
Is It Still Possible to Think about HSP70 as a Therapeutic Target in Onco-Hematological Diseases?
 , , , , , , and
, , , , , , and
Abstract
:1. State of the Art of Treatment in Onco-Hematological Diseases
2. The Heat Shock Protein of 70kDa (HSP70)
2.1. The Heat Shock Proteins
2.2. The HSP70 Family
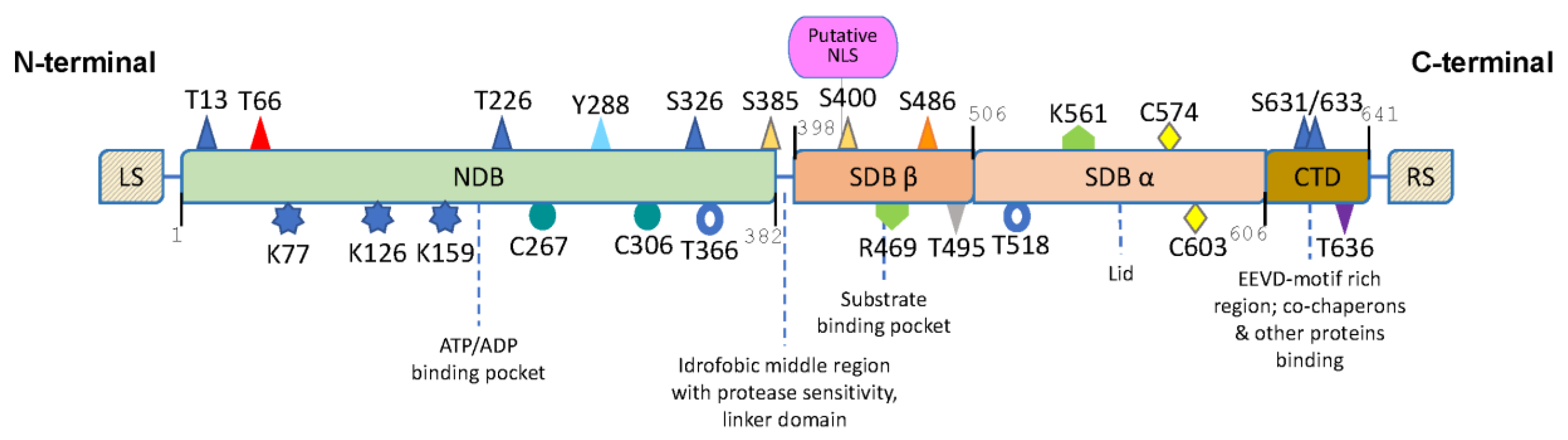
2.3. HSP70 Structure and Chaperone Cycle
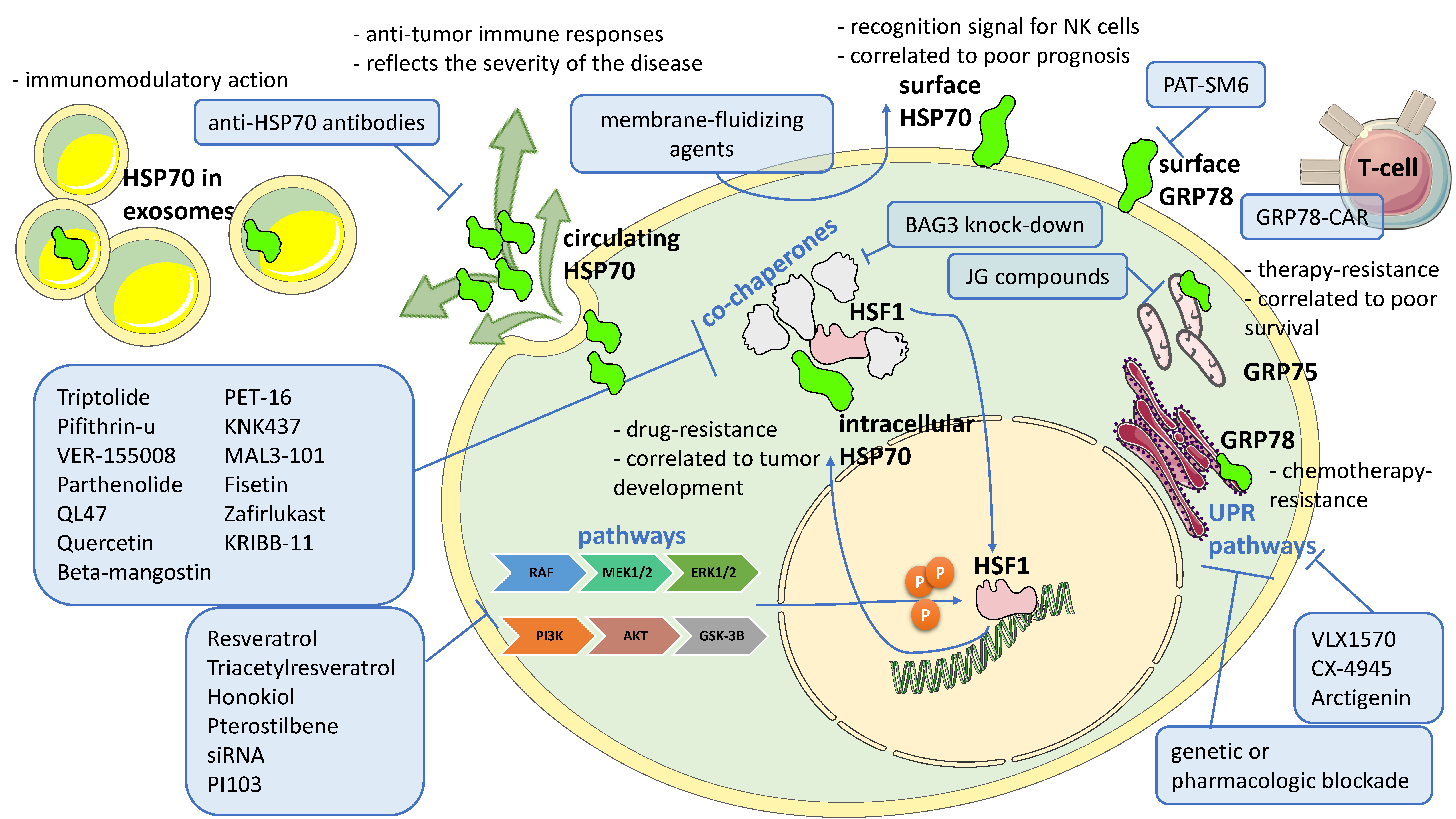
3. HSP70 and Its Targeting in Onco-Hematological Diseases
3.1. Acute and Chronic Leukemias
3.2. Chronic Lymphocytic Leukemia
3.3. Multiple Myeloma
3.4. Lymphomas
3.4.1. Diffuse Large B-Cell Lymphoma
3.4.2. Mantle Cell Lymphoma
3.4.3. Hodgkin’s Lymphoma
3.4.4. Other Lymphomas
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pandrala, M.; Bruyneel, A.A.N.; Hnatiuk, A.P.; Mercola, M.; Malhotra, S.V. Designing Novel BCR-ABL Inhibitors for Chronic Myeloid Leukemia with Improved Cardiac Safety. J. Med. Chem. 2022, 65, 10898–10919. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Barreca, M.; Spanò, V.; Raimondi, M.V.; Poma, P.; Notarbartolo, M.; Barraja, P.; Montalbano, A. Novel insights on [1,2]oxazolo[5,4-e]isoindoles on multidrug resistant acute myeloid leukemia cell line. Drug Dev. Res. 2022, 83, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Abel, A.-C.; Maruca, A.; Montalbano, A.; Raimondi, M.V.; Tarantelli, C.; Gaudio, E.; et al. Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: Further chemical modifications and potential therapeutic effects against lymphomas. Eur. J. Med. Chem. 2022, 243, 114744. [Google Scholar] [CrossRef]
- Cao, S.; Ma, L.; Liu, Y.; Wei, M.; Yao, Y.; Li, C.; Wang, R.; Liu, N.; Dong, Z.; Li, X.; et al. Proteolysis-Targeting Chimera (PROTAC) Modification of Dovitinib Enhances the Antiproliferative Effect against FLT3-ITD-Positive Acute Myeloid Leukemia Cells. J. Med. Chem. 2021, 64, 16497–16511. [Google Scholar] [CrossRef] [PubMed]
- Cilibrasi, V.; Spanò, V.; Bortolozzi, R.; Barreca, M.; Raimondi, M.V.; Rocca, R.; Maruca, A.; Montalbano, A.; Alcaro, S.; Ronca, R.; et al. Synthesis of 2H-Imidazo[2′,1’:2,3] [1,3]thiazolo[4,5-e]isoindol-8-yl-phenylureas with promising therapeutic features for the treatment of acute myeloid leukemia (AML) with FLT3/ITD mutations. Eur. J. Med. Chem. 2022, 235, 114292. [Google Scholar] [CrossRef]
- Gu, H.; He, J.; Li, Y.; Mi, D.; Guan, T.; Guo, W.; Liu, B.; Chen, Y. B-cell Lymphoma 6 Inhibitors: Current Advances and Prospects of Drug Development for Diffuse Large B-cell Lymphomas. J. Med. Chem. 2022, 65, 15559–15583. [Google Scholar] [CrossRef]
- Palacios, D.S. Drug Hunting at the Nexus of Medicinal Chemistry and Chemical Biology and the Discovery of Novel Therapeutic Modalities. J. Med. Chem. 2022, 65, 13594–13613. [Google Scholar] [CrossRef]
- Hou, Y.; Kuang, W.; Min, W.; Liu, Z.; Zhang, F.; Yuan, K.; Wang, X.; Sun, C.; Cheng, H.; Wang, L.; et al. Design, Synthesis, and Biological Evaluation of Icaritin Derivatives as Novel Putative DEPTOR Inhibitors for Multiple Myeloma Treatment. J. Med. Chem. 2021, 64, 14942–14954. [Google Scholar] [CrossRef]
- Smith, A.L.; Eiken, A.P.; Skupa, S.A.; Moore, D.Y.; Umeta, L.T.; Smith, L.M.; Lyden, E.R.; D’Angelo, C.R.; Kallam, A.; Vose, J.M.; et al. A Novel Triple-Action Inhibitor Targeting B-Cell Receptor Signaling and BRD4 Demonstrates Preclinical Activity in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2022, 23, 6712. [Google Scholar] [CrossRef]
- Chen, R.; Tsai, J.; Thompson, P.A.; Chen, Y.; Xiong, P.; Liu, C.; Burrows, F.; Sivina, M.; Burger, J.A.; Keating, M.J.; et al. The multi-kinase inhibitor TG02 induces apoptosis and blocks B-cell receptor signaling in chronic lymphocytic leukemia through dual mechanisms of action. Blood Cancer J. 2021, 11, 57. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Srivastava, P. Heat-Shock Proteins. Curr. Protoc. Immunol. 2003, 58, A.1T.1–A.1T.6. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.J.; Giménez, G.; Prince, T.L.; Ackerman, A.; Bonorino, C.; Calderwood, S.K. Heat Shock Proteins Are Essential Components in Transformation and Tumor Progression: Cancer Cell Intrinsic Pathways and Beyond. Int. J. Mol. Sci. 2019, 20, 4507. [Google Scholar] [CrossRef] [Green Version]
- Haslbeck, M.; Vierling, E. A First Line of Stress Defense: Small Heat Shock Proteins and Their Function in Protein Homeostasis. J. Mol. Biol. 2015, 427, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Asea, A. Heat shock proteins and toll-like receptors. Handb. Exp. Pharmacol. 2008, 183, 111. [Google Scholar] [CrossRef]
- Hassan, F.; Nawaz, A.; Rehman, M.S.; Ali, M.A.; Dilshad, S.M.; Yang, C. Prospects of HSP70 as a genetic marker for thermo-tolerance and immuno-modulation in animals under climate change scenario. Anim. Nutr. 2019, 5, 340–350. [Google Scholar] [CrossRef]
- Albakova, Z.; Mangasarova, Y.; Albakov, A.; Gorenkova, L. HSP70 and HSP90 in Cancer: Cytosolic, Endoplasmic Reticulum and Mitochondrial Chaperones of Tumorigenesis. Front. Oncol. 2022, 12, 829520. [Google Scholar] [CrossRef]
- De Maio, A.; Vazquez, D. Extracellular Heat Shock Proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Linder, M.; von Strandmann, E.P. The Role of Extracellular HSP70 in the Function of Tumor-Associated Immune Cells. Cancers 2021, 13, 4721. [Google Scholar] [CrossRef]
- Broquet, A.H.; Thomas, G.; Masliah, J.; Trugnan, G.; Bachelet, M. Expression of the Molecular Chaperone Hsp70 in Detergent-resistant Microdomains Correlates with Its Membrane Delivery and Release. J. Biol. Chem. 2003, 278, 21601–21606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, V.L.; Rodriíguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 Translocates into the Plasma Membrane after Stress and Is Released into the Extracellular Environment in a Membrane-Associated Form that Activates Macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mambula, S.S.; Stevenson, M.A.; Ogawa, K.; Calderwood, S.K. Mechanisms for Hsp70 secretion: Crossing membranes without a leader. Methods 2007, 43, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mambula, S.S.; Calderwood, S.K. Heat Shock Protein 70 Is Secreted from Tumor Cells by a Nonclassical Pathway Involving Lysosomal Endosomes. J. Immunol. 2006, 177, 7849–7857. [Google Scholar] [CrossRef] [Green Version]
