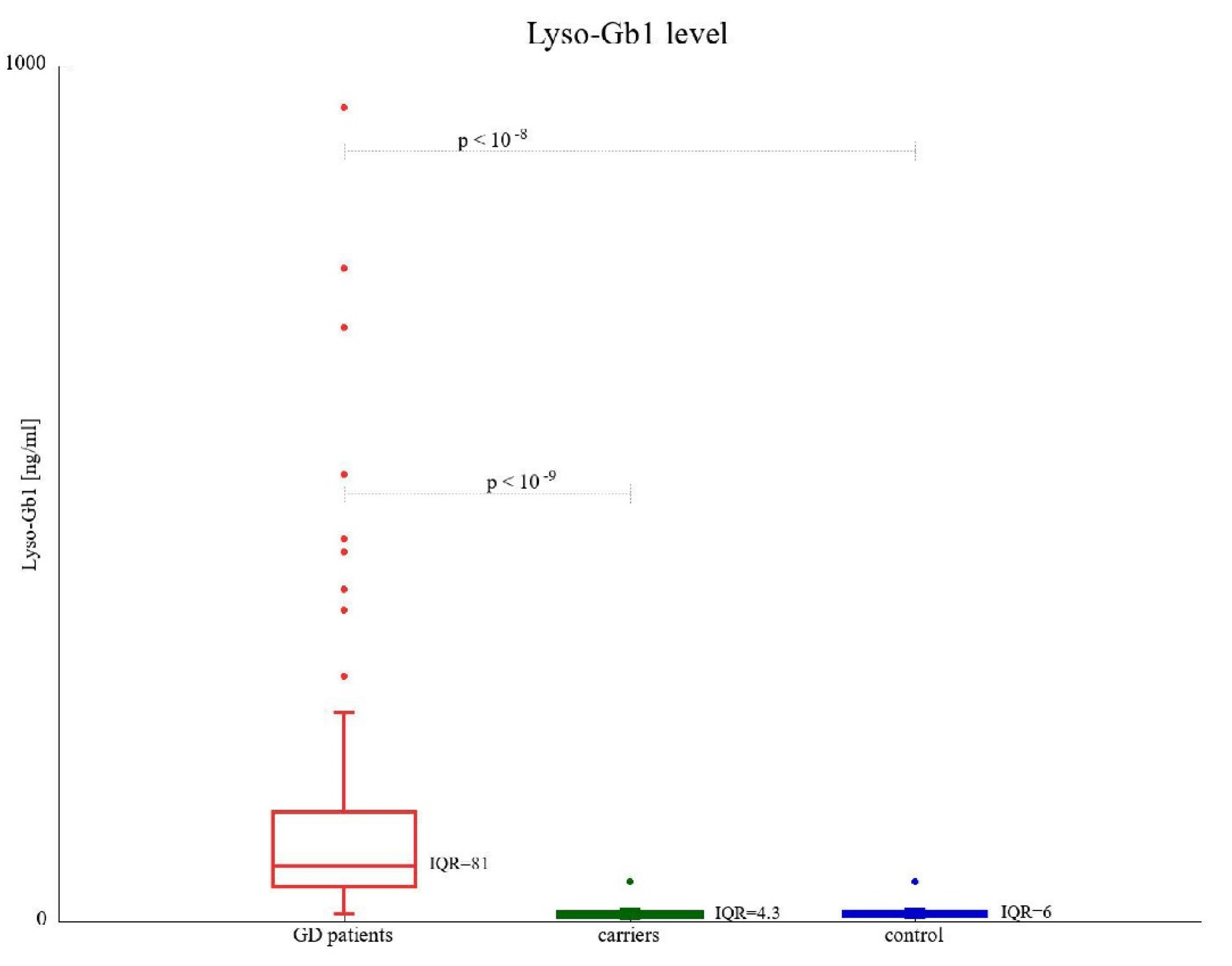
3.2. Lyso-Gb1 Concentration
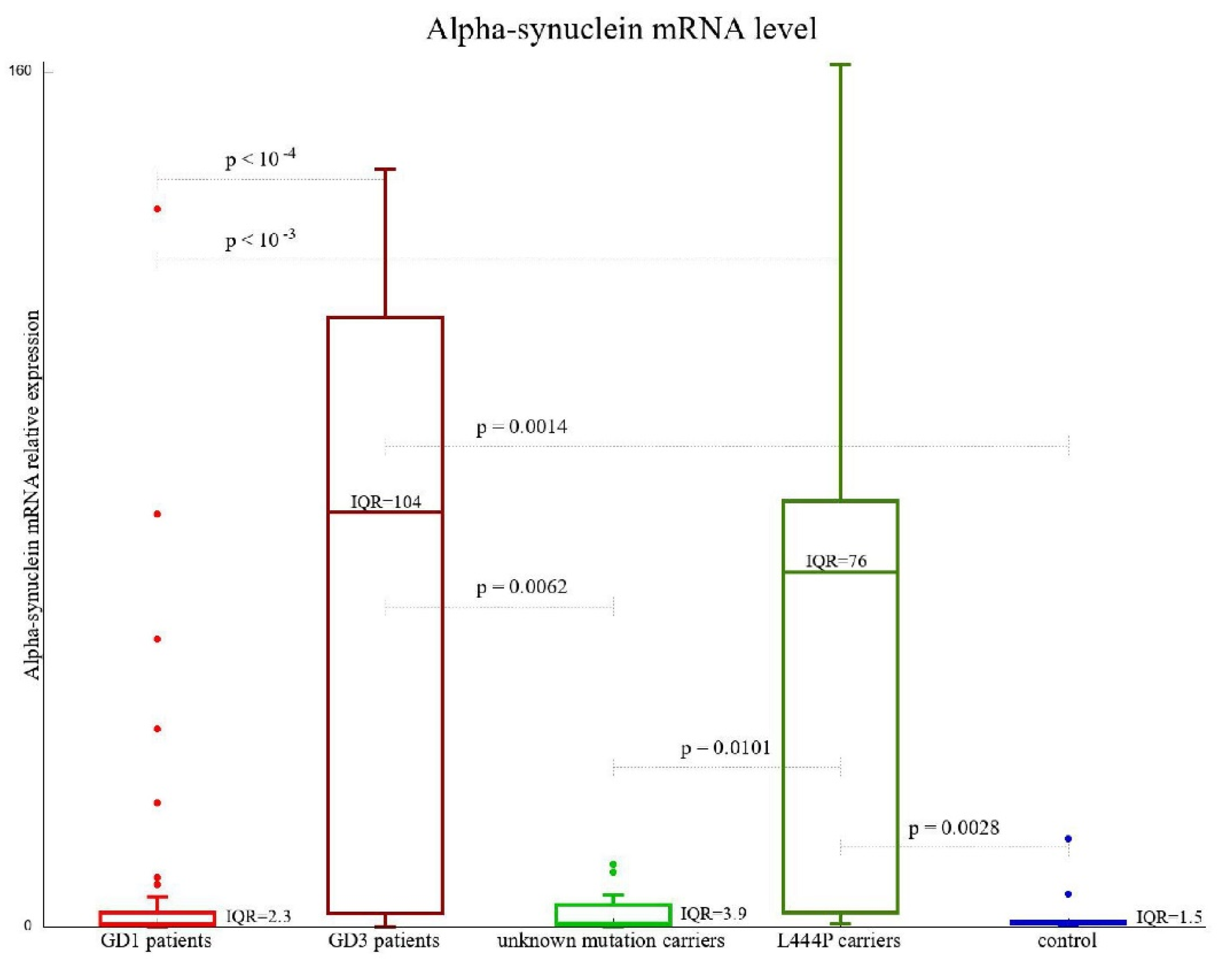
3.3. SNCA Gene Expression
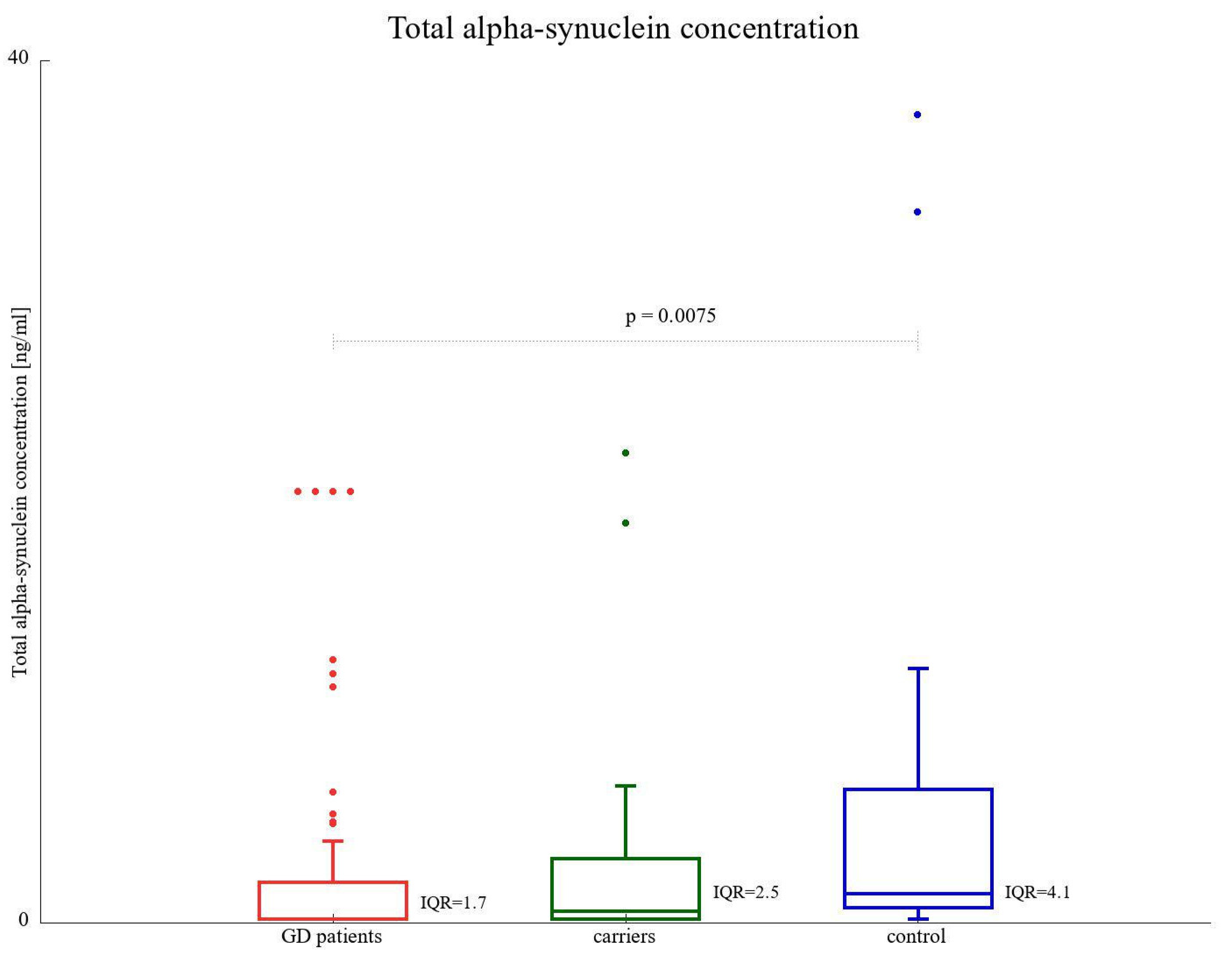
3.4. α-SNCA Total Protein Level
3.5. α-SNCA Oligomer Concentration
3.6. Correlations with Age
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and Classification of 304 Mutant Alleles in Patients with Type 1 and Type 3 Gaucher Disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, M.; Wilder, S.; Horowitz, Z.; Reiner, O.; Gelbart, T.; Beutler, E. The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 1989, 4, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Investig. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar]
- Dekker, N.; van Dussen, L.; Hollak, C.E.M.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.M.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine Is a Highly Sensitive and Specific Biomarker for Primary Diagnostic and Follow-Up Monitoring in Gaucher Disease in a Non-Jewish, Caucasian Cohort of Gaucher Disease Patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Brooks, D.J. The early diagnosis of Parkinson’s disease. Ann. Neurol. 1998, 44, S10–S18. [Google Scholar] [CrossRef]
- Bergman, H.; Deuschl, G. Pathophysiology of Parkinson’s disease: From clinical neurology to basic neuroscience and back. Mov. Disord. 2002, 17, S28–S40. [Google Scholar] [CrossRef] [PubMed]
- Wolters, E.C. Non-motor extranigral signs and symptoms in Parkinson’s disease. Park. Relat. Disord. 2009, 15, S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; Lees, A.; Stern, M. Milestones in Parkinson’s disease-Clinical and pathologic features. Mov. Disord. 2011, 26, 1015–1021. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathology of sporadic Parkinson’s disease: Evaluation and changes of concepts. Mov. Disord. 2012, 27, 8–30. [Google Scholar] [CrossRef]
