
scTransSort: Transformers for Intelligent Annotation of Cell Types by Gene Embeddings

Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
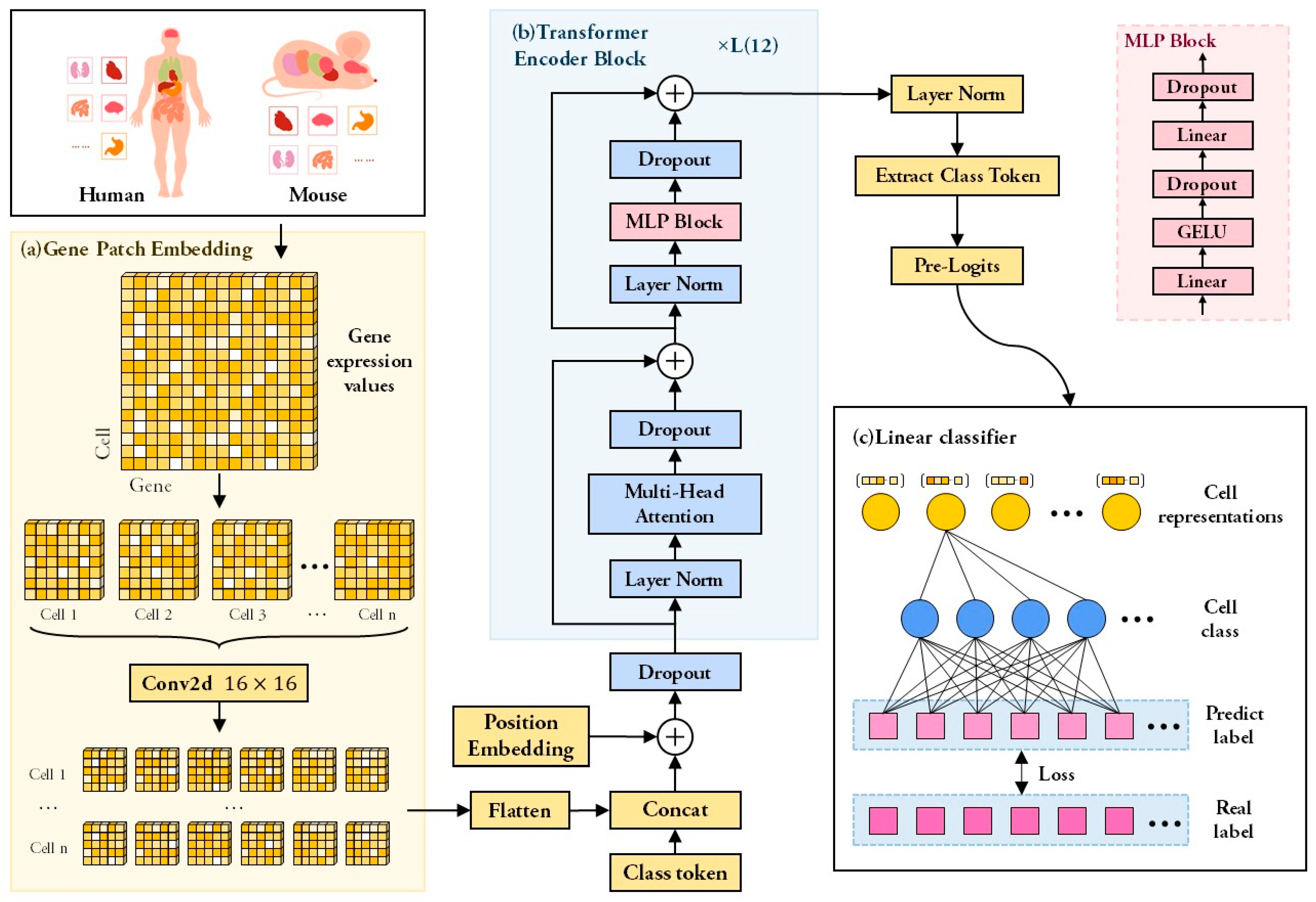
2.2. Framework of scTransSort
2.3. Loss Function and Parameters Setting
3. Results
3.1. Evaluation Metrics
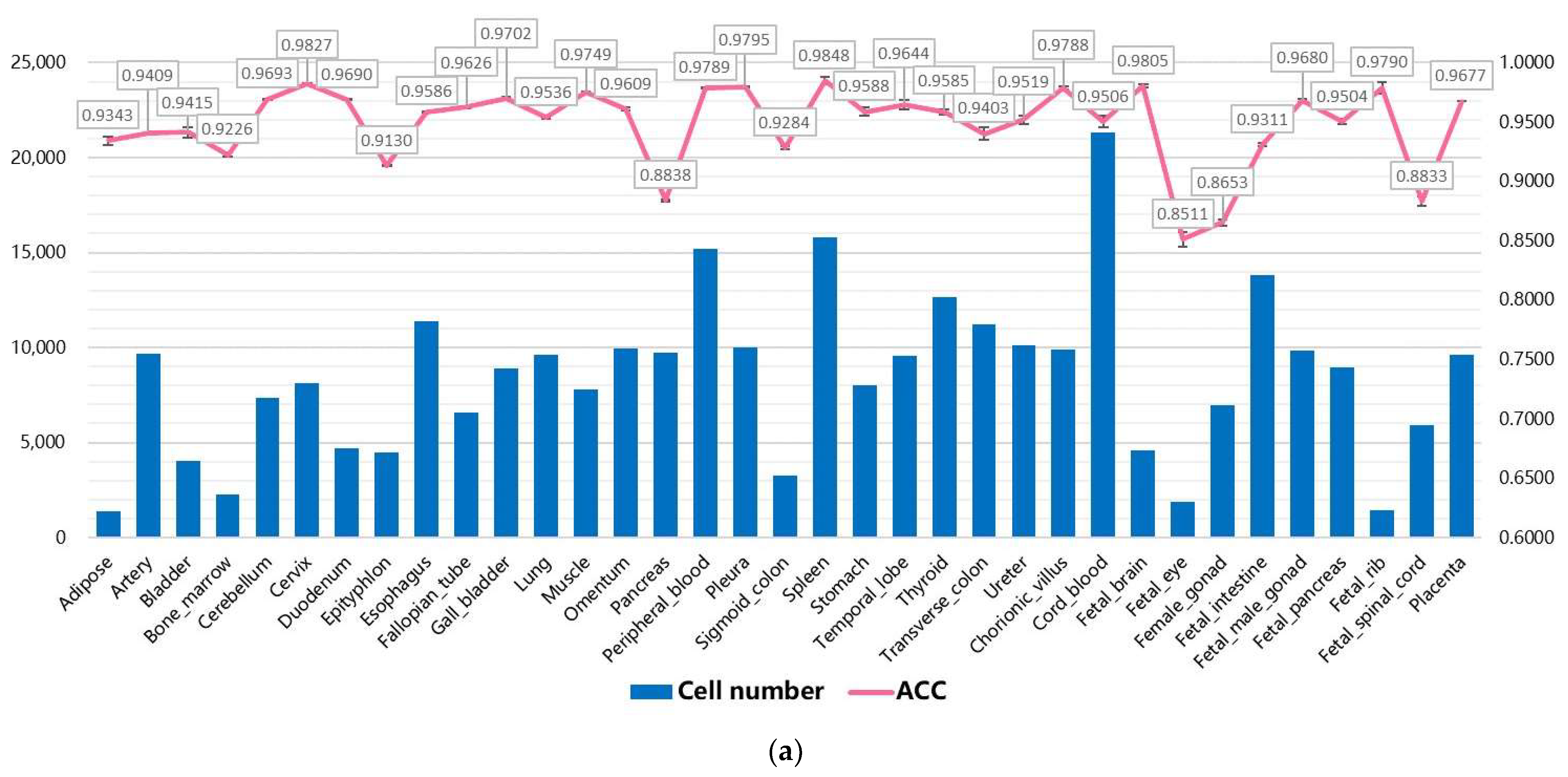
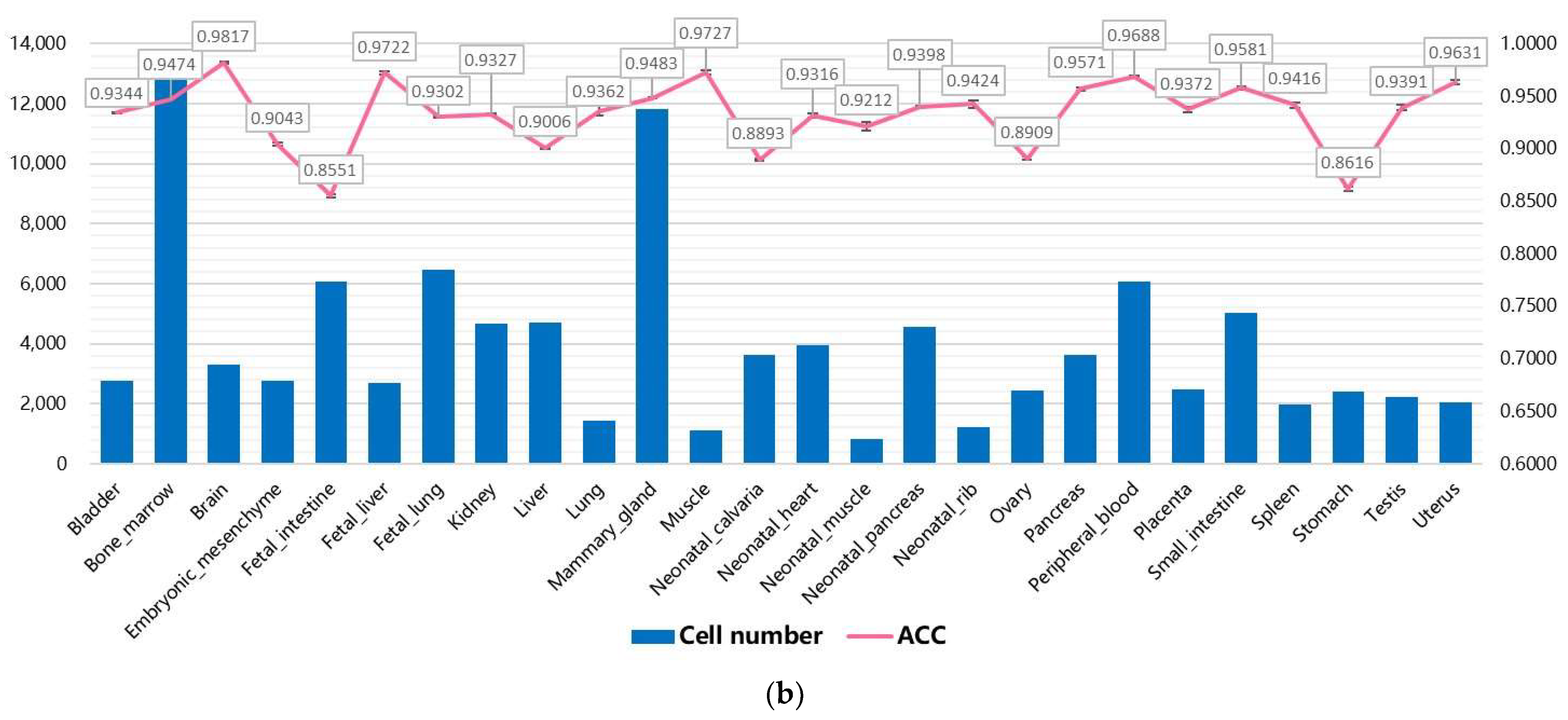
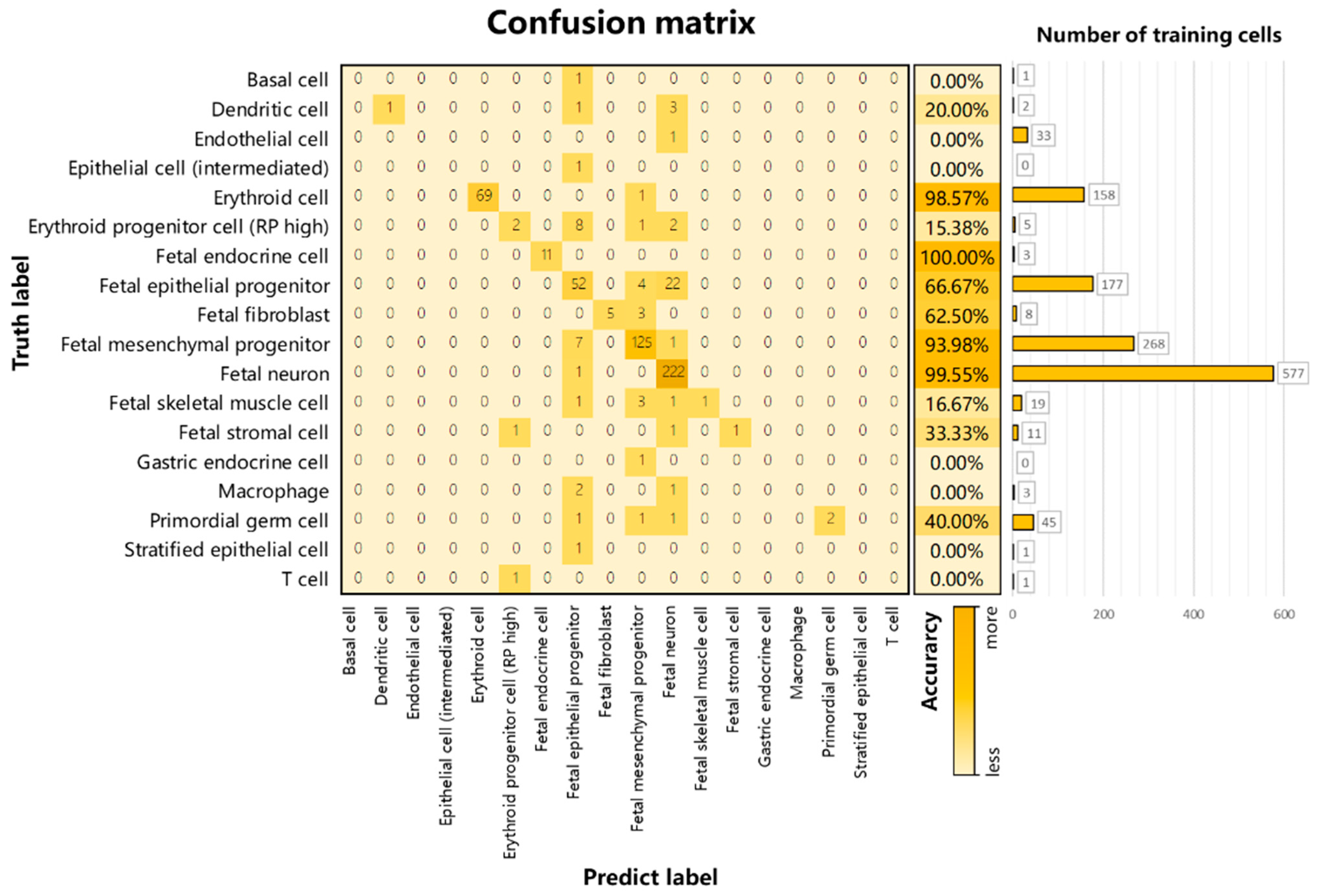
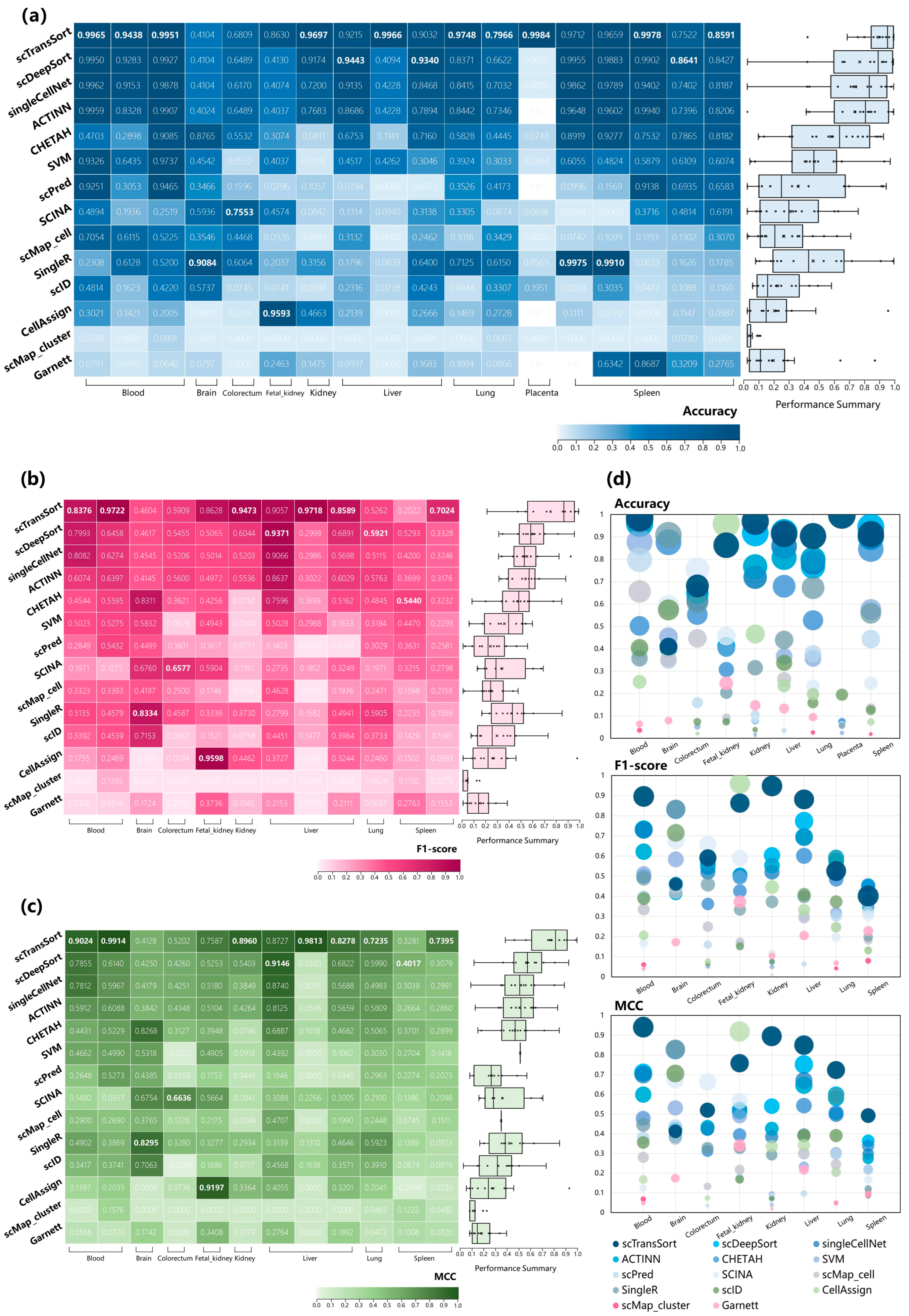
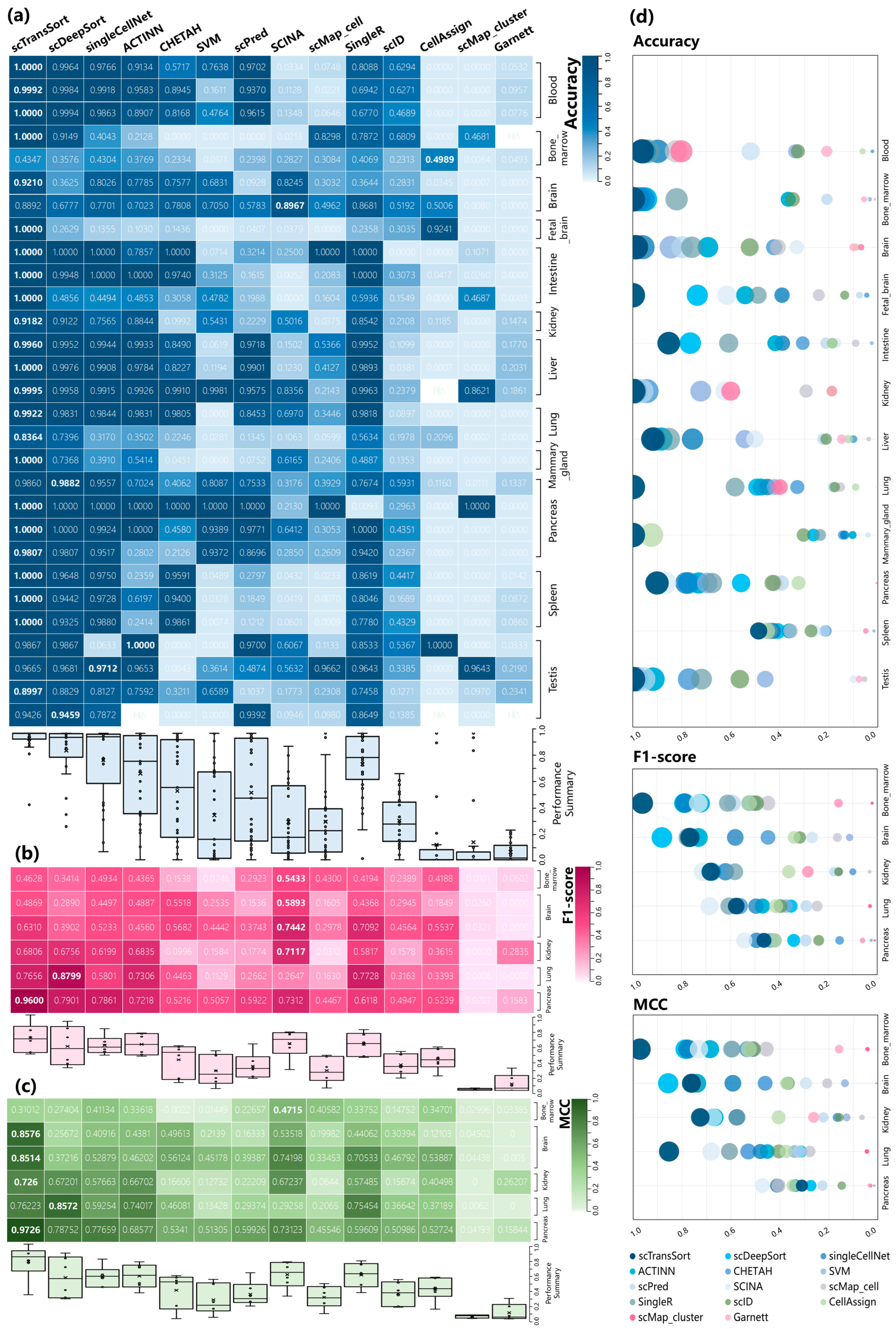
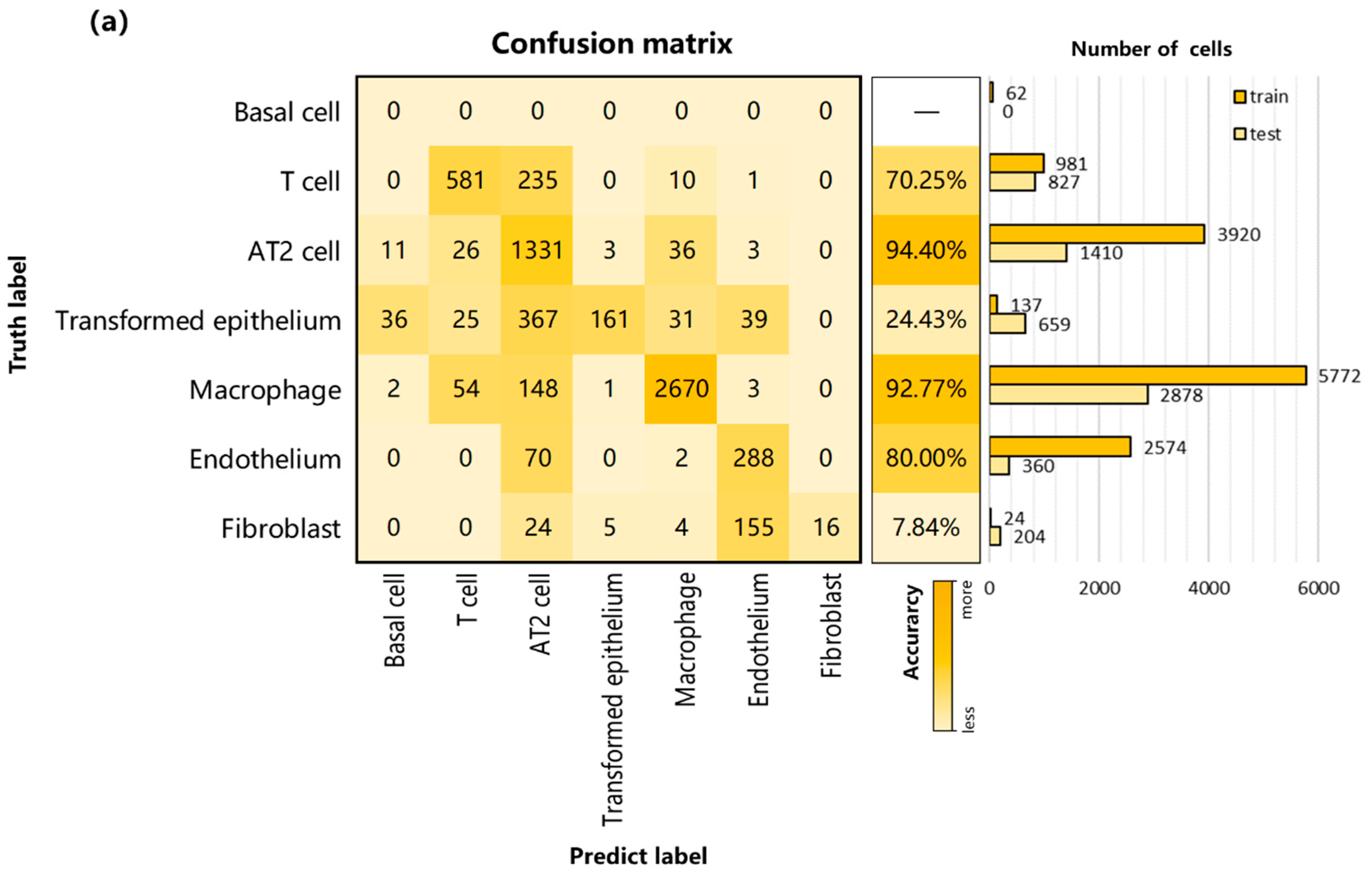
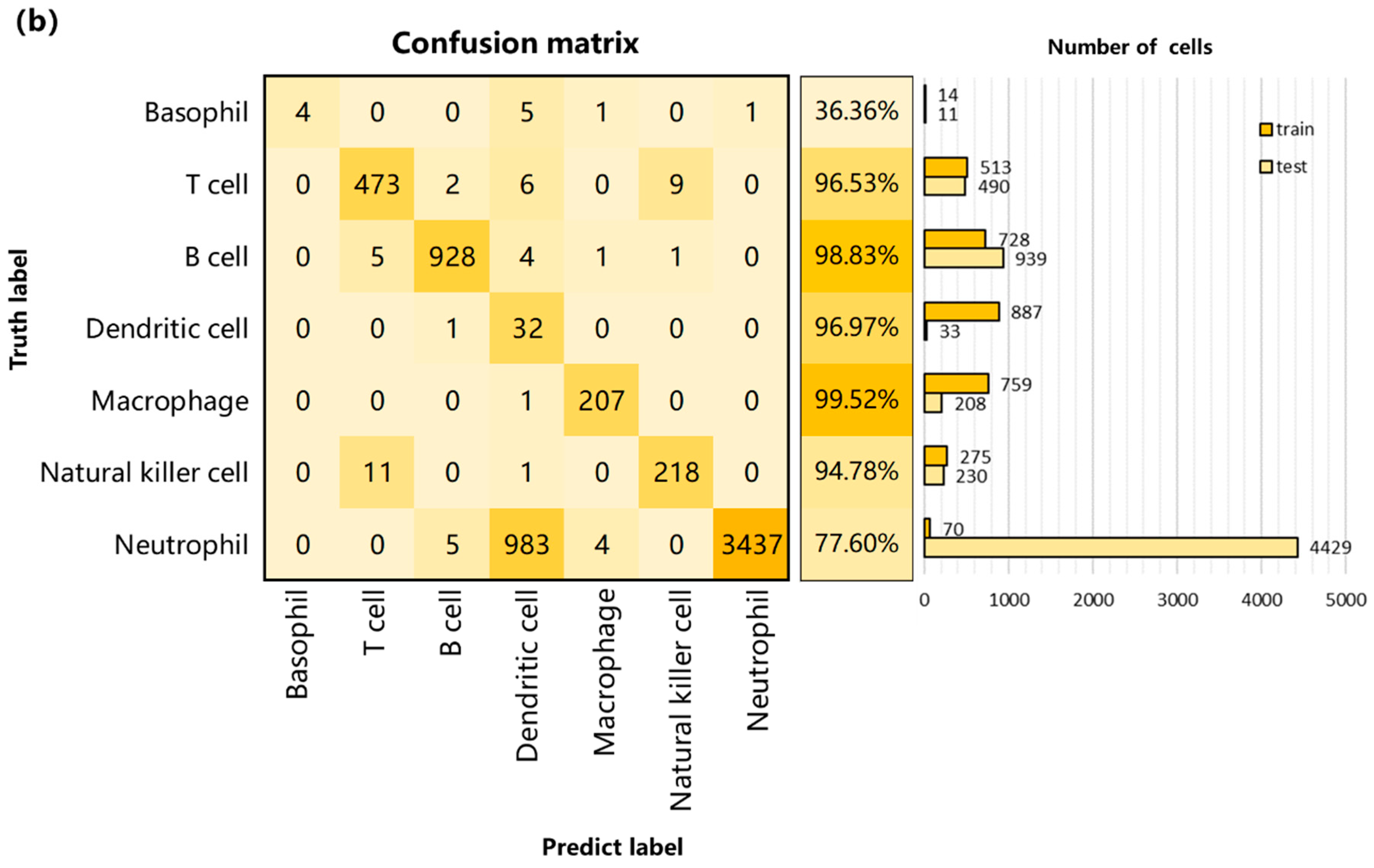
3.2. Performance on Internal Datasets
3.3. Performance and Robustness Compared with Other Methods
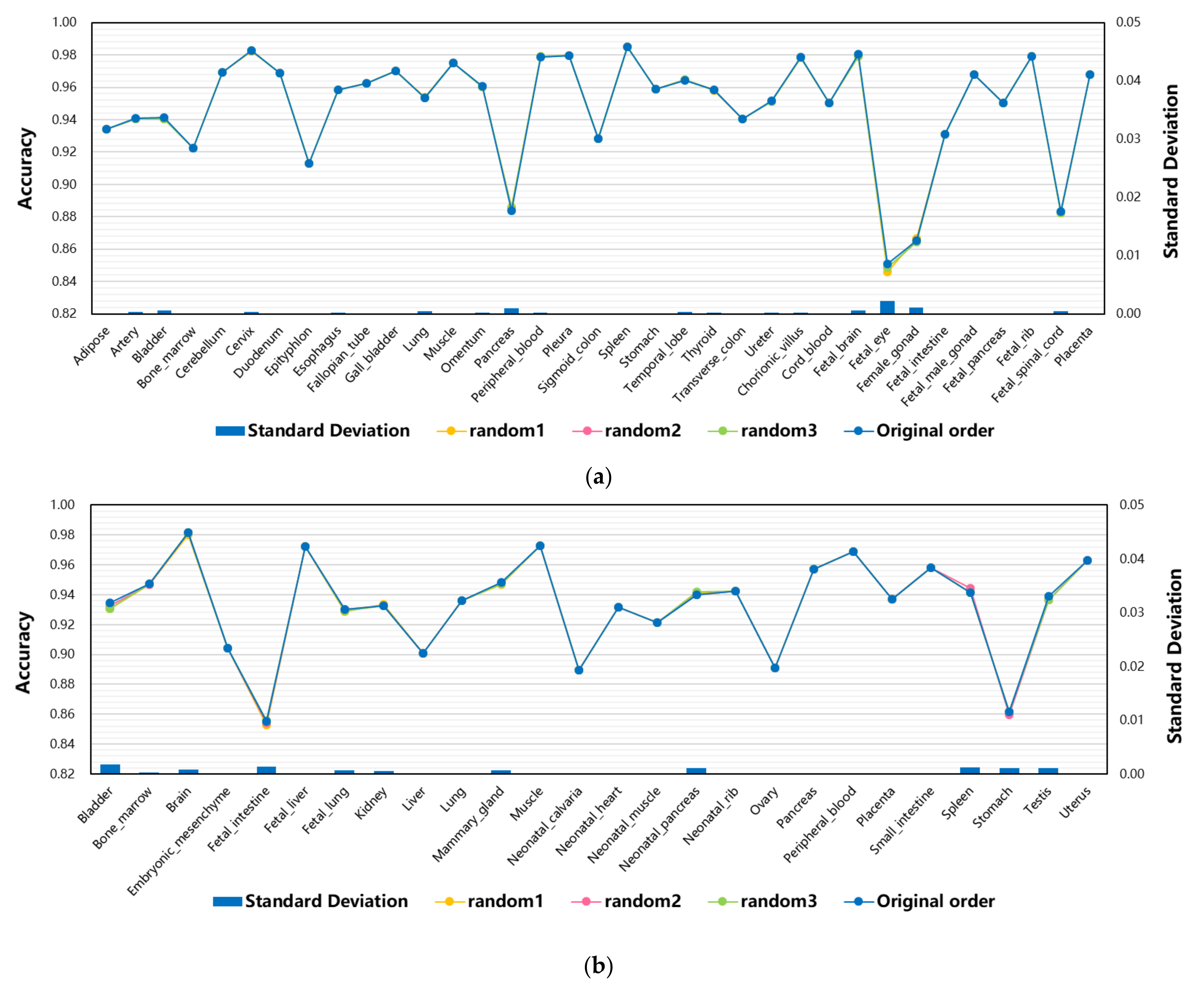
3.4. The Effect of Feature Order of Input Data on Model Performance
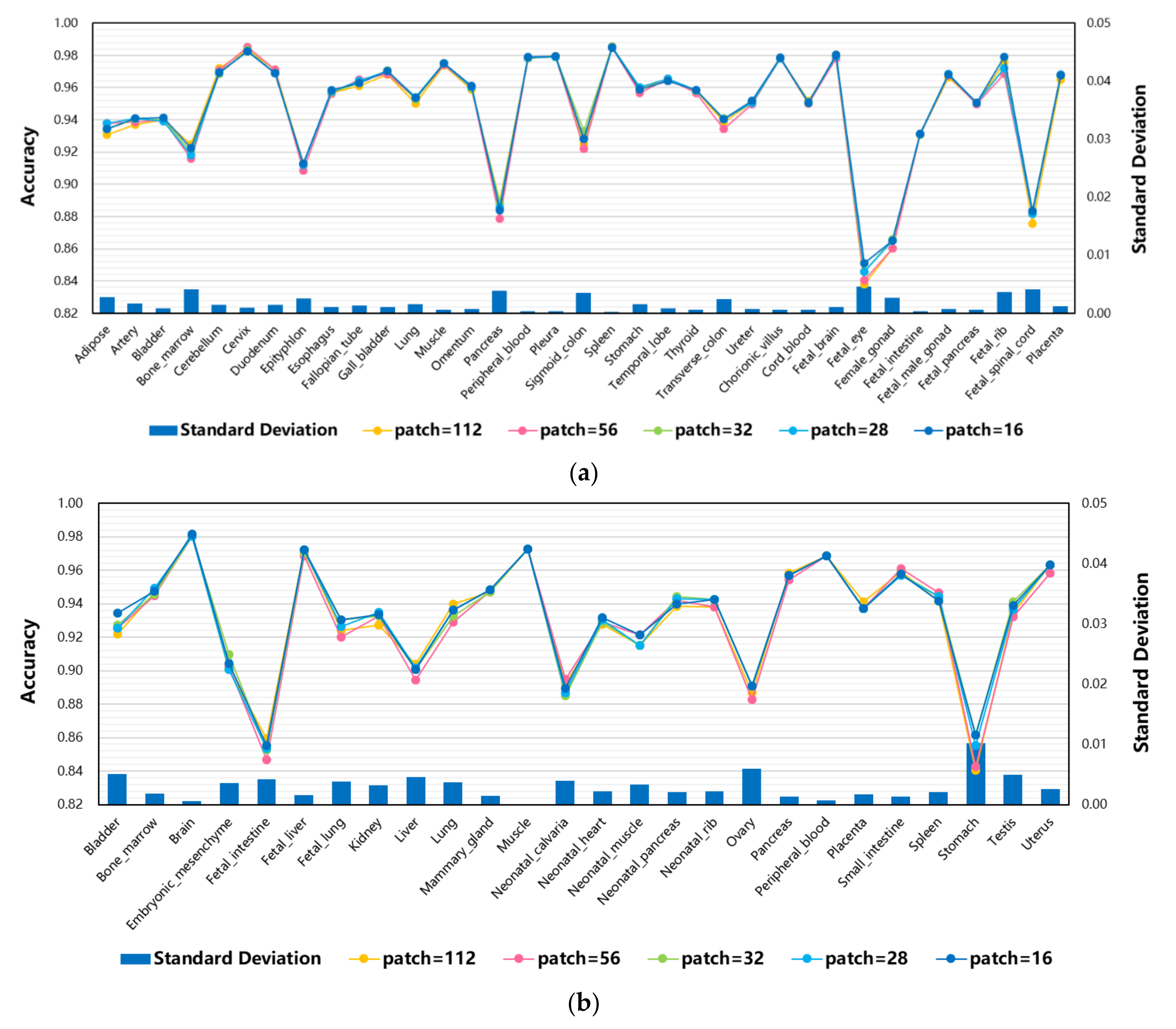
3.5. The Effect of Different Patches on Model Performance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shao, X.; Lu, X.; Liao, J.; Chen, H.; Fan, X. New avenues for systematically inferring cell-cell communication: Through single-cell transcriptomics data. Protein Cell 2020, 11, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Healey, H.M.; Bassham, S.; Cresko, W.A. Single-cell