- Gehrmann, M.; Kimm, M.; Stangl, S.; Schmid, T.; Noël, P.; Rummeny, E.; Multhoff, G. Imaging of Hsp70-positive tumors with cmHsp70.1 antibody-conjugated gold nanoparticles. Int. J. Nanomed. 2015, 10, 5687–5700. [Google Scholar] [CrossRef] [Green Version]
- Ferrarini, M.; Heltai, S.; Zocchi, M.R.; Rugarli, C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 1992, 51, 613–619. [Google Scholar] [CrossRef]
- Multhoff, G.; Botzler, C.; Wiesnet, M.; Müller, E.; Meier, T.; Wilmanns, W.; Issels, R.D. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 1995, 61, 272–279. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and Function of Sphingolipid- and Cholesterol-rich Membrane Rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [Green Version]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat Shock Protein 70 Surface-Positive Tumor Exosomes Stimulate Migratory and Cytolytic Activity of Natural Killer Cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, G.I.; Febbraio, M.A. Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef] [Green Version]
- Finka, A.; Sharma, S.K.; Goloubinoff, P. Multi-layered molecular mechanisms of polypeptide holding, unfolding and disaggregation by HSP70/HSP110 chaperones. Front. Mol. Biosci. 2015, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [Green Version]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [Green Version]
- Tavaria, M.; Gabriele, T.; Kola, I.; Anderson, R.L. A hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones 1996, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Mizzen, L.; Chang, C.; Garrels, J.; Welch, W. Identification, characterization, and purification of two mammalian stress proteins present in mitochondria, grp 75, a member of the hsp 70 family and hsp 58, a homolog of the bacterial groEL protein. J. Biol. Chem. 1989, 264, 20664–20675. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Tang, J.; Zhang, B.; Lin, Y.; Hanai, J.-I.; Galloway, J.; Bedell, V.; Bahary, N.; Han, Z.; Ramchandran, R.; et al. A novel endothelial-specific heat shock protein HspA12B is required in both zebrafish development and endothelial functions in vitro. J. Cell Sci. 2006, 119, 4117–4126. [Google Scholar] [CrossRef] [Green Version]
- Otterson, G.; Flynn, G.; Kratzke, R.; Coxon, A.; Johnston, P.; Kaye, F. Stch encodes the ‘ATPase core’ of a microsomal stress 70 protein. EMBO J. 1994, 13, 1216–1225. [Google Scholar] [CrossRef]
- Vostakolaei, M.A.; Hatami-Baroogh, L.; Babaei, G.; Molavi, O.; Kordi, S.; Abdolalizadeh, J. Hsp70 in cancer: A double agent in the battle between survival and death. J. Cell. Physiol. 2020, 236, 3420–3444. [Google Scholar] [CrossRef]
- Espinoza, M.F.; Nguyen, K.K.; Sycks, M.M.; Lyu, Z.; Quanrud, G.M.; Montoya, M.R.; Genereux, J.C. Heat shock protein Hspa13 regulates endoplasmic reticulum and cytosolic proteostasis through modulation of protein translocation. J. Biol. Chem. 2022, 298, 102597. [Google Scholar] [CrossRef]
- Wan, T.; Zhou, X.; Chen, G.; An, H.; Chen, T.; Zhang, W.; Liu, S.; Jiang, Y.; Yang, F.; Wu, Y.; et al. Novel heat shock protein Hsp70L1 activates dendritic cells and acts as a Th1 polarizing adjuvant. Blood 2004, 103, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.; Schröder, H.; Rüdiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Struct. Biol. 2000, 7, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef] [PubMed]
- Liberek, K.; Marszalek, J.; Ang, D.; Georgopoulos, C.; Zylicz, M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc. Natl. Acad. Sci. USA 1991, 88, 2874–2878. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670. [Google Scholar] [CrossRef] [Green Version]
- Mecha, M.F.; Hutchinson, R.B.; Lee, J.H.; Cavagnero, S. Protein folding in vitro and in the cell: From a solitary journey to a team effort. Biophys. Chem. 2022, 287, 106821. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Subjeck, J.R. High molecular weight stress proteins: Identification, cloning and utilisation in cancer immunotherapy. Int. J. Hyperth. 2013, 29, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabaud-Gibouin, V.; Durand, M.; Quéré, R.; Girodon, F.; Garrido, C.; Jego, G. Heat-Shock Proteins in Leukemia and Lymphoma: Multitargets for Innovative Therapeutic Approaches. Cancers 2023, 15, 984. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. Medcomm 2022, 3, e161. [Google Scholar] [CrossRef]
- Kumar, V.; Mapa, K. Hsp70: A Multi-Tasking Chaperone at the Crossroad of Cellular Proteostasis. In Regulation of Heat Shock Protein Responses; Asea, A., Kaur, P., Eds.; Heat Shock Proteins; Springer: Cham, Germany, 2018; Volume 13. [Google Scholar] [CrossRef]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- Kliková, K.; Pilchova, I.; Štefaniková, A.; Hatok, J.; Dobrota, D.; Račay, P. The Role of Heat Shock Proteins in Leukemia. Klin. Onkol. 2016, 29, 29–38. [Google Scholar] [CrossRef]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in Bcr-Abl–mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Lanneau, D.; De Thonel, A.; Maurel, S.; Didelot, C.; Garrido, C. Apoptosis Versus Cell Differentiation: Role of heat shock protein HSP90, HSP70 and HSP27. Prion 2007, 1, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Lee, J.J.; Seo, J.S. HSP70 deficiency results in activation of c-Jun N-terminal kinase, extracellular signal-regulated kinase, and caspase-3 in hyperosmolarity-induced apoptosis. J. Biol. Chem. 2005, 280, 6634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature 2000, 2, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Nylandsted, J.; Jäättelä, M. Heat Shock Protein 70 Promotes Cancer Cell Viability by Safeguarding Lysosomal Integrity. Cell Cycle 2004, 3, 1484–1485. [Google Scholar] [CrossRef]
