
Ablation of GPR56 Causes β-Cell Dysfunction by ATP Loss through Mistargeting of Mitochondrial VDAC1 to the Plasma Membrane

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. INS-1 832/13 Cell Culture
2.4. EndoC βH1 Cell Culture
2.5. Knockdown of GPR56 in Mouse Islets, INS-1 832/13, and EndoC βH1 Cells
2.6. Determination of Intracellular Pathways
2.7. Immunostaining and Confocal Imaging
2.8. Quantitative Polymerase Chain Reaction (qPCR)
2.9. Western Blots
2.10. Insulin Secretion
2.11. cAMP Determination
2.12. ATP Determination
2.13. Statistics
3. Results
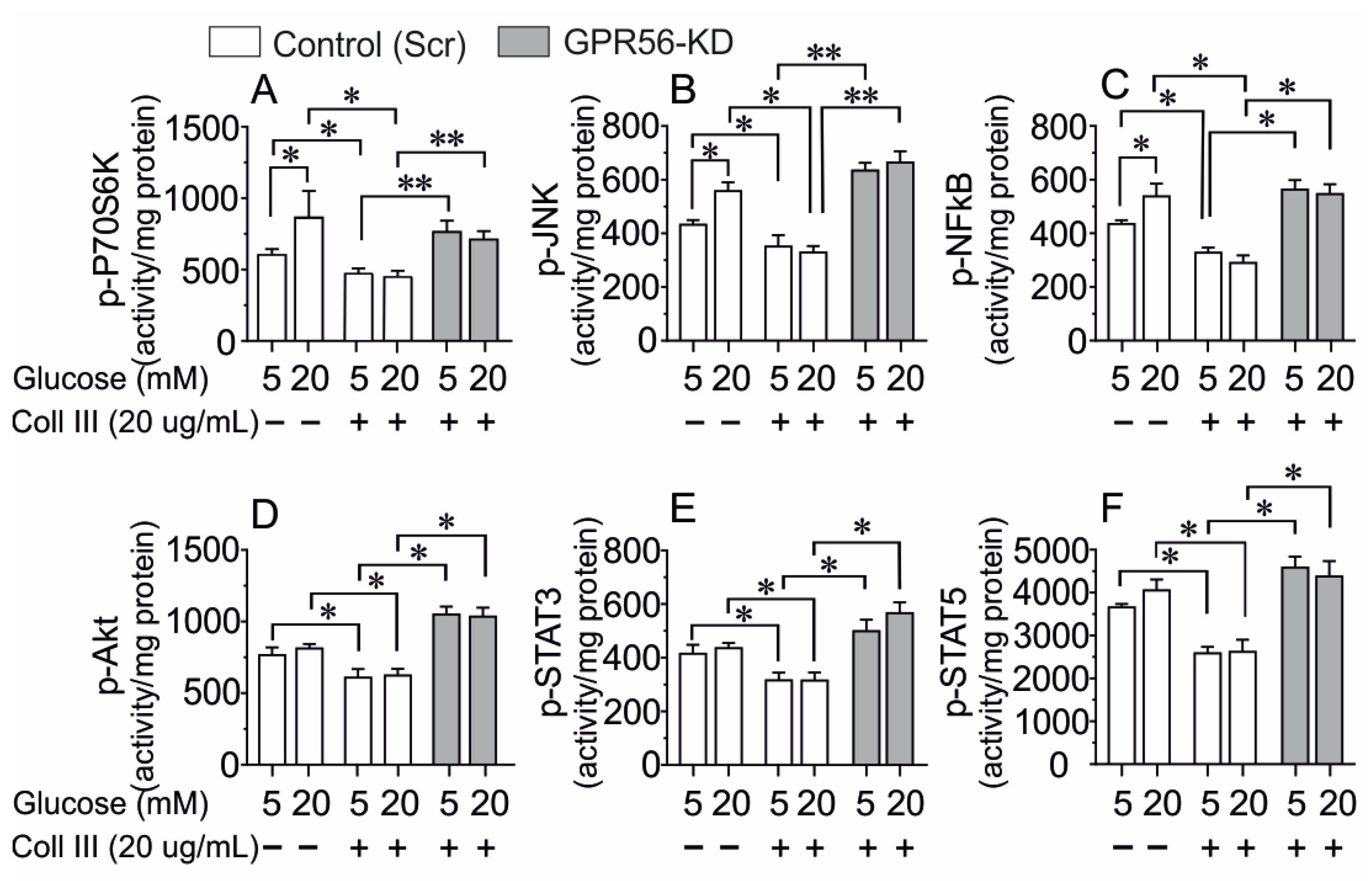
3.1. The Consequence of GPR56-KD on the Activation of Several Intracellular Pathways
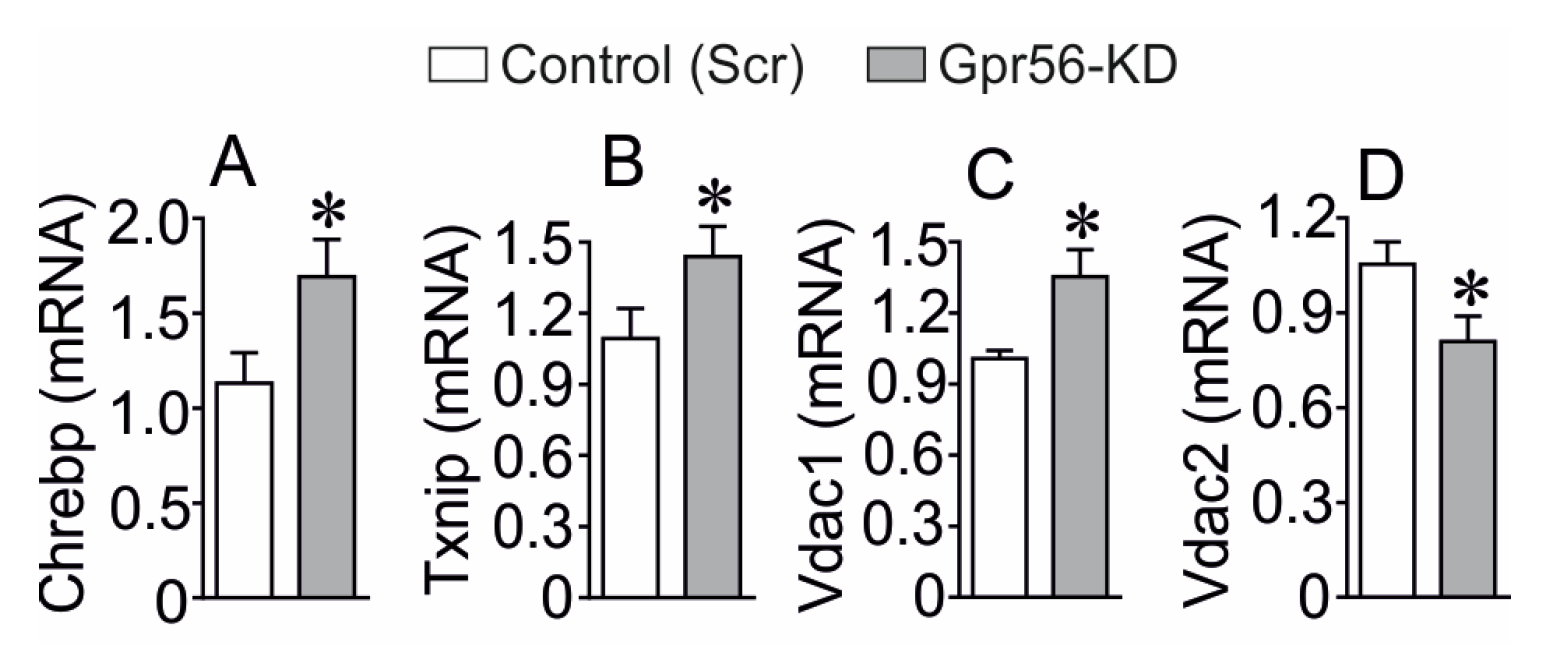
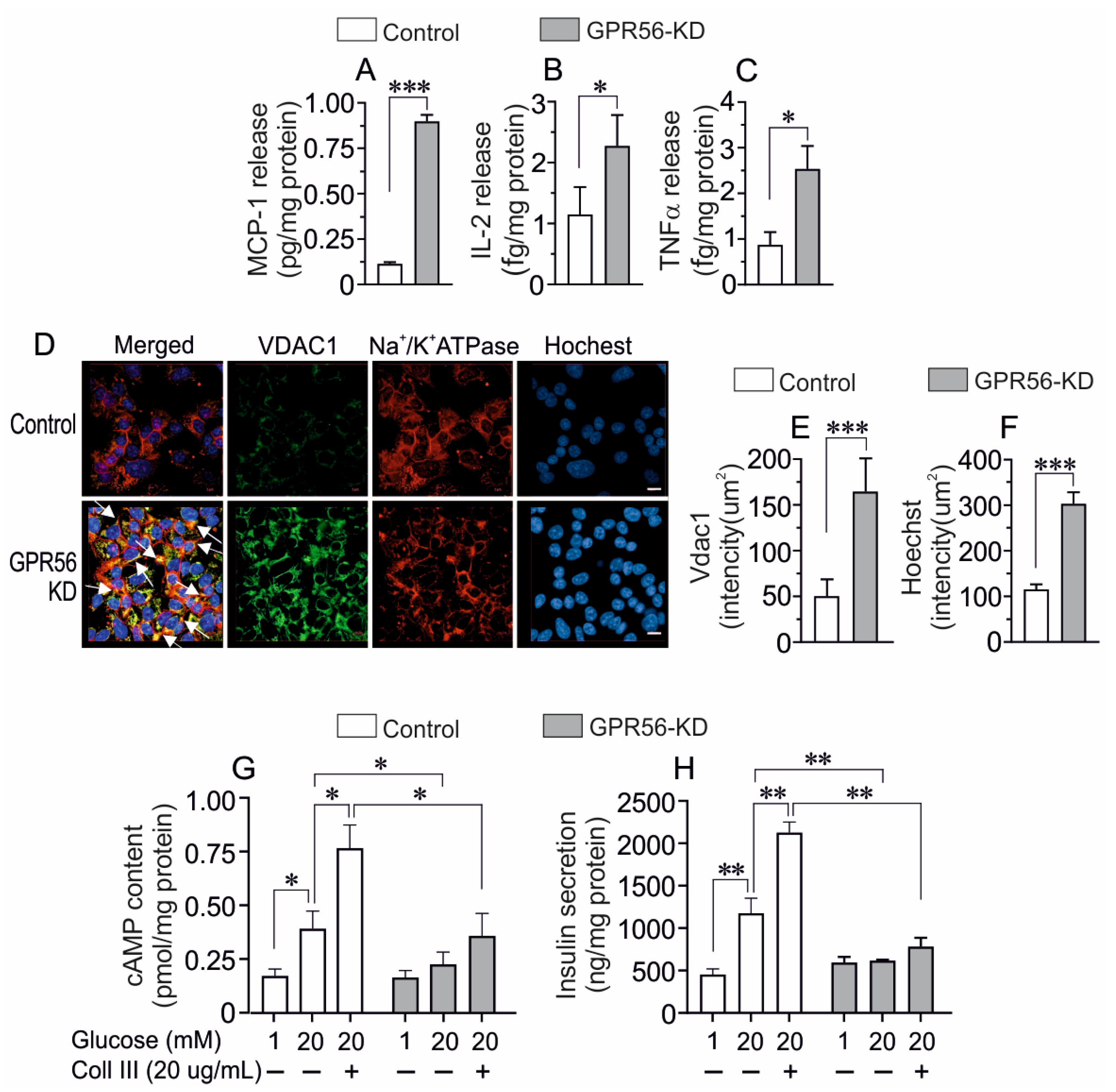
3.2. The Effect of GPR56-KD on the Expression of Chrebp, Txnip, Vdac1 and Vdac2
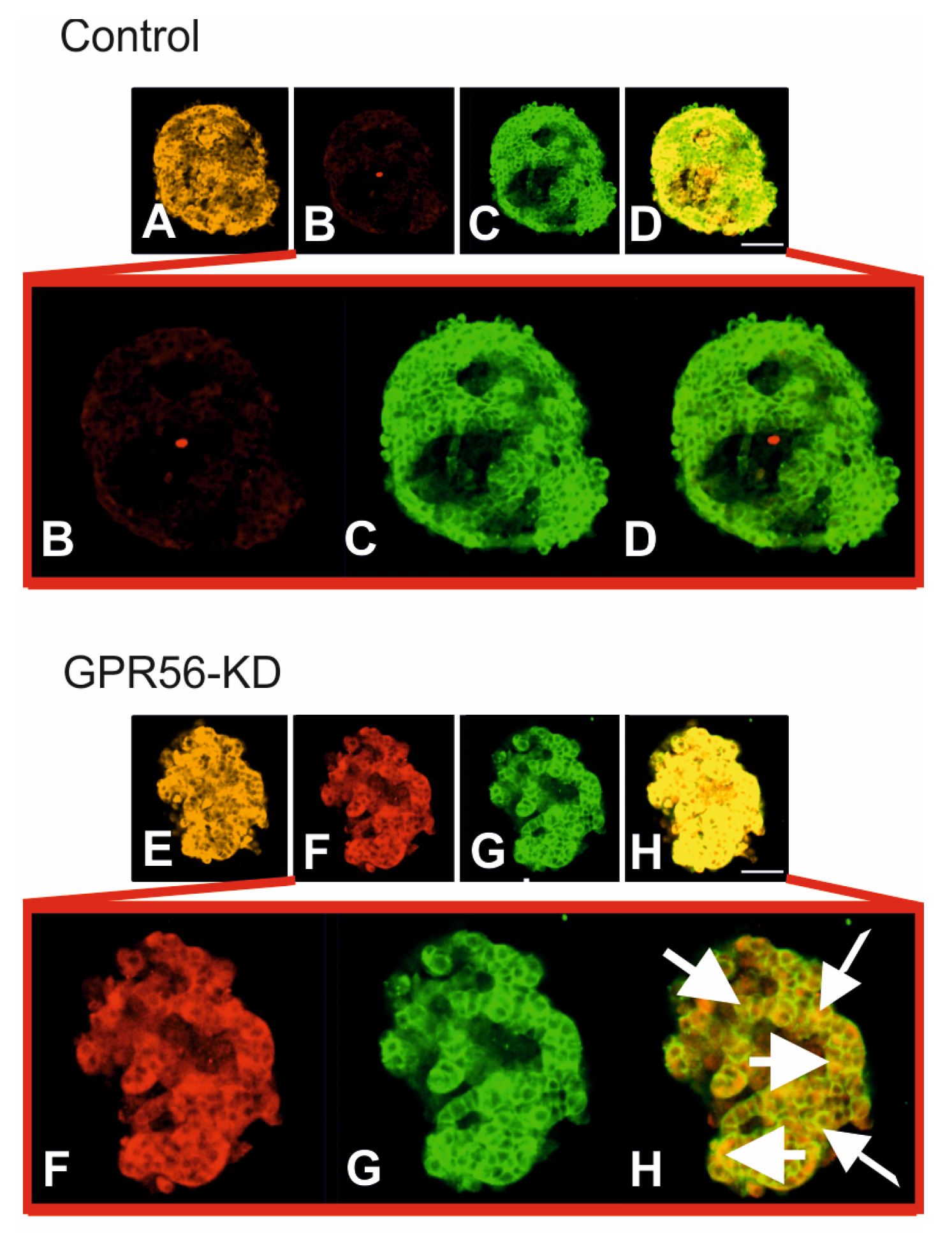