- Buddhala, C.; Loftin, S.K.; Kuley, B.M.; Cairns, N.J.; Campbell, M.C.; Perlmutter, J.S.; Kotzbauer, P.T. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann. Clin. Transl. Neurol. 2015, 2, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.; Hardy, J. The Evolution of Genetics: Alzheimer’s and Parkinson’s Diseases. Neuron 2016, 90, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Ann. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R. α-Synuclein and neuronal cell death. Mol. Neurodegener. 2009, 4, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Coon, E.A.; Singer, W. Synucleinopathies. Continuum 2020, 26, 72–92. [Google Scholar] [CrossRef]
- Hallacli, E.; Kayatekin, C.; Nazeen, S.; Wang, X.H.; Sheinkopf, Z.; Sathyakumar, S.; Sarkar, S.; Jiang, X.; Dong, X.; Di Maio, R.; et al. The Parkinson’s disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability. Cell 2022, 185, 2035–2056.e33. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Par-kinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, U.; Singh, S.; Pal, S.; Agrawal, B.K.; Saini, R.V.; Chakrabarti, S. Alpha-synuclein as a Biomarker of Parkinson’s Disease: Good, but Not Good Enough. Front. Aging Neurosci. 2021, 13, 702639. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Pfefferkorn, C.M.; Jiang, Z.; Lee, J.C. Biophysics of α-synuclein membrane interactions. Biochim. Biophys. Acta 2012, 1818, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Amyloidogenesis of Natively Unfolded Proteins. Curr. Alzheimer Res. 2008, 5, 260–287. [Google Scholar] [CrossRef]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 2012, 28, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Tylki-Szymańska, A.; Vellodi, A.; El-Beshlawy, A.; Cole, J.A.; Kolodny, E. Neuronopathic Gaucher disease: Demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J. Inherit. Metab. Dis. 2010, 33, 339–346. [Google Scholar] [CrossRef]