- Bivik, C.; Rosdahl, I.; Ollinger, K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis 2007, 28, 537–544. [Google Scholar] [CrossRef]
- Mjahed, H.; Girodon, F.; Fontenay, M.; Garrido, C. Heat shock proteins in hematopoietic malignancies. Exp. Cell Res. 2012, 318, 1946–1958. [Google Scholar] [CrossRef]
- Pui, C.-H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Ferrara, F.; Schiffer, C.A. Acute myeloid leukaemia in adults. Lancet 2013, 381, 484–495. [Google Scholar] [CrossRef]
- Ochi, Y. Genetic landscape of chronic myeloid leukemia. Int. J. Hematol. 2023, 117, 30. [Google Scholar] [CrossRef]
- Zhang, W.; Drach, J.; Andreeff, M.; Deisseroth, A. Proliferation of hematopoietic cells is accompanied by suppressed expression of heat shock protein 70. Biochem. Biophys. Res. Commun. 1992, 183, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Chant, I.D.; Rose, P.E.; Morris, A.G. Analysis of heat-shock protein expression in myeloid leukaemia cells by flow cytometry. Br. J. Haematol. 1995, 90, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Chant, I.D.; Rose, P.E.; Morris, A.G. Susceptibility of AML cells to in vitro apoptosis correlates with heat shock protein 70 (hsp70) expression. Br. J. Haematol. 1996, 93, 898–902. [Google Scholar] [CrossRef]
- Gehrmann, M.; Schmetzer, H.; Eissner, G.; Haferlach, T.; Hiddemann, W.; Multhoff, G. Membrane-bound heat shock protein 70 (Hsp70) in acute myeloid leukemia: A tumor specific recognition structure for the cytolytic activity of autologous NK cells. Haematologica 2003, 88, 474. [Google Scholar] [PubMed]
- Hantschel, M.; Pfister, K.; Jordan, A.; Scholz, R.; Andreesen, R.; Schmitz, G.; Schmetzer, H.; Hiddemann, W.; Multhoff, G. Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chaperones 2000, 5, 438–442. [Google Scholar] [CrossRef]
- Stangl, S.; Gross, C.; Pockley, A.G.; Asea, A.A.; Multhoff, G. Influence of Hsp70 and HLA-E on the killing of leukemic blasts by cytokine/Hsp70 peptide-activated human natural killer (NK) cells. Cell Stress Chaperones 2008, 13, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Steiner, K.; Graf, M.; Hecht, K.; Reif, S.; Rossbacher, L.; Pfister, K.; Kolb, H.-J.; Schmetzer, H.M.; Multhoff, G. High HSP70-membrane expression on leukemic cells from patients with acute myeloid leukemia is associated with a worse prognosis. Leukemia 2006, 20, 2076–2079. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.-H.; Tseng, R.; Hannah, A.; Estrov, Z.; Estey, E.; Kantarjian, H.; Albitar, M. Clinical correlation of circulating heat shock protein 70 in acute leukemia. Leuk. Res. 2010, 34, 605–609. [Google Scholar] [CrossRef] [Green Version]
- Piszcz, J.; Bolkun, L.; Cichocka, E.; Galar, M.; Hołownia, A.; Kłoczko, J. Prognostic relevance of HSP70 antigen and antibody measurement in patients with acute myeloid leukemia of intermediate and unfavorable cytogenetic risk. Pol. Arch. Intern. Med. 2014, 124, 165–172. [Google Scholar] [CrossRef]
- Li, J.; Ge, Z. High HSPA8 expression predicts adverse outcomes of acute myeloid leukemia. BMC Cancer 2021, 21, 475. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Ozer, Z.; Garner, A.L.; Pitt, J.; Ma, H.; Janda, K.D. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br. J. Haematol. 2011, 153, 741–752. [Google Scholar] [CrossRef]
- Masouleh, B.K.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E.; et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E2219–E2228. [Google Scholar] [CrossRef] [Green Version]
- Angeles-Floriano, T.; Rivera-Torruco, G.; García-Maldonado, P.; Juárez, E.; Gonzalez, Y.; Parra-Ortega, I.; Vilchis-Ordoñez, A.; Lopez-Martinez, B.; Arriaga-Pizano, L.; Orozco-Ruíz, D.; et al. Cell surface expression of GRP78 and CXCR4 is associated with childhood high-risk acute lymphoblastic leukemia at diagnostics. Sci. Rep. 2022, 12, 2322. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zhang, A.; Huang, J.; Suo, M.; Zhong, Y.; Liang, Y. Suppression of HSP70 inhibits the development of acute lymphoblastic leukemia via TAK1/Egr-1. Biomed. Pharmacother. 2019, 119, 109399. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.H.; Kim, M.K.; Choi, E.Y.; Kim, S.H.; Sohn, H.W.; Ham, D.I.; Chung, D.H.; Kim, T.J.; Lee, W.J.; Park, C.K.; et al. CD99 (MIC2) regulates the LFA-1/ICAM-1-mediated adhesion of lymphocytes, and its gene encodes both positive and negative regulators of cellular adhesion. J. Immunol. 1997, 159, 2250–2258. [Google Scholar] [CrossRef]
- Alberti, I.; Bernard, G.; Rouquette-Jazdanian, A.K.; Pelassy, C.; Pourtein, M.; Aussel, C.; Bernard, A. CD99 isoform expression dictates T-cell functional outcomes. FASEB J. 2002, 16, 1946–1948. [Google Scholar] [CrossRef]
- Schenkel, A.; Mamdouh, Z.; Chen, X.; Liebman, R.M.; Muller, W.A. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 2002, 3, 143–150. [Google Scholar] [CrossRef]
