
Blood–Brain Barrier Integrity Is Perturbed in a Mecp2-Null Mouse Model of Rett Syndrome

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Evaluation of BBB Integrity and Analysis of FITC-Albumin/Laminin by Immunohistochemistry
2.3. Immunoblottings
2.4. RNA Extraction and Reverse-Transcription qPCR
2.5. Statistics
3. Results
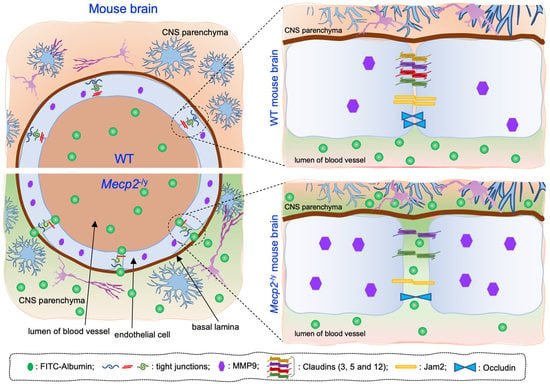
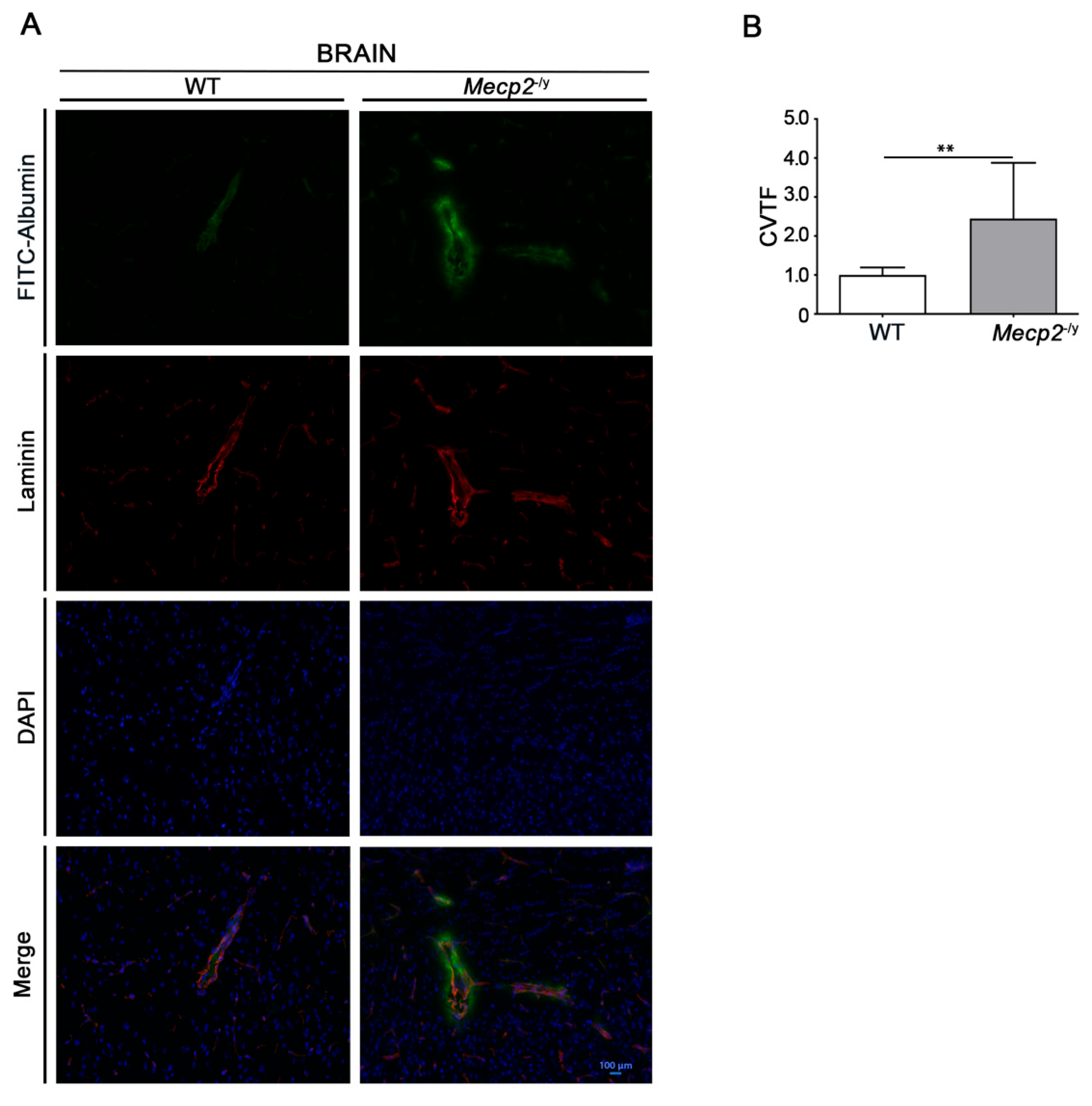
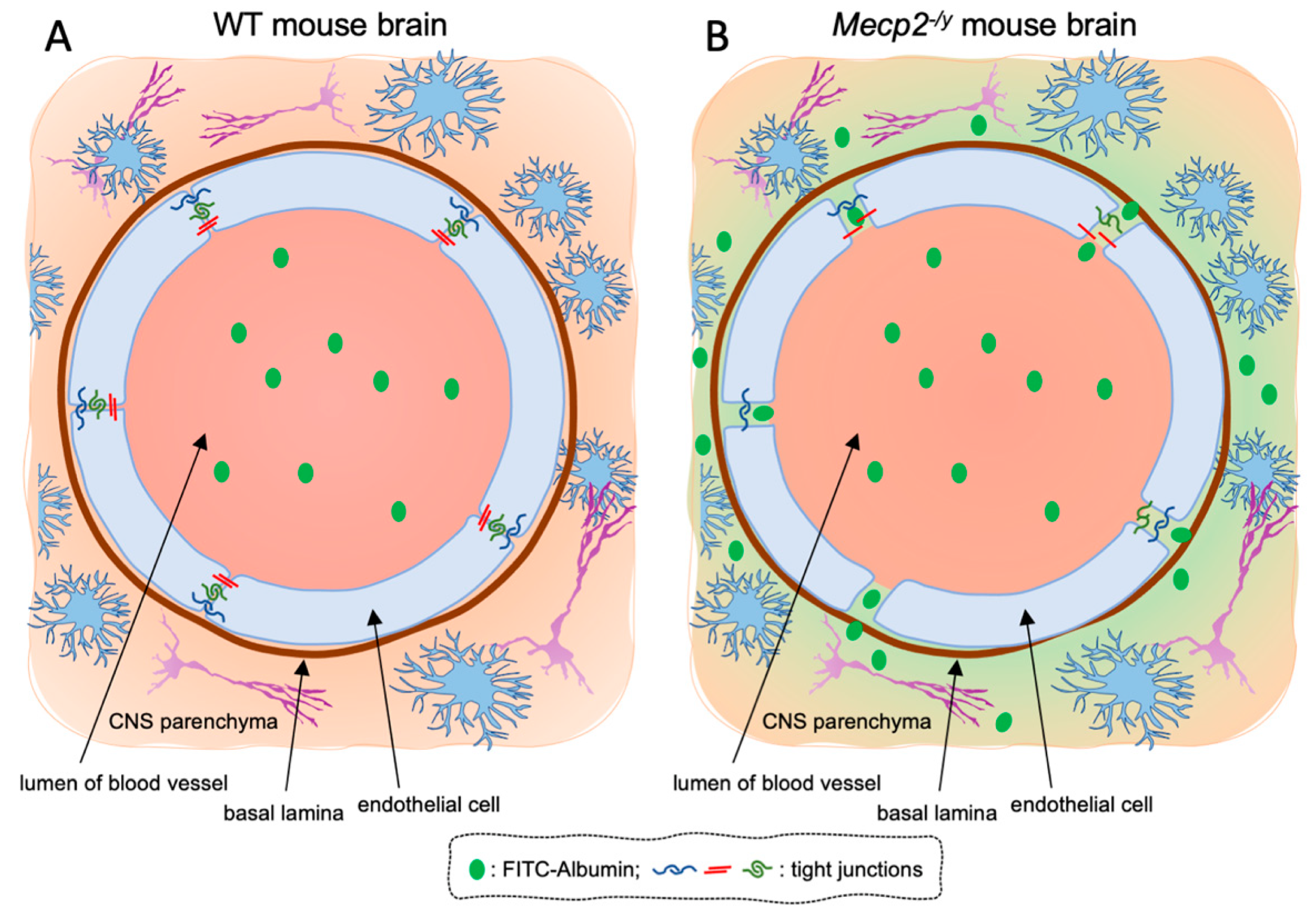
3.1. Blood–Brain Barrier Permeability Is Altered in Symptomatic RTT Mecp2-/y Mice
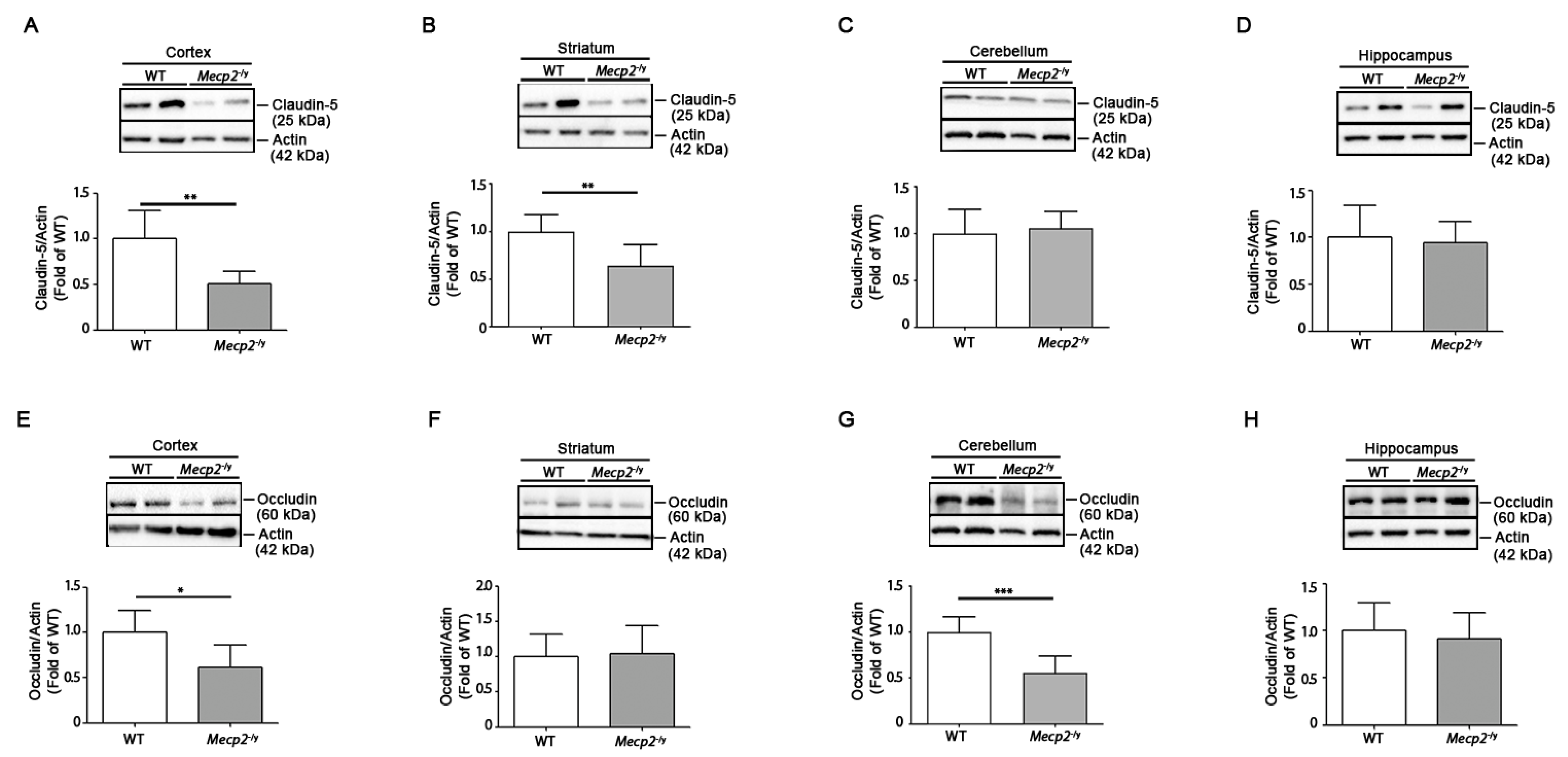
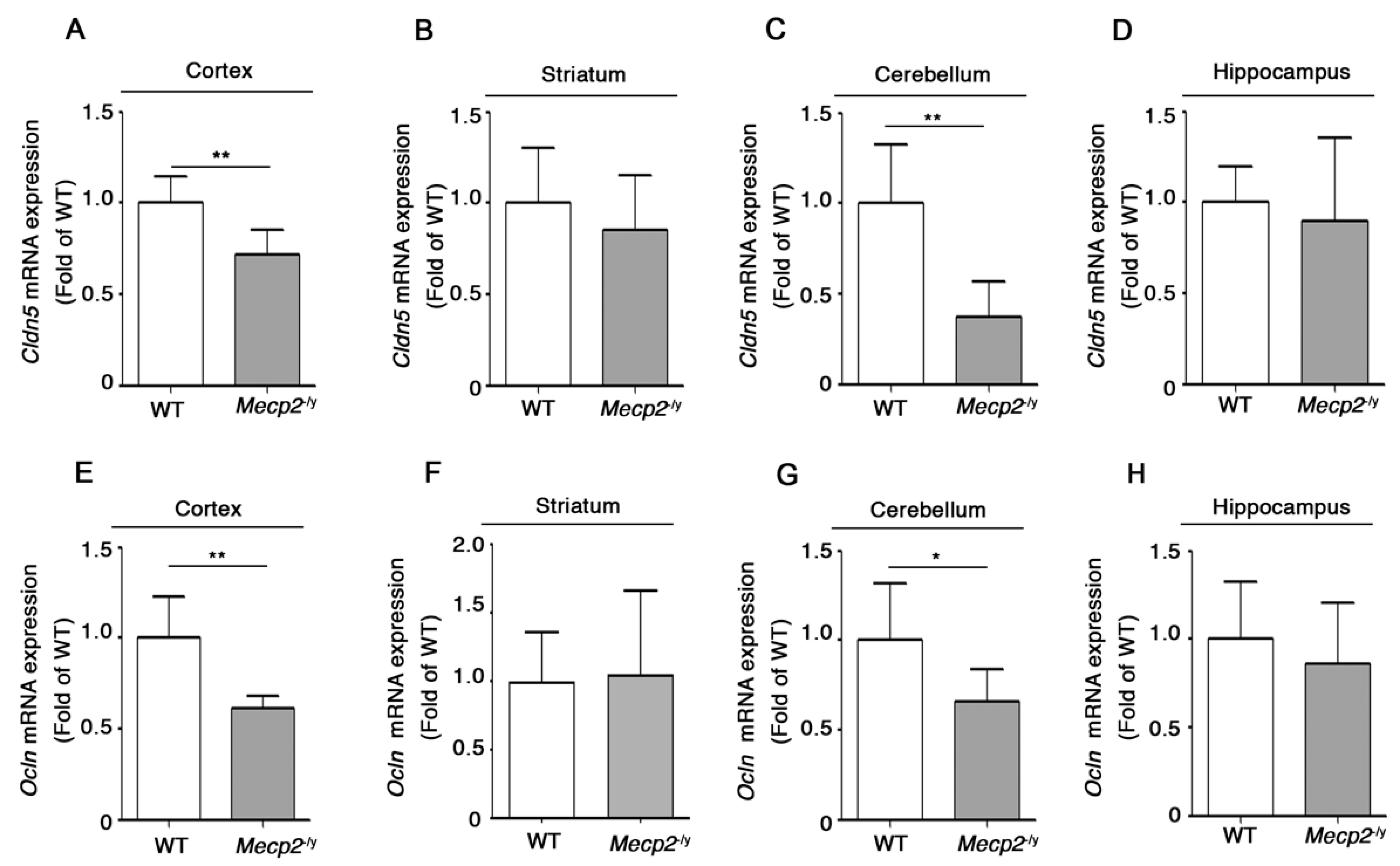
3.2. Expression of Claudin-5 and Occludin Proteins Is Decreased in Brain Tissues of Symptomatic Mecp2-/y Mice
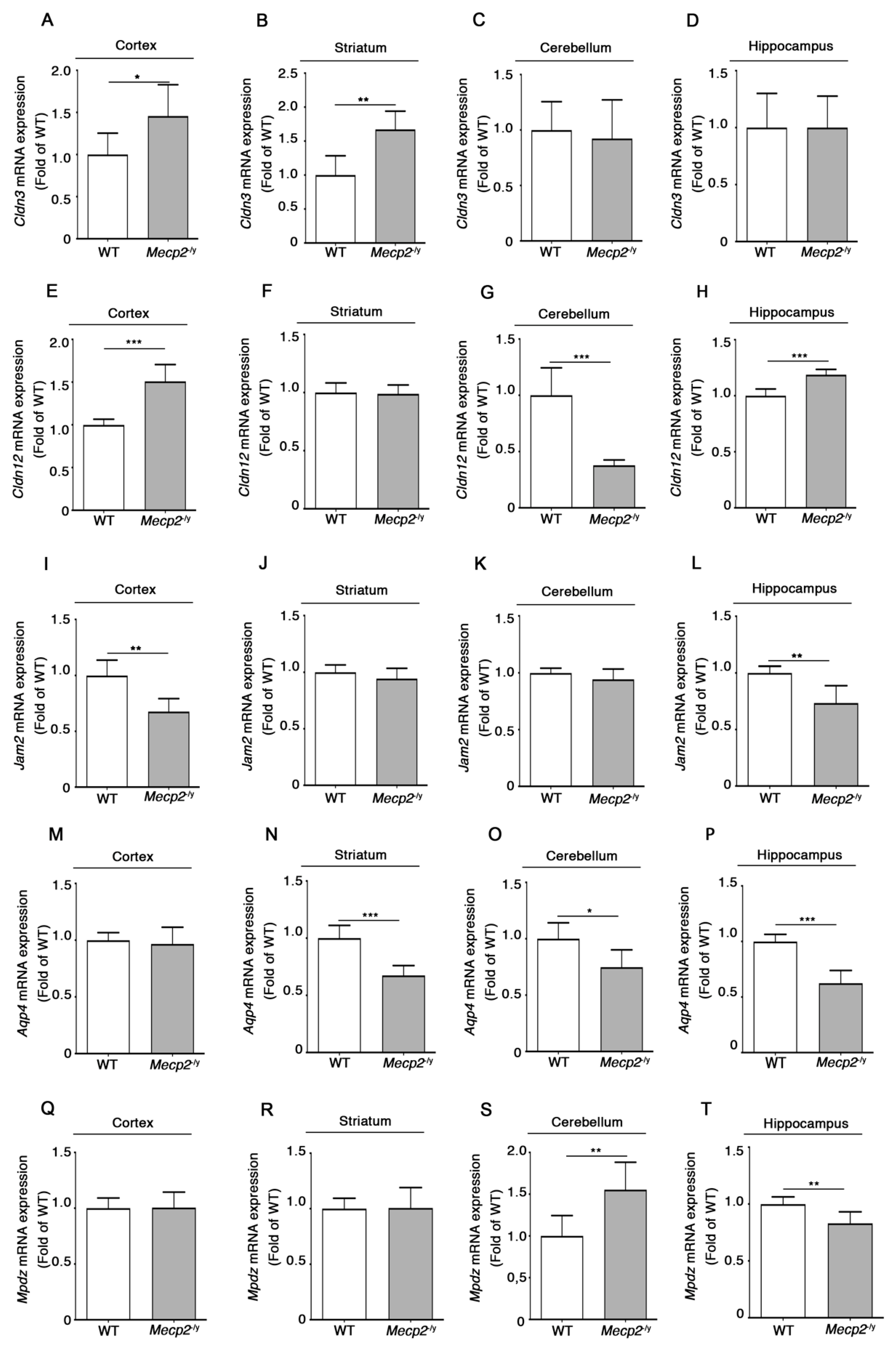
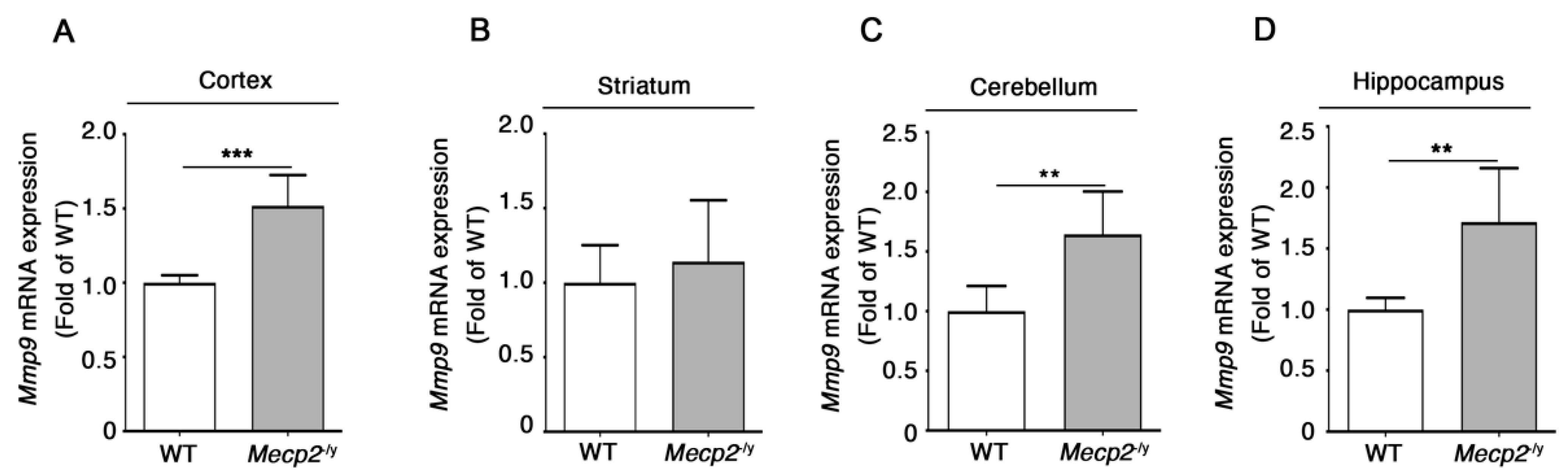
3.3. Expression of Additional Genes Involved in BBB Structure and Function Is Aberrant in Symptomatic Mecp2-/y Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hagberg, B. Rett’s syndrome: Prevalence and impact on progressive severe mental retardation in girls. Acta Paediatr. Scand 1985, 74, 405–408. [Google Scholar] [CrossRef]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neul, j.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- D’Esposito, M.; Quaderi, N.A.; Ciccodicola, A.; Bruni, P.; Esposito, T.; D’Urso, M.; Brown, S.D. Isolation, physical mapping, and northern analysis of the X-linked human gene encoding methyl CpG-binding protein, MECP2. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 1996, 7, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, H.Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Della Ragione, F.; Filosa, S.; Scalabri, F.; D’Esposito, M. MeCP2 as a genome-wide modulator: The renewal of an old story. Front. Genet. 2012, 3, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fioriniello, S.; Csukonyi, E.; Marano, D.; Brancaccio, A.; Madonna, M.; Zarrillo, C.; Romano, A.; Marracino, F.; Matarazzo, M.R.; D’Esposito, M.; et al. MeCP2 and Major Satellite Forward RNA Cooperate for Pericentric Heterochromatin Organization. Stem Cell Rep. 2020, 15, 1317–1332. [Google Scholar] [CrossRef] [PubMed]
- Fioriniello, S.; Marano, D.; Fiorillo, F.; D’Esposito, M.; Della Ragione, F. Epigenetic Factors That Control Pericentric Heterochromatin Organization in Mammals. Genes 2020, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Marano, D.; Fioriniello, S.; Fiorillo, F.; Gibbons, R.J.; D’Esposito, M.; Della Ragione, F. ATRX Contributes to MeCP2-Mediated Pericentric Heterochromatin Organization during Neural Differentiation. Int. J. Mol. Sci. 2019, 20, 5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, N.L.; Heaven, M.R.; Holt, L.M.; Crossman, D.K.; Boggio, K.J.; Shaffer, S.A.; Flint, D.L.; Olsen, M.L. RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome. Mol. Autism 2017, 8, 56. [Google Scholar] [CrossRef]
- Panighini, A.; Duranti, E.; Santini, F.; Maffei, M.; Pizzorusso, T.; Funel, N.; Taddei, S.; Bernardini, N.; Ippolito, C.; Virdis, A.; et al. Vascular dysfunction in a mouse model of Rett syndrome and effects of curcumin treatment. PLoS ONE 2013, 8, e64863. [Google Scholar] [CrossRef] [Green Version]