Iso-Sequencing enables rapid genome annotation for scRNAseq analysis. Genetics 2022, 220, iyac017. [Google Scholar] [CrossRef] [PubMed]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Asp. Med. 2018, 59, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, G.; Arias, J.E.R.; Schäfer, P.; Busskamp, V. Automated methods for cell type annotation on scRNA-seq data. Comput. Struct. Biotechnol. J. 2021, 19, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.; Tian, X.; Xu, J. Single-cell transcriptome analysis in plants: Advances and challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Wang, B.; Zhu, J.; Pierson, E.; Ramazzotti, D.; Batzoglou, S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 2017, 14, 414–416. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef] [Green Version]
- Plass, M.; Solana, J.; Wolf, F.A.; Ayoub, S.; Misios, A.; Glažar, P.; Obermayer, B.; Theis, F.J.; Kocks, C.; Rajewsky, N. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 2018, 360, eaaq1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fincher, C.T.; Wurtzel, O.; de Hoog, T.; Kravarik, K.M.; Reddien, P.W. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science 2018, 360, eaaq1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the mouse cell atlas by microwell-seq. Cell 2018, 172, 1091–1107.e17. [Google Scholar] [CrossRef] [Green Version]
- Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ozturk, K.; Contreras, R.L.; Simon, J.; McCann, S.; Chen, W.J.; Carter, H.; Fraley, S.I. Phenotypically supervised single-cell sequencing parses within-cell-type heterogeneity. iScience 2021, 24, 101991. [Google Scholar] [CrossRef]
- McKellar, D.W.; Walter, L.D.; Song, L.T.; Mantri, M.; Wang, M.F.; De Vlaminck, I.; Cosgrove, B.D. Large-scale integration of single-cell transcriptomic data captures transitional progenitor states in mouse skeletal muscle regeneration. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, T.; Michielsen, L.; Cats, D.; Hoogduin, D.; Mei, H.; Reinders, M.J.T.; Mahfouz, A. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Aevermann, B.D.; Bakken, T.E.; Miller, J.A.; Hodge, R.D.; Lein, E.S.; Scheuermann, R.H. FR-Match: Robust matching of cell type clusters from single cell RNA sequencing data using the Friedman–Rafsky non-parametric test. Brief. Bioinform. 2021, 22, bbaa339. [Google Scholar] [CrossRef]