- Husak, Z.; Dworzak, M.N. CD99 ligation upregulates HSP70 on acute lymphoblastic leukemia cells and concomitantly increases NK cytotoxicity. Cell Death Dis. 2012, 3, e425. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.M.; et al. Abrogation of Heat Shock Protein 70 Induction as a Strategy to Increase Antileukemia Activity of Heat Shock Protein 90 Inhibitor 17-Allylamino-Demethoxy Geldanamycin. Cancer Res. 2005, 65, 10536–10544. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, S.; Rao, I.; Earp, J.C.; Jusko, W.J.; Wetzler, M. Down-regulation of heat shock protein 70 improves arsenic trioxide and 17-DMAG effects on constitutive signal transducer and activator of transcription 3 activity. Cancer Chemother. Pharmacol. 2009, 66, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Kühnl, A.A.; Reins, J.; Fischer, S.; Ortiz-Tanchez, J.; Schlee, C.; Mochmann, L.H.; Heesch, S.; Benlasfer, O.O.; Hofmann, W.-K.; et al. Antileukemic activity of the HSP70 inhibitor pifithrin-μ in acute leukemia. Blood Cancer J. 2011, 1, e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reikvam, H.; Nepstad, I.; Sulen, A.; Gjertsen, B.T.; Hatfield, K.J.; Bruserud, Ø. Increased antileukemic effects in human acute myeloid leukemia by combining HSP70 and HSP90 inhibitors. Expert Opin. Investig. Drugs 2013, 22, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Omer, F.A.A.; Hashim, N.B.M.; Ibrahim, M.Y.; Dehghan, F.; Yahayu, M.; Karimian, H.; Salim, L.Z.A.; Mohan, S. Beta-mangostin from Cratoxylum arborescens activates the intrinsic apoptosis pathway through reactive oxygen species with downregulation of the HSP70 gene in the HL60 cells associated with a G0/G1 cell-cycle arrest. Tumor Biol. 2017, 39, 1010428317731451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.; McGowan, A.; DiNatale, G.J.; Chiramanewong, T.; Cai, T.; Connor, R.E. Hsp72 Is an Intracellular Target of the α,β-Unsaturated Sesquiterpene Lactone, Parthenolide. ACS Omega 2017, 2, 7267–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zou, F.; Wang, A.; Miao, W.; Liang, Q.; Weisberg, E.L.; Wang, Y.; Liu, J.; Wang, W.; Liu, Q. Targeting chaperon protein HSP70 as a novel therapeutic strategy for FLT3-ITD-positive acute myeloid leukemia. Signal Transduct. Target. Ther. 2021, 6, 334. [Google Scholar] [CrossRef]
- Pellegrini, P.; Selvaraj, K.; Faustini, E.; Mofers, A.; Zhang, X.; Ternerot, J.; Schubert, A.; Linder, S.; D′arcy, P. Induction of ER Stress in Acute Lymphoblastic Leukemia Cells by the Deubiquitinase Inhibitor VLX1570. Int. J. Mol. Sci. 2020, 21, 4757. [Google Scholar] [CrossRef]
- Kurozumi, N.; Tsujioka, T.; Ouchida, M.; Sakakibara, K.; Nakahara, T.; Suemori, S.; Takeuchi, M.; Kitanaka, A.; Shibakura, M.; Tohyama, K. VLX1570 induces apoptosis through the generation of ROS and induction of ER stress on leukemia cell lines. Cancer Sci. 2021, 112, 3302–3313. [Google Scholar] [CrossRef]
- Hebbar, N.; Epperly, R.; Vaidya, A.; Thanekar, U.; Moore, S.E.; Umeda, M.; Ma, J.; Patil, S.L.; Langfitt, D.; Huang, S.; et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat. Commun. 2022, 13, 587. [Google Scholar] [CrossRef]
- Li, X.-L.; Sun, L.-R.; Wang, Z.; Sun, X.-F. The preparation of leukemia cell vaccine expressing BCG heat shock protein 70 and anti-leukemia effect in vitro. Int. Immunopharmacol. 2012, 14, 235–242. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Melchionda, F.; Pession, A.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2013, 28, 543–553. [Google Scholar] [CrossRef]
- Hallek, M.; Al-Sawaf, O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, N.C.; Leoni, F.; Ireland, H.E.; Hoyle, C.; Williams, J.H.H. Differential heat shock protein localization in chronic lymphocytic leukemia. J. Leukoc. Biol. 2009, 87, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huergo-Zapico, L.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez, E.; López-Soto, A.; Fernandez-Guizan, A.; Acebes-Huerta, A.; Toyos, J.R.D.L.; Lopez-Larrea, C.; Groh, V.; et al. Expression of ERp5 and GRP78 on the membrane of chronic lymphocytic leukemia cells: Association with soluble MICA shedding. Cancer Immunol. Immunother. 2012, 61, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Madden, L.A.; Hayman, Y.A.; Underwood, C.; Vince, R.V.; Greenman, J.; Allsup, D.; Ali, S. Increased inducible heat shock protein 72 expression associated with PBMC isolated from patients with haematological tumours. Scand. J. Clin. Lab. Investig. 2012, 72, 380–386. [Google Scholar] [CrossRef]
- Dempsey, N.C.; Ireland, H.E.; Smith, C.M.; Hoyle, C.F.; Williams, J.H. Heat Shock Protein translocation induced by membrane fluidization increases tumor-cell sensitivity to chemotherapeutic drugs. Cancer Lett. 2010, 296, 257–267. [Google Scholar] [CrossRef]
- Frezzato, F.; Accordi, B.; Trimarco, V.; Gattazzo, C.; Martini, V.; Milani, G.; Bresolin, S.; Severin, F.; Visentin, A.; Basso, G.; et al. Profiling B cell chronic lymphocytic leukemia by reverse phase protein array: Focus on apoptotic proteins. J. Leukoc. Biol. 2016, 100, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Frezzato, F.; Raggi, F.; Martini, V.; Severin, F.; Trimarco, V.; Visentin, A.; Scomazzon, E.; Accordi, B.; Bresolin, S.; Piazza, F.; et al. HSP70/HSF1 axis, regulated via a PI3K/AKT pathway, is a druggable target in chronic lymphocytic leukemia. Int. J. Cancer 2019, 145, 3089–3100. [Google Scholar] [CrossRef]
- Frezzato, F.; Visentin, A.; Severin, F.; Pizzo, S.; Ruggeri, E.; Mouawad, N.; Martinello, L.; Pagnin, E.; Trimarco, V.; Tonini, A.; et al. Targeting of HSP70/HSF1 Axis Abrogates In Vitro Ibrutinib-Resistance in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 5453. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Liu, P.; Sun, M.; Wu, L.-Y.; Zhu, H.-Y.; Qiao, C.; Dong, H.-J.; Zhu, D.-X.; Xu, W.; Li, J.-Y. Bag3 gene expression in chronic lymphocytic leukemia and its association with patients’ prognosis. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2010, 18, 838. [Google Scholar]
- Rosati, A.; Basile, A.; Falco, A.; D’Avenia, M.; Festa, M.; Graziano, V.; De Laurenzi, V.; Arra, C.; Pascale, M.; Turco, M.C. Role of BAG3 protein in leukemia cell survival and response to therapy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2012, 1826, 365–369. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, W.; Fu, Y.; Shen, W.; Miao, K.; Hong, M.; Xu, W.; Young, K.H.; Liu, P.; Li, J. Overexpressed BAG3 is a potential therapeutic target in chronic lymphocytic leukemia. Ann. Hematol. 2014, 93, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Ijabi, R.; Roozehdar, P.; Afrisham, R.; Moradi-Sardareh, H.; Kaviani, S.; Ijabi, J.; Sahebkar, A. Association of GRP78, HIF-1α and BAG3 Expression with the Severity of Chronic Lymphocytic Leukemia. Anti-Cancer Agents Med. Chem. 2020, 20, 429–436. [Google Scholar] [CrossRef]