- Kapasi, A.; Schneider, J.A. Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim Biophys. Acta 2016, 1862, 878–886. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Csiszar, A.; Ungvari, Z. Functional vascular contributions to cognitive impairment and dementia: Mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1–H20. [Google Scholar] [CrossRef] [Green Version]
- Schiro, G.; Balistreri, C.R. The close link between brain vascular pathological conditions and neurodegenerative diseases: Focus on some examples and potential treatments. Vasc. Pharmacol. 2022, 142, 106951. [Google Scholar] [CrossRef]
- Berger-Sweeney, J. Cognitive deficits in Rett syndrome: What we know and what we need to know to treat them. Neurobiol. Learn. Mem. 2011, 96, 637–646. [Google Scholar] [CrossRef]
- Loffler, G.; Gordon, G.E. Cognitive function in Rett syndrome: Profoundly impaired or near normal? Eur. J. Paediatr. Neurol. 2018, 22, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Pelka, G.J.; Watson, C.M.; Radziewic, T.; Hayward, M.; Lahooti, H.; Christodoulou, J.; Tam, P.P. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain 2006, 129, 887–898. [Google Scholar] [CrossRef]
- Schaevitz, L.R.; Moriuchi, J.M.; Nag, N.; Mellot, T.J.; Berger-Sweeney, J. Cognitive and social functions and growth factors in a mouse model of Rett syndrome. Physiol. Behav. 2010, 100, 255–263. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Vojdani, A.; Blaurock-Busch, E.; Busch, Y.; Friedle, A.; Franco-Lira, M.; Sarathi-Mukherjee, P.; Martinez-Aguirre, X.; Park, S.B.; Torres-Jardon, R.; et al. Air pollution and children: Neural and tight junction antibodies and combustion metals, the role of barrier breakdown and brain immunity in neurodegeneration. J. Alzheimers Dis. 2015, 43, 1039–1058. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Scalabri, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagace, M.; Kuan, W.L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism. 2016, 7, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Sharma, B. Minocycline ameliorates prenatal valproic acid induced autistic behaviour, biochemistry and blood brain barrier impairments in rats. Brain Res. 2016, 1630, 83–97. [Google Scholar] [CrossRef]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crockett, A.M.; Ryan, S.K.; Vasquez, A.H.; Canning, C.; Kanyuch, N.; Kebir, H.; Ceja, G.; Gesualdi, J.; Zackai, E.; McDonald-McGinn, D.; et al. Disruption of the blood-brain barrier in 22q11.2 deletion syndrome. Brain 2021, 144, 1351–1360. [Google Scholar] [CrossRef]
- Zaragoza, R. Transport of Amino Acids Across the Blood-Brain Barrier. Front. Physiol. 2020, 11, 973. [Google Scholar] [CrossRef] [PubMed]
- Jian, W.X.; Zhang, Z.; Chu, S.F.; Peng, Y.; Chen, N.H. Potential roles of brain barrier dysfunctions in the early stage of Alzheimer’s disease. Brain Res. Bull. 2018, 142, 360–367. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Di Pardo, A.; Castaldo, S.; Capocci, L.; Amico, E.; Vittorio, M. Assessment of Blood-brain Barrier Permeability by Intravenous Infusion of FITC-labeled Albumin in a Mouse Model of Neurodegenerative Disease. J. Vis. Exp. 2017, 129, e56389. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [Green Version]