- Heydari, A.A.; Davalos, O.A.; Zhao, L.; Hoyer, K.K.; Sindi, S.S. ACTIVA: Realistic single-cell RNA-seq generation with automatic cell-type identification using introspective variational autoencoders. Bioinformatics 2022, 38, 2194–2201. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, P. Evaluation of machine learning approaches for cell-type identification from single-cell transcriptomics data. Brief. Bioinform. 2021, 22, bbab035. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chowdhury, S.; Victor, U.; Li, X.; Qian, L. Semi-supervised Deep Learning for Cell Type Identification from Single-Cell Transcriptomic Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- De Kanter, J.K.; Lijnzaad, P.; Candelli, T.; Margaritis, T.; Holstege, F.C. CHETAH: A selective, hierarchical cell type identification method for single-cell RNA sequencing. Nucleic Acids Res. 2019, 47, e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. scmap: Projection of single-cell RNA-seq data across data sets. Nat. Methods 2018, 15, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Boufea, K.; Seth, S.; Batada, N.N. scID uses discriminant analysis to identify transcriptionally equivalent cell types across single-cell RNA-seq data with batch effect. iScience 2020, 23, 100914. [Google Scholar] [CrossRef]
- Alquicira-Hernandez, J.; Sathe, A.; Ji, H.P.; Nguyen, Q.; Powell, J.E. scPred: Accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019, 20, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Pellegrini, M. ACTINN: Automated identification of cell types in single cell RNA sequencing. Bioinformatics 2020, 36, 533–538. [Google Scholar] [CrossRef]
- Zhang, A.W.; O’Flanagan, C.; Chavez, E.A.; Lim, J.L.; Ceglia, N.; McPherson, A. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 2019, 16, 1007–1015. [Google Scholar] [CrossRef]
- Pliner, H.A.; Shendure, J.; Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods 2019, 16, 983–986. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, D.; Zhong, X.; Choi, J.H.; Ma, Y.; Wang, S.; Mahrt, E.; Guo, W.; Stawiski, E.W.; Modrusan, Z.; et al. SCINA: A semi-supervised subtyping algorithm of single cells and bulk samples. Genes 2019, 10, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Cahan, P. SingleCellNet: A computational tool to classify single cell RNA-Seq data across platforms and across species. Cell Syst. 2019, 9, 207–213.e2. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Yang, H.; Zhuang, X.; Liao, J.; Yang, P.; Cheng, J.; Lu, X.; Chen, H.; Fan, X. scDeepSort: A pre-trained cell-type annotation method for single-cell transcriptomics using deep learning with a weighted graph neural network. Nucleic Acids Res. 2021, 49, e122. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, M.; Pang, S.; Song, L.; Qiao, S. Intelligent fault diagnosis and visual interpretability of rotating machinery based on residual neural network. Measurement 2022, 196, 111228. [Google Scholar] [CrossRef]
- Yu, S.; Wang, M.; Pang, S.; Song, L.; Zhai, X.; Zhao, Y. TDMSAE: A transferable decoupling multi-scale autoencoder for mechanical fault diagnosis. Mech. Syst. Signal Process. 2023, 185, 109789. [Google Scholar] [CrossRef]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901. [Google Scholar]
- Lepikhin, D.; Lee, H.; Xu, Y.; Chen, D.; Firat, O.; Huang, Y. Gshard: Scaling giant models with conditional computation and automatic sharding. arXiv 2020, arXiv:2006.16668. [Google Scholar]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T. An image is worth 16×16 words: Transformers for image recognition at scale. arXiv 2020, arXiv:2010.11929. [Google Scholar]
- Khan, S.; Naseer, M.; Hayat, M.; Zamir, S.W.; Khan, F.S.; Shah, M. Transformers in vision: A survey. ACM Comput. Surv. 2022, 54, 1–41. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, A.; Gomez, A.; Kaiser, Ł.; Polosukhin, L. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar]
- Wang, Q.; Li, B.; Xiao, T.; Zhu, J.; Li, C.; Wong, D.F.; Chao, L.S. Learning deep transformer models for machine translation. arXiv 2019, arXiv:1906.01787. [Google Scholar]
- Baevski, A.; Auli, M. Adaptive input representations for neural language modeling. arXiv 2018, arXiv:1809.10853. [Google Scholar]
- Rosenblatt, F. The Perceptron, a Perceiving and Recognizing Automaton Project Para. Master’s Thesis, Cornell Aeronautical Laboratory, Buffalo, NY, USA, 1957. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Range |
|---|---|
| patch_size | 16 |
| batch_size | 64 |
| epoch | 50 |
| initial_lr | 1 × 10−3 |
| end_lr | 1 × 10−5 |