- Castaneda, O.; Baz, R. Multiple Myeloma Genomics—A Concise Review. Acta Medica Acad. 2019, 48, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Nakaki, T.; Deans, R.J.; Lee, A.S. Enhanced transcription of the 78,000-dalton glucose-regulated protein (GRP78) gene and association of GRP78 with immunoglobulin light chains in a nonsecreting B-cell myeloma line (NS-1). Mol. Cell. Biol. 1989, 9, 2233. [Google Scholar] [CrossRef]
- Nimmanapalli, R.; Gerbino, E.; Dalton, W.S.; Gandhi, V.; Alsina, M. HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br. J. Haematol. 2008, 142, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cultrara, C.N.; Kozuch, S.D.; Ramasundaram, P.; Heller, C.J.; Shah, S.; Beck, A.E.; Sabatino, D.; Zilberberg, J. GRP78 modulates cell adhesion markers in prostate Cancer and multiple myeloma cell lines. BMC Cancer 2018, 18, 1263. [Google Scholar] [CrossRef] [Green Version]
- Dakappagari, N.; Neely, L.; Tangri, S.; Lundgren, K.; Hipolito, L.; Estrellado, A.; Burrows, F.; Zhang, H. An investigation into the potential use of serum Hsp70 as a novel tumour biomarker for Hsp90 inhibitors. Biomarkers 2010, 15, 31–38. [Google Scholar] [CrossRef]
- Davenport, E.L.; Zeisig, A.A.; Aronson, L.I.; Moore, H.E.; Hockley, S.; Gonzalez, D.; Smith, E.M.; Powers, M.V.; Sharp, S.Y.; Workman, P.; et al. Targeting heat shock protein 72 enhances Hsp90 inhibitor-induced apoptosis in myeloma. Leukemia 2010, 24, 1804–1807. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Andrulis, M.; Stühmer, T.; Müller, E.; Hofmann, C.; Steinbrunn, T.; Heimberger, T.; Schraud, H.; Kressmann, S.; Einsele, H.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Y.; Bai, J.; Yang, Y.; Wang, F.; Feng, Y.; Zhang, R.; Li, F.; Zhang, P.; Lv, N.; et al. Blockade of HSP70 by VER-155008 synergistically enhances bortezomib-induced cytotoxicity in multiple myeloma. Cell Stress Chaperones 2020, 25, 357–367. [Google Scholar] [CrossRef]
- Heimberger, T.; Andrulis, M.; Riedel, S.; Stühmer, T.; Schraud, H.; Beilhack, A.; Bumm, T.; Bogen, B.; Einsele, H.; Bargou, R.C.; et al. The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br. J. Haematol. 2013, 160, 465–476. [Google Scholar] [CrossRef]
- Bustany, S.; Cahu, J.; Descamps, G.; Pellat-Deceunynck, C.; Sola, B. Heat shock factor 1 is a potent therapeutic target for enhancing the efficacy of treatments for multiple myeloma with adverse prognosis. J. Hematol. Oncol. 2015, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, M.J.; Scott, S.S.; Scott, C.M.; Behrman, S.; Walter, P.; Wipf, P.; Coplan, J.D.; Chrico, W.; Joseph, D.; Brodsky, J.L.; et al. Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3-101. J. Oncol. 2011, 2011, 232037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Fok, J.J.; Mirabella, F.; Aronson, L.I.; Fryer, R.A.; Workman, P.; Morgan, G.J.; Davies, F.E. Hsp70 inhibition induces myeloma cell death via the intracellular accumulation of immunoglobulin and the generation of proteotoxic stress. Cancer Lett. 2013, 339, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eugênio, A.I.P.; Fook-Alves, V.L.; De Oliveira, M.B.; Fernando, R.C.; Zanatta, D.B.; Strauss, B.; Silva, M.R.R.; Porcionatto, M.A.; Colleoni, G.W.B. Proteasome and heat shock protein 70 (HSP70) inhibitors as therapeutic alternative in multiple myeloma. Oncotarget 2017, 8, 114698–114709. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.K.; Budina-Kolomets, A.; Murphy, M.; Nefedova, Y. Efficacy of the HSP70 inhibitor PET-16 in multiple myeloma. Cancer Biol. Ther. 2015, 16, 1422–1426. [Google Scholar] [CrossRef]
- Malek, M.A.A.; Jagannathan, S.; Malek, E.; Sayed, D.M.; Elgammal, S.A.; El-Azeem, H.G.A.; Thabet, N.M.; Driscoll, J.J. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015, 6, 3098–3110. [Google Scholar] [CrossRef] [Green Version]
- Adomako, A.; Calvo, V.; Biran, N.; Osman, K.; Chari, A.; Paton, J.C.; Paton, A.W.; Moore, K.; Schewe, D.M.; Aguirre-Ghiso, J.A. Identification of markers that functionally define a quiescent multiple myeloma cell sub-population surviving bortezomib treatment. BMC Cancer 2015, 15, 444. [Google Scholar] [CrossRef] [Green Version]
- Rasche, L.; Menoret, E.; Dubljevic, V.; Menu, E.; Vanderkerken, K.; Lapa, C.; Steinbrunn, T.; Chatterjee, M.; Knop, S.; Duell, J.; et al. A GRP78-Directed Monoclonal Antibody Recaptures Response in Refractory Multiple Myeloma with Extramedullary Involvement. Clin. Cancer Res. 2016, 22, 4341–4349. [Google Scholar] [CrossRef] [Green Version]
- Ninkovic, S.; Harrison, S.J.; Quach, H. Glucose-regulated protein 78 (GRP78) as a potential novel biomarker and therapeutic target in multiple myeloma. Expert Rev. Hematol. 2020, 13, 1201–1210. [Google Scholar] [CrossRef]
- Vulpis, E.; Cecere, F.; Molfetta, R.; Soriani, A.; Fionda, C.; Peruzzi, G.; Caracciolo, G.; Palchetti, S.; Masuelli, L.; Simonelli, L.; et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis. Oncoimmunology 2017, 6, e1279372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.-T.; Wu, Y.; Niu, T. Human DKK1 and human HSP70 fusion DNA vaccine induces an effective anti-tumor efficacy in murine multiple myeloma. Oncotarget 2017, 9, 178–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Bashir, Q.; Qazilbash, M.H.; Andersson, B.S. Panobinostat and venetoclax enhance the cytotoxicity of gemcitabine, busulfan, and melphalan in multiple myeloma cells. Exp. Hematol. 2020, 81, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, I.D.; Lin, Y.-H.T.; Lam, C.; Shao, H.; Tharp, K.M.; Hale, M.; Kasap, C.; Mariano, M.C.; Kishishita, A.; Escobar, B.P.; et al. Allosteric HSP70 inhibitors perturb mitochondrial proteostasis and overcome proteasome inhibitor resistance in multiple myeloma. Cell Chem. Biol. 2022, 29, 1288–1302.e7. [Google Scholar] [CrossRef]