- Di Pardo, A.; Castaldo, S.; Amico, E.; Pepe, G.; Marracino, F.; Capocci, L.; Giovannelli, A.; Madonna, M.; van Bergeijk, J.; Buttari, F.; et al. Stimulation of S1PR5 with A-971432, a selective agonist, preserves blood-brain barrier integrity and exerts therapeutic effect in an animal model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 2490–2501. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.; Kim, M.; Park, W.; Lim, J.S.; Lee, E.; Cha, H.; Ahn, J.S.; Kim, J.H.; Hong, S.H.; Park, J.E.; et al. Upregulation of AQP4 Improves Blood-Brain Barrier Integrity and Perihematomal Edema Following Intracerebral Hemorrhage. Neurotherapeutics 2021, 18, 2692–2706. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139 (Suppl. S2), 91–114. [Google Scholar] [CrossRef] [Green Version]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [Green Version]
- Komada, M.; Nishimura, Y. Epigenetics and Neuroinflammation Associated With Neurodevelopmental Disorders: A Microglial Perspective. Front. Cell Dev. Biol. 2022, 10, 852752. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Cervellati, C.; Cordone, V.; Hayek, J.; Valacchi, G. Compromised immune/inflammatory responses in Rett syndrome. Free. Radic. Biol. Med. 2020, 152, 100–106. [Google Scholar] [CrossRef]
- Feldman, G.J.; Mullin, J.M.; Ryan, M.P. Occludin: Structure, function and regulation. Adv. Drug Deliv. Rev. 2005, 57, 883–917. [Google Scholar] [CrossRef] [PubMed]