- Halperin, A.; Elstein, D.; Zimran, A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol. Dis. 2006, 36, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.; Filocamo, M.; Horowitz, M. ITCH regulates degradation of mutant glucocerebrosidase: Implications to Gaucher disease. Hum. Mol. Genet. 2013, 22, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.-H.; Xu, K.; Sun, Y.; Liou, B.; Quinn, B.; Li, R.-H.; Xue, L.; Zhang, W.; Setchell, K.D.; Witte, D.; et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 2014, 23, 3943–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. J. Clin. Med. 2019, 8, 1662. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Stojkovska, I.; Krainc, D.; Mazzulli, J.R. Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res. 2018, 373, 51–60. [Google Scholar] [CrossRef]
- Argyriou, A.; Dermentzaki, G.; Papasilekas, T.; Moraitou, M.; Stamboulis, E.; Vekrellis, K.; Michelakakis, H.; Stefanis, L. Increased dimerization of alpha-synuclein in erythrocytes in Gaucher disease and aging. Neurosci. Lett. 2012, 528, 205–209. [Google Scholar] [CrossRef]
- Pchelina, S.; Nuzhnyi, E.; Emelyanov, A.; Boukina, T.; Usenko, T.; Nikolaev, M.; Salogub, G.; Yakimovskii, A.; Zakharova, E.Y. Increased plasma oligomeric alpha-synuclein in patients with lysosomal storage diseases. Neurosci. Lett. 2014, 583, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerche, S.; Wurster, I.; Roeben, B.; Zimmermann, M.; Riebenbauer, B.; Deuschle, C.; Hauser, A.K.; Schulte, C.; Berg, D.; Maetzler, W.; et al. Parkinson’s Disease: Glucocerebrosidase 1 Mutation Severity Is Associated with CSF Alpha-synuclein Profiles. Mov. Disord. 2020, 35, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Chiba-Falek, O.; Lopez, G.J.; Nussbaum, R.L. Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov. Disord. 2006, 21, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Bras, J.; Paisan-Ruiz, C.; Guerreiro, R.; Ribeiro, M.H.; Morgadinho, A.; Januario, C.; Sidransky, E.; Oliveira, C.; Singleton, A. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol. Aging 2009, 30, 1515–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinderi, K.; Bostantjopoulou, S.; Paisan-Ruiz, C.; Katsarou, Z.; Hardy, J.; Fidani, L. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci. Lett. 2009, 452, 87–89. [Google Scholar] [CrossRef]