- Oroń, M.; Grochowski, M.; Jaiswar, A.; Legierska, J.; Jastrzębski, K.; Nowak-Niezgoda, M.; Kołos, M.; Kaźmierczak, W.; Olesiński, T.; Lenarcik, M.; et al. The molecular network of the proteasome machinery inhibition response is orchestrated by HSP70, revealing vulnerabilities in cancer cells. Cell Rep. 2022, 40, 111428. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Teras, L.R.; De Santis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA A Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef]
- Kienle, D.; Kröber, A.; Katzenberger, T.; Ott, G.; Leupolt, E.; Barth, T.F.E.; Möller, P.; Benner, A.; Habermann, A.; Müller-Hermelink, H.K.; et al. VH mutation status and VDJ rearrangement structure in mantle cell lymphoma: Correlation with genomic aberrations, clinical characteristics, and outcome. Blood 2003, 102, 3003–3009. [Google Scholar] [CrossRef] [Green Version]
- Hromadnikova, I.; Sedlackova, L. Analysis of cell surface and relative gene expression of heat shock protein 70 in human leukemia cell lines. Leuk. Lymphoma 2008, 49, 570–576. [Google Scholar] [CrossRef]
- Kwiecińska, A.; Porwit, A.; Souchelnytskyi, N.; Kaufeldt, A.; Larsson, C.; Bajalica-Lagercrantz, S.; Souchelnytskyi, S. Proteomic Profiling of Diffuse Large B-Cell Lymphomas. Pathobiology 2018, 85, 211–219. [Google Scholar] [CrossRef]
- Mozos, A.; Roué, G.; López-Guillermo, A.; Jares, P.; Campo, E.; Colomer, D.; Martinez, A. The Expression of the Endoplasmic Reticulum Stress Sensor BiP/GRP78 Predicts Response to Chemotherapy and Determines the Efficacy of Proteasome Inhibitors in Diffuse Large B-Cell Lymphoma. Am. J. Pathol. 2011, 179, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-F.; Yan, L.; Liu, Z.; Liu, L.-X.; Lin, J.; Liu, Z.-Y.; Chen, X.-P.; Zhang, W.; Xu, Z.-Z.; Shi, T.; et al. HSP70-Hrd1 axis precludes the oncorepressor potential of N-terminal misfolded Blimp-1s in lymphoma cells. Nat. Commun. 2017, 8, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutt, S.; Atallah, M.B.; Minamida, Y.; Filatenkov, A.; Jensen, K.P.; Iliopoulou, B.P.; Tamosiuniene, R.; Waters, J.; Engleman, E.G.; Strober, S. Accelerated, but not conventional, radiotherapy of murine B-cell lymphoma induces potent T cell–mediated remissions. Blood Adv. 2018, 2, 2568–2580. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Ye, Y.; Gui, A.; Sun, X.; Xie, S.; Zhan, Y.; Chen, R.; Yan, Y.; Gu, J.; Qiu, S.; et al. MORTALIN-Ca2+ axis drives innate rituximab resistance in diffuse large B-cell lymphoma. Cancer Lett. 2022, 537, 215678. [Google Scholar] [CrossRef]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Di Maggio, S.A.; Cabrelle, A.; et al. Protein Kinase CK2 Inhibition Down Modulates the NF-κB and STAT3 Survival Pathways, Enhances the Cellular Proteotoxic Stress and Synergistically Boosts the Cytotoxic Effect of Bortezomib on Multiple Myeloma and Mantle Cell Lymphoma Cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef] [Green Version]
- Sekihara, K.; Saitoh, K.; Han, L.; Ciurea, S.; Yamamoto, S.; Kikkawa, M.; Kazuno, S.; Taka, H.; Kaga, N.; Arai, H.; et al. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget 2017, 8, 34552–34564. [Google Scholar] [CrossRef] [Green Version]
- Vekaria, P.H.; Kumar, A.; Subramaniam, D.; Dunavin, N.; Vallurupalli, A.; Schoenen, F.; Ganguly, S.; Anant, S.; McGuirk, J.P.; Jensen, R.A.; et al. Functional cooperativity of p97 and histone deacetylase 6 in mediating DNA repair in mantle cell lymphoma cells. Leukemia 2019, 33, 1675–1686. [Google Scholar] [CrossRef]
- Takahashi, H.; Fujita, S.; Shibata, Y.; Tsuda, N.; Okabe, H. Expression of heat shock protein 70 (HSP70) and EBV latent membrane protein 1 (LMP1) in Reed-Sternberg cells of Hodgkin’s disease. Anal. Cell. Pathol. 1996, 12, 71. [Google Scholar]
- Quijano, S.M.; Saavedra, C.; Bravo, M.M.; Fiorentino, S.; Orozco, O. Expression of heat shock proteins HSP72 and HSP73 in Colombian patients with Hodgkin lymphoma positive and negative for Epstein Barr virus. Rev. Med. Chil. 2003, 13, 1375. [Google Scholar]
- Santón, A.; García-Cosío, M.; Cristóbal, E.; Pascual, A.; Muriel, A.; García-Laraña, J. Expression of heat shock proteins in classical Hodgkin lymphoma: Correlation with apoptotic pathways and prognostic significance. Histopathology 2011, 58, 1072–1080. [Google Scholar] [CrossRef]
- Chang, K.-C.; Chen, P.C.-H.; Chen, Y.-P.; Chang, Y.; Su, I.-J. Dominant expression of survival signals of endoplasmic reticulum stress response in Hodgkin lymphoma. Cancer Sci. 2011, 102, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.; Terrinca, J.; Rodríguez, A.B.; Espino, J.; Pariente, J.A. Extracellular heat shock proteins protect U937 cells from H2O2-induced apoptotic cell death. Mol. Cell. Biochem. 2016, 412, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Zhou, D.-J.; Cui, Z.-G.; Sun, L.; Feng, Q.-W.; Zakki, S.A.; Hiraku, Y.; Wu, C.-A.; Inadera, H. The molecular mechanism of a novel derivative of BTO-956 induced apoptosis in human myelomonocytic lymphoma cells. Apoptosis 2021, 26, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Zhuang, Y.; Fu, J.; Li, X.; Shi, Q.; Ju, X. Exosomes derived from bone marrow stromal cells decrease the sensitivity of leukemic cells to etoposide. Oncol. Lett. 2017, 14, 3082–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staquicini, D.I.; D’Angelo, S.; Ferrara, F.; Karjalainen, K.; Sharma, G.; Smith, T.L.; Tarleton, C.A.; E. Jaalouk, D.; Kuniyasu, A.; Baze, W.B.; et al. Therapeutic targeting of membrane-associated GRP78 in leukemia and lymphoma: Preclinical efficacy in vitro and formal toxicity study of BMTP-78 in rodents and primates. Pharmacogen. J. 2017, 18, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Baba, Y.; Shigemi, Z.; Hara, N.; Moriguchi, M.; Ikeda, M.; Watanabe, T.; Fujimuro, M. Arctigenin induces the apoptosis of primary effusion lymphoma cells under conditions of glucose deprivation. Int. J. Oncol. 2018, 52, 505–517. [Google Scholar] [CrossRef]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D’Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef] [Green Version]