- Piontek, J.; Winkler, L.; Wolburg, H.; Muller, S.L.; Zuleger, N.; Piehl, C.; Wiesner, B.; Krause, G.; Blasig, I.E. Formation of tight junction: Determinants of homophilic interaction between classic claudins. FASEB J. 2008, 22, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welcome, M.O. Cellular mechanisms and molecular signaling pathways in stress-induced anxiety, depression, and blood-brain barrier inflammation and leakage. Inflammopharmacology 2020, 28, 643–665. [Google Scholar] [CrossRef]
- Haj-Yasein, N.N.; Vindedal, G.F.; Eilert-Olsen, M.; Gundersen, G.A.; Skare, O.; Laake, P.; Klungland, A.; Thoren, A.E.; Burkhardt, J.M.; Ottersen, O.P.; et al. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. USA 2011, 108, 17815–17820. [Google Scholar] [CrossRef] [Green Version]
- Meshorer, E.; Biton, I.E.; Ben-Shaul, Y.; Ben-Ari, S.; Assaf, Y.; Soreq, H.; Cohen, Y. Chronic cholinergic imbalances promote brain diffusion and transport abnormalities. FASEB J. 2005, 19, 910–922. [Google Scholar] [CrossRef]
- Nicchia, G.P.; Nico, B.; Camassa, L.M.; Mola, M.G.; Loh, N.; Dermietzel, R.; Spray, D.C.; Svelto, M.; Frigeri, A. The role of aquaporin-4 in the blood-brain barrier development and integrity: Studies in animal and cell culture models. Neuroscience 2004, 129, 935–945. [Google Scholar] [CrossRef]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Quondamatteo, F.; Herken, R.; Errede, M.; Ribatti, D.; Svelto, M.; Roncali, L. Role of aquaporin-4 water channel in the development and integrity of the blood-brain barrier. J. Cell Sci. 2001, 114, 1297–1307. [Google Scholar] [CrossRef]
- Bauer, A.T.; Burgers, H.F.; Rabie, T.; Marti, H.H. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J. Cereb. Blood Flow Metab. 2010, 30, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Jiang, C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef]