- Mao, X.-Y.; Burgunder, J.-M.; Zhang, Z.-J.; An, X.-K.; Zhang, J.-H.; Yang, Y.; Li, T.; Wang, Y.-C.; Chang, X.-L.; Peng, R. Association between GBA L444P mutation and sporadic Parkinson’s disease from Mainland China. Neurosci. Lett. 2010, 469, 256–259. [Google Scholar] [CrossRef]
- Mitsui, J.; Mizuta, I.; Toyoda, A.; Ashida, R.; Takahashi, Y.; Goto, J.; Fukuda, Y.; Date, H.; Iwata, A.; Yamamoto, M.; et al. Mutations for Gaucher Disease Confer High Susceptibility to Parkinson Disease. Arch. Neurol. 2009, 66, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Morgan, A.; Lang, A.E.; Salehi-Rad, S.; Kawarai, T.; Meng, Y.; Ray, P.N.; Farrer, L.A.; George-Hyslop, P.S.; Rogaeva, E. Analysis of the glucocerebrosidase gene in Parkinson’s disease. Mov. Disord. 2005, 20, 367–370. [Google Scholar] [CrossRef]
- Sun, Q.-Y.; Guo, J.-F.; Wang, L.; Yu, R.-H.; Zuo, X.; Yao, L.-Y.; Pan, Q.; Xia, K.; Tang, B.-S. Glucocerebrosidase Gene L444P mutation is a risk factor for Parkinson’s disease in Chinese population. Mov. Disord. 2010, 25, 1005–1011. [Google Scholar] [CrossRef]
- Tan, E.-K.; Tong, J.; Fook-Chong, S.; Yih, Y.; Wong, M.-C.; Pavanni, R.; Zhao, Y. Glucocerebrosidase Mutations and Risk of Parkinson Disease in Chinese Patients. Arch. Neurol. 2007, 64, 1056–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-R.; Chen, C.-M.; Chao, C.-Y.; Ro, L.-S.; Lyu, R.-K.; Chang, K.-H.; Lee-Chen, G.-J. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J. Neurol. Neurosurg. Psychiatry 2007, 78, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, S.G.; Eblan, M.J.; Gutti, U.; Hruska, K.S.; Stubblefield, B.K.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol. Genet. Metab. 2007, 91, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Spitz, M.; Rozenberg, R.; Pereira Lda, V.; Reis Barbosa, E. Association between Parkinson’s disease and glucocerebrosidase mutations in Brazil. Parkinsonism Relat. Disord. 2008, 14, 58–62. [Google Scholar] [CrossRef]