- Chao, C.C.K.; Yam, W.C.; Chen, L.K.; Lin-Chao, S. Cloning of a functional Burkitt’s lymphoma polypeptide-binding protein/78 kDa glucose-regulated protein (BiP/GRP78) gene promoter by the polymerase chain reaction, and its interaction with inducible cellular factors. Biochem. J. 1992, 286, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Severin, F.; Frezzato, F.; Visentin, A.; Martini, V.; Trimarco, V.; Carraro, S.; Tibaldi, E.; Brunati, A.M.; Piazza, F.; Semenzato, G.; et al. In Chronic Lymphocytic Leukemia the JAK2/STAT3 Pathway Is Constitutively Activated and Its Inhibition Leads to CLL Cell Death Unaffected by the Protective Bone Marrow Microenvironment. Cancers 2019, 11, 1939. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.-W.; Chen, Y.; Liu, W.; Chen, Y.-J.; Fan, Z.-M.; Liu, M.; Li, L.-J. Inhibition of JAK2/STAT3 Signaling Pathway Suppresses Proliferation of Burkitt’s Lymphoma Raji Cells via Cell Cycle Progression, Apoptosis, and Oxidative Stress by Modulating HSP70. Med. Sci. Monit. 2018, 24, 6255. [Google Scholar] [CrossRef]
- Yan, Y.; Gao, Y.-Y.; Liu, B.-Q.; Niu, X.-F.; Zhuang, Y.; Wang, H.-Q. Resveratrol-induced cytotoxicity in human Burkitt’s lymphoma cells is coupled to the unfolded protein response. BMC Cancer 2010, 10, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aissani, B.; Martinez-Maza, O.; Kaslow, R.A.; Wiener, H.W.; Bream, J.H.; Stosor, V.; Martinson, J.J.; Jacobson, L.P.; Shrestha, S. Increasing Levels of Serum Heat Shock Protein 70 Precede the Development of AIDS-Defining Non-Hodgkin Lymphoma Among Carriers of HLA-B8-DR3. J. Acquir. Immune Defic. Syndr. 2019, 81, 266. [Google Scholar] [CrossRef] [PubMed]
- Bonvini, P.; Rosa, H.D.; Vignes, N.; Rosolen, A. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: Role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res. 2004, 64, 3256–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonvini, P.; Zorzi, E.; Mussolin, L.; Pillon, M.; Romualdi, C.; Peron, M.; D’Amore, E.S.G.; Lamant, L.; Rosolen, A. Consequences of heat shock protein 72 (Hsp72) expression and activity on stress-induced apoptosis in CD30+ NPM–ALK+ anaplastic large-cell lymphomas. Leukemia 2012, 26, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zhao, Z.; Menke, D.M.; Rizzo, K.A. Correlation of BAG-3 and Heat Shock Protein 70 with CD30 expression in T-cell Lymphomas. Sci. Rep. 2014, 4, 3952. [Google Scholar] [CrossRef] [Green Version]
- Fujii, K.; Idogawa, M.; Suzuki, N.; Iwatsuki, K.; Kanekura, T. Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78. Int. J. Mol. Sci. 2021, 22, 11258. [Google Scholar] [CrossRef]
- Fujii, K.; Suzuki, N.; Ikeda, K.; Hamada, T.; Yamamoto, T.; Kondo, T.; Iwatsuki, K. Proteomic study identified HSP 70kDa protein 1A as a possible therapeutic target, in combination with histone deacetylase inhibitors, for lymphoid neoplasms. J. Proteom. 2012, 75, 1401–1410. [Google Scholar] [CrossRef]
- Gaudio, E.; Paduano, F.; Ngankeu, A.; Lovat, F.; Fabbri, M.; Sun, H.-L.; Gasparini, P.; Efanov, A.; Peng, Y.; Zanesi, N.; et al. Heat shock protein 70 regulates Tcl1 expression in leukemia and lymphomas. Blood 2013, 121, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Baglietto, L.; Qiao, Y.; Liu, B.; Laska, E.J.; Chakravarthi, P.; Kulko, J.M.; Bona, R.D.; Fang, M.; Hegde, U.; Moyo, V.; et al. Combination of Imatinib Mesylate with Autologous Leukocyte-Derived Heat Shock Protein and Chronic Myelogenous Leukemia. Clin. Cancer Res. 2005, 11, 4460–4468. [Google Scholar] [CrossRef] [Green Version]
- Fredly, H.; Reikvam, H.; Gjertsen, B.T.; Bruserud, Ø. Disease-stabilizing treatment with all-trans retinoic acid and valproic acid in acute myeloid leukemia: Serum hsp70 and hsp90 levels and serum cytokine profiles are determined by the disease, patient age, and anti-leukemic treatment. Am. J. Hematol. 2012, 87, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Ryningen, A.; Stapnes, C.; Lassalle, P.; Corbascio, M.; Gjertsen, B.-T.; Bruserud, Ø. A subset of patients with high-risk acute myelogenous leukemia shows improved peripheral blood cell counts when treated with the combination of valproic acid, theophylline and all-trans retinoic acid. Leuk. Res. 2009, 33, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Ryningen, A.; Stapnes, C.; Paulsen, K.; Lassalle, P.; Gjertsen, B.T.; Bruserud, Ø. In vivo biological effects of ATRA in the treatment of AML. Expert Opin. Investig. Drugs 2008, 17, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Fredly, H.; Ersvær, E.; Kittang, A.O.; Tsykunova, G.; Gjertsen, B.T.; Bruserud, Ø. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin. Epigenet. 2013, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, S.H.; Karp, J.E.; Litzow, M.R.; Mesa, R.A.; Hogan, W.; Steensma, D.P.; Flatten, K.S.; Loegering, D.A.; Schneider, P.A.; Peterson, K.L.; et al. Phase I and pharmacological study of cytarabine and tanespimycin in relapsed and refractory acute leukemia. Haematologica 2011, 96, 1619–1626. [Google Scholar] [CrossRef] [Green Version]
- Yong, K.; Cavet, J.; Johnson, P.; Morgan, G.; Williams, C.; Nakashima, D.; Akinaga, S.; Oakervee, H.; Cavenagh, J. Phase I study of KW-2478, a novel Hsp90 inhibitor, in patients with B-cell malignancies. Br. J. Cancer 2016, 114, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Babi, A.; Menlibayeva, K.; Bex, T.; Doskaliev, A.; Akshulakov, S.; Shevtsov, M. Targeting Heat Shock Proteins in Malignant Brain Tumors: From Basic Research to Clinical Trials. Cancers 2022, 14, 5435. [Google Scholar] [CrossRef]