3.3. The Impact of Gpr56-KD on the Vdac1 Protein Expression and its Mistargeting to the Cell Surface in Mouse Islets
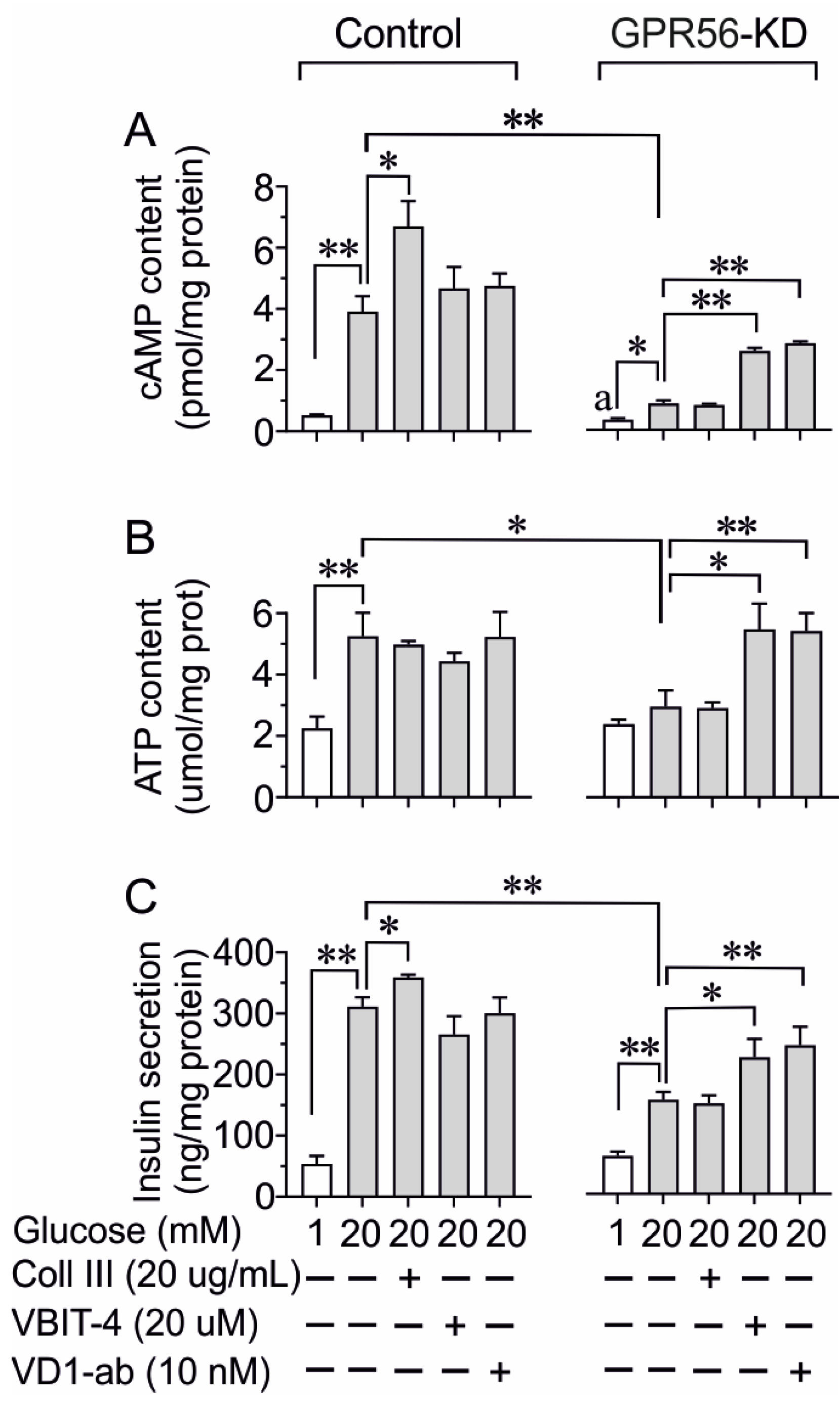
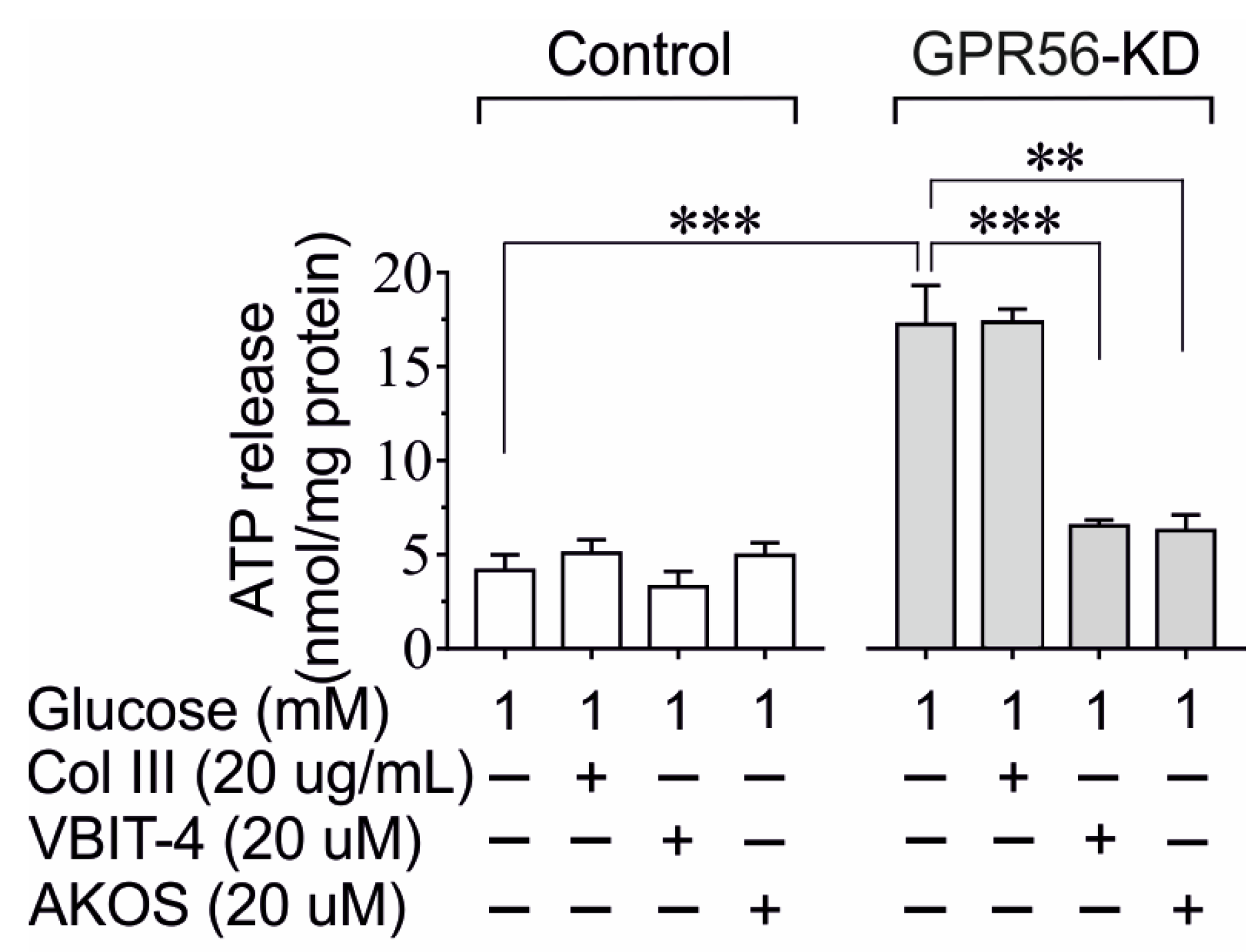
3.4. The Impact of Gpr56-KD on cAMP, ATP Content, and Insulin Secretion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53 (Suppl. 3), S16–S21. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.; Bain, S.; Kanamarlapudi, V. A Review of Current Trends with Type 2 Diabetes Epidemiology, Aetiology, Pathogenesis, Treatments