- De Marco, E.V.; Annesi, G.; Tarantino, P.; Rocca, F.E.; Provenzano, G.; Civitelli, D.; Cirò Candiano, I.C.; Annesi, F.; Carrideo, S.; Condino, F.; et al. Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov. Disord. 2008, 23, 460–463. [Google Scholar] [CrossRef]
- Becker-Cohen, M.; Zimran, A.; Dinur, T.; Tiomkin, M.; Cozma, C.; Rolfs, A.; Arkadir, D.; Shulman, E.; Manor, O.; Paltiel, O.; et al. A Comprehensive Assessment of Qualitative and Quantitative Prodromal Par-kinsonian Features in Carriers of Gaucher Disease-Identifying Those at the Greatest Risk. Int. J. Mol. Sci. 2022, 23, 12211. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Xiong, J.; Zhang, X.; Chen, Z.; Yu, L.; Chen, C.; Huang, J.; Zhang, Z.; Mohmed, A.A.; et al. Glucocerebrosidase L444P mutation confers genetic risk for Parkinson’s disease in central China. Behav. Brain Funct. 2012, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red Blood Cells Are the Major Source of Alpha-Synuclein in Blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef]
- Zanzoni, A.; Marchese, D.; Agostini, F.; Bolognesi, B.; Cirillo, D.; Botta-Orfila, M.; Livi, C.M.; Rodriguez-Mulero, S.; Tartaglia, G.G. Principles of self-organization in biological pathways: A hypothesis on the autogenous association of alpha-synuclein. Nucleic Acids Res. 2013, 41, 9987–9998. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, S.; Kam, T.-I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients | Common Variant Name | DNA Nucleotide Change | Protein Change |
|---|---|---|---|
| 28 | N370S/L444P | c.1226A>G c.1448T>C | p.Asn409Ser p.Leu483Pro |
| 6 | N370S/ others | c.1226A>G | p.Asn409Ser |
| 2 | N370S/N370S | c.1226A>G | p.Asn409Ser |
| 2 | G377S/G377S | c.1246G>A | p.Gly416Ser |
| 1 | N370S/ G377S | c.1226A>G c.1246G>A | p.Asn409Ser p.Gly416Ser |
| 1 | N370S/84GG | c.1226A>G c.84dupG | p.Asn409Ser p.Leu29AlafsTer18 |
| 1 | N370S/R496H | c.1226A>G c.1604G>A | p.Asn409Ser p.Arg535His |
| 1 | R433S/R433S | c.1416A>T | p.Arg444Ser |
| 1 | D438N/R87W | c.1312G>A c.259C>T | p.Asp438Asn p.Arg87Trp |
| 1 | D448G/R202X | c.1343A>G c.604C>T | p.Asp448Gly p.Arg202X |