- Specht, H.M.; Ahrens, N.; Blankenstein, C.; Duell, T.; Fietkau, R.; Gaipl, U.S.; Günther, C.; Gunther, S.; Habl, G.; Hautmann, H.; et al. Heat Shock Protein 70 (Hsp70) Peptide Activated Natural Killer (NK) Cells for the Treatment of Patients with Non-Small Cell Lung Cancer (NSCLC) after Radiochemotherapy (RCTx)—From Preclinical Studies to a Clinical Phase II Trial. Front. Immunol. 2015, 6, 162. [Google Scholar] [CrossRef] [Green Version]
- Javid, H.; Hashemian, P.; Yazdani, S.; Mashhad, A.S.; Karimi-Shahri, M. The role of heat shock proteins in metastatic colorectal cancer: A review. J. Cell. Biochem. 2022, 123, 1704–1735. [Google Scholar] [CrossRef]
- Fu, X.; Liu, J.; Yan, X.; DiSanto, M.E.; Zhang, X. Heat Shock Protein 70 and 90 Family in Prostate Cancer. Life 2022, 12, 1489. [Google Scholar] [CrossRef]
- Sabbadini, R.; Pesce, E.; Parodi, A.; Mustorgi, E.; Bruzzone, S.; Pedemonte, N.; Casale, M.; Millo, E.; Cichero, E. Probing Allosteric Hsp70 Inhibitors by Molecular Modelling Studies to Expedite the Development of Novel Combined F508del CFTR Modulators. Pharmaceuticals 2021, 14, 1296. [Google Scholar] [CrossRef]
- Liso, A.; Benedetti, R.; Fagioli, M.; Mariano, A.; Falini, B. Modulatory effects of mycobacterial heat-shock protein 70 in DNA vaccination against lymphoma. Haematologica 2005, 90, 60–65. [Google Scholar] [PubMed]
- Juric, M.K.; Shevtsov, M.; Mozes, P.; Ogonek, J.; Crossland, R.E.; Dickinson, A.M.; Greinix, H.T.; Holler, E.; Weissinger, E.M.; Multhoff, G. B-Cell-Based and Soluble Biomarkers in Body Liquids for Predicting Acute/Chronic Graft-versus-Host Disease after Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2017, 7, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| CHROMOSOME | GENE | PROTEIN (and Alternative Names) | LOCALIZATION | INDUCIBLE |
|---|---|---|---|---|
| 6p21.3 | HSPA1A | HSPA1A, HSP70-1, HSP72, HSPA1, HSP70-1A, HSP70i | Cytosol, nucleus, cell. membrane, exosomes | Yes |
| 6p21.3 | HSPA1B | HSPA1B, HSP70-2, HSP70-1B | Cytosol, nucleus, exosomes | Yes |
| 6p21.3 | HSPA1L | HSPA1L, HSP70-1L, Hsp-hom, HSP70-1t, hum70t | Cytosol, nucleus | No |
| 14q24.1 | HSPA2 | HSPA2, Heat shock 70kD protein-2, HSP70.2 | Cytosol, nucleus, cell. membrane, exosomes | No |
| 9q33.3 | HSPA5 | HSPA5, HSP70-5, BIP, GRP78, MIF2 | ER, exosomes | No |
| 1q23 | HSPA6 | HSPA6, HSP70-6, Heat shock 70KD protein 6 (HSP70B’) | Cytosol, exosomes | Yes |
| 1q23.3 | HSPA7 | HSPA7, HSP70-7, Heat shock 70KD protein 7 (HSP70B) | Blood microparticles, exosomes | Yes |
| 11q24.1 | HSPA8 | HSPA8, Hsp70-8, HSC70, HSC71, HSP71, HSP73 | Cytosol, nucleus, cell. membrane, exosomes | No |
| 5q31.1 | HSPA9 | HSPA9, HSP70-9, GRP75, HSPA9B, MOT, MOT2, PBP74, mot-2, mtHSP70, mortalin | Mitochondria, nucleus | No |
| 10q26.12 | HSPA12A | HSPA12A, HSP70-12A, FLJ13874, KIAA0417 | Intracellular, exosomes | No |
| 20p13 | HSPA12B | HSPA12B, HSP70-12B, RP23-32L15.1, 2700081N06RIK | Endothelial cells, intracellular, plasma | No |
| 21q11 | HSPA13 | HSPA13, HSP70-13, Stch | ER, exosomes, microsomes | No |
| 10p13 | HSPA14 | HSPA14, HSP70-14, HSP70L1, MCG131990 | Cytosol, cell. membrane | Yes |
| CT.gov Identifier | Study Phase | Disease | Status | Aims | Location | Publication |
|---|---|---|---|---|---|---|
| NCT00030303 # | I | CML | C | Determine the feasibility and toxicity of vaccination with autologous HSP70 in patients with chronic phase CML. | US | [160] |
| NCT00027144 # | I | CML | C | Determine the response of the immune system of patients with CML to a vaccine made from their own tumor. | US | NA |
| NCT00058747 # | II | CML | T | Investigate AG-858 (Autologous HSP70 Protein-Peptide Complex), in CML patients who are cytogenetically positive after treatment with Gleevec. | US/UK | NA |
| NCT00096005 # | I | Lymphoma and Solid tumors | T | Determine the dose-limiting toxicity and maximum tolerated dose of 17-AAG and bortezomib in patients with advanced solid tumors or lymphomas and determine changes in biomarkers (e.g., HSP70) | US | NA |
| NCT00957736 # | I | cGVHD in patients after donor stem cell transplant | T | Determine and define the biological basis of different subtypes of cGVHD using a targeted SNP (including HSP70) approach. | US | NA |
| NCT00175812 * | I/II | AML | C | Primary outcome measure: survival. | Norway | [161,162,163] |
| NCT00995332 * | I/II | AML | C | Determine survival as primary outcome measure. | Norway | [161,164] |
| NCT00098423 * | I | R/R Acute and Chronic Leukemias | C | Determine the maximum tolerated dose and toxicity of 17-AAG when administered with cytarabine and determine the effect of this regimen on client proteins in vivo and ex vivo in leukemic blasts. | US | [165] |
| NCT00457782 * | I | MM, CLL and NHLs | C | Determine the safety, tolerability and dose-limiting toxicities of KW-2478 and determine the maximum tolerated dose and recommended Phase 2 dose for patients. | UK | [166] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouawad, N.; Capasso, G.; Ruggeri, E.; Martinello, L.; Severin, F.; Visentin, A.; Facco, M.; Trentin, L.; Frezzato, F. Is It Still Possible to Think about HSP70 as a Therapeutic Target in Onco-Hematological Diseases? Biomolecules 2023, 13, 604. https://doi.org/10.3390/biom13040604
Mouawad N, Capasso G, Ruggeri E, Martinello L, Severin F, Visentin A, Facco M, Trentin L, Frezzato F. Is It Still Possible to Think about HSP70 as a Therapeutic Target in Onco-Hematological Diseases? Biomolecules. 2023; 13(4):604. https://doi.org/10.3390/biom13040604
Chicago/Turabian StyleMouawad, Nayla, Guido Capasso, Edoardo Ruggeri, Leonardo Martinello, Filippo Severin, Andrea Visentin, Monica Facco, Livio Trentin, and Federica Frezzato. 2023. "Is It Still Possible to Think about HSP70 as a Therapeutic Target in Onco-Hematological Diseases?" Biomolecules 13, no. 4: 604. https://doi.org/10.3390/biom13040604