| weight_decay | 1 × 10−4 |
| Optimizer | SGD |
| Activation | GeLU |
| Actual Positive | Actual Negative | |
|---|---|---|
| Predicted Positive | TP | FP |
| Predicted Negative | FN | TN |
| Precision | TP/(TP+FP) | |
| Recall | TP/(TP+FN) | |
| Accuracy (ACC) | (TP+TN)/(TP+FP+FN+TN) | |
| Matthews correlation coefficient (MCC) | MCC = | |
| F1-score | F1-score = 2 | |
| Standard Deviation | s = where n is the number of data, the i-th data, and ) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, L.; Wang, G.; Dai, H.; Li, X.; Wang, S.; Song, T. scTransSort: Transformers for Intelligent Annotation of Cell Types by Gene Embeddings. Biomolecules 2023, 13, 611. https://doi.org/10.3390/biom13040611
Jiao L, Wang G, Dai H, Li X, Wang S, Song T. scTransSort: Transformers for Intelligent Annotation of Cell Types by Gene Embeddings. Biomolecules. 2023; 13(4):611. https://doi.org/10.3390/biom13040611
Chicago/Turabian StyleJiao, Linfang, Gan Wang, Huanhuan Dai, Xue Li, Shuang Wang, and Tao Song. 2023. "scTransSort: Transformers for Intelligent Annotation of Cell Types by Gene Embeddings" Biomolecules 13, no. 4: 611. https://doi.org/10.3390/biom13040611