| 1 | R48W/84GG | c.259C>T c.84dupG | p.Arg87Trp p.Leu29AlafsTer18 |
| 1 | R48W/R48W | c.259C>T | p.Arg87Trp |
| 1 | 84GG/ Other | c.84dupG | p.Leu29AlafsTer18 |
| Healthy Controls | GBA1 Mutation Carriers | GD1 | GD3 | |
|---|---|---|---|---|
| n | 16 | 19 | 47 | 18 |
| Sex, F/M | 10/6 | 14/5 | 28/19 | 12/8 |
| Age in years, median (range) | 44.5 | 52 (13–64) | 40 (9–71) | 30 (22–62) |
| Lyso-Gb1 (ng/mL), median (range) | 8.49 (4.82–46.73) | 7.51 (4.26–46.73) | 71.5 (22.4–950.9) | 64.55 (9.14–693.8) |
| SNCA mRNA level (2−ΔcT), median (range) | 0.83 (0.08–16.38) | 4.16 (0.02–161.42) | 0.47 (0.01–134.42) | 77.48 (0.04–141.72) |
| Total α-SNCA concentration (ng/mL), median (range) | 1.34 (0.16–37.48) | 0.53 (0.16–21.76) | 0.16 (0.16–20) | 0.92 (0.16–20) |
| Oligomer α-SNCA concentration (ng/mL), median (range) | 2.08 (0.96–4.73) | 1.69 (1.03–3.33) | 1.76 (0.76–4.01) | 1.857(0.76–3.03) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubiela, P.; Szymańska-Rożek, P.; Eljaszewicz, A.; Lipiński, P.; Hasiński, P.; Giersz, D.; Walewska, A.; Tynecka, M.; Moniuszko, M.; Tylki-Szymańska, A. Alpha-Synuclein mRNA Level Found Dependent on L444P Variant in Carriers and Gaucher Disease Patients on Enzyme Replacement Therapy. Biomolecules 2023, 13, 644. https://doi.org/10.3390/biom13040644
Dubiela P, Szymańska-Rożek P, Eljaszewicz A, Lipiński P, Hasiński P, Giersz D, Walewska A, Tynecka M, Moniuszko M, Tylki-Szymańska A. Alpha-Synuclein mRNA Level Found Dependent on L444P Variant in Carriers and Gaucher Disease Patients on Enzyme Replacement Therapy. Biomolecules. 2023; 13(4):644. https://doi.org/10.3390/biom13040644
Chicago/Turabian StyleDubiela, Paweł, Paulina Szymańska-Rożek, Andrzej Eljaszewicz, Patryk Lipiński, Piotr Hasiński, Dorota Giersz, Alicja Walewska, Marlena Tynecka, Marcin Moniuszko, and Anna Tylki-Szymańska. 2023. "Alpha-Synuclein mRNA Level Found Dependent on L444P Variant in Carriers and Gaucher Disease Patients on Enzyme Replacement Therapy" Biomolecules 13, no. 4: 644. https://doi.org/10.3390/biom13040644