- Mun-Bryce, S.; Rosenberg, G.A. Matrix metalloproteinases in cerebrovascular disease. J. Cereb. Blood Flow Metab. 1998, 18, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, H.; Dansie, L.E.; Hickmott, P.W.; Ethell, D.W.; Ethell, I.M. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J. Neurosci. 2014, 34, 9867–9879. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- De Felice, C.; Ciccoli, L.; Leoncini, S.; Signorini, C.; Rossi, M.; Vannuccini, L.; Guazzi, G.; Latini, G.; Comporti, M.; Valacchi, G.; et al. Systemic oxidative stress in classic Rett syndrome. Free. Radic. Biol. Med. 2009, 47, 440–448. [Google Scholar] [CrossRef]
- Signorini, C.; De Felice, C.; Leoncini, S.; Giardini, A.; D’Esposito, M.; Filosa, S.; Della Ragione, F.; Rossi, M.; Pecorelli, A.; Valacchi, G.; et al. F(4)-neuroprostanes mediate neurological severity in Rett syndrome. Clin. Chim. Acta 2011, 412, 1399–1406. [Google Scholar] [CrossRef]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef]
- Golubiani, G.; Lagani, V.; Solomonia, R.; Muller, M. Metabolomic Fingerprint of Mecp2-Deficient Mouse Cortex: Evidence for a Pronounced Multi-Facetted Metabolic Component in Rett Syndrome. Cells 2021, 10, 2494. [Google Scholar] [CrossRef]
- Maezawa, I.; Jin, L.W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.W.; Horiuchi, M.; Wulff, H.; Liu, X.B.; Cortopassi, G.A.; Erickson, J.D.; Maezawa, I. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: A mechanism for mitochondrial dysfunction and neurotoxicity. J. Neurosci. 2015, 35, 2516–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Forward Sequence (5′–3′) | Primer Reverse Sequence (5′–3′) |
|---|---|---|
| Cldn5 | CAGTTAAGGCACGGGTAGCA | GGCACCGTCGGATCATAGAA |
| Occludin | AGACCTGATGAATTCAAACCCAAT | ATGCATCTCTCCGCCATACAT |
| Cldn3 | TACAAGACGAGACGGCCAAG | GGGCACCAACGGGTTATAGA |
| Jam2 | GGCAAATGGGTTTTCTGCATC | TGGAGAGCCTGTTGGTAGTAGA |
| Mpdz | GTCCTTTTAGTTGGTGTTTGGCA | CGCGTTCTTTCAGCTTGCTT |
| Aqp4 | ATCCTCTACCTGGTCACACCT | ATAGTGAACACCAACTGGAAAGTG |
| Cldn12 | ACGGCCTTCAATTCTTCCGT | AGACCGGCTCAAACTTCCTG |
| Mmp9 | GACGACATAGACGGCATCCAG | GGATAGGCCGTGGGAGGTAT |
| Gapdh | CCAGGAGCGAGACCCCACTA | GGGCGGAGATGATGACCCTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepe, G.; Fioriniello, S.; Marracino, F.; Capocci, L.; Maglione, V.; D’Esposito, M.; Di Pardo, A.; Della Ragione, F. Blood–Brain Barrier Integrity Is Perturbed in a Mecp2-Null Mouse Model of Rett Syndrome. Biomolecules 2023, 13, 606. https://doi.org/10.3390/biom13040606
Pepe G, Fioriniello S, Marracino F, Capocci L, Maglione V, D’Esposito M, Di Pardo A, Della Ragione F. Blood–Brain Barrier Integrity Is Perturbed in a Mecp2-Null Mouse Model of Rett Syndrome. Biomolecules. 2023; 13(4):606. https://doi.org/10.3390/biom13040606
Chicago/Turabian StylePepe, Giuseppe, Salvatore Fioriniello, Federico Marracino, Luca Capocci, Vittorio Maglione, Maurizio D’Esposito, Alba Di Pardo, and Floriana Della Ragione. 2023. "Blood–Brain Barrier Integrity Is Perturbed in a Mecp2-Null Mouse Model of Rett Syndrome" Biomolecules 13, no. 4: 606. https://doi.org/10.3390/biom13040606