and Future Perspectives. Diabetes Metab. Syndr. Obes. 2021, 14, 3567–3602. [Google Scholar] [CrossRef]
- Wigger, L.; Barovic, M.; Brunner, A.D.; Marzetta, F.; Schöniger, E.; Mehl, F.; Kipke, N.; Friedland, D.; Burdet, F.; Kessler, C.; et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab. 2021, 3, 1017–1031. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, Y.; Meng, Z.X. Transcriptional control of pancreatic beta-cell identity and plasticity during the pathogenesis of type 2 diabetes. J. Genet. Genom. 2022, 49, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Wajchenberg, B.L. Beta-cell failure in diabetes and preservation by clinical treatment. Endocr. Rev. 2007, 28, 187–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retnakaran, R.; Choi, H.; Ye, C.; Kramer, C.K.; Zinman, B. Two-year trial of intermittent insulin therapy vs metformin for the preservation of beta-cell function after initial short-term intensive insulin induction in early type 2 diabetes. Diabetes Obes. Metab. 2018, 20, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Wollheim, C.B. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol. Cell. Endocrinol. 2012, 353, 128–137. [Google Scholar] [CrossRef]
- Las, G.; Oliveira, M.F.; Shirihai, O.S. Emerging roles of beta-cell mitochondria in type-2-diabetes. Mol. Aspects Med. 2020, 71, 100843. [Google Scholar] [CrossRef]
- Madec, A.M.; Perrier, J.; Panthu, B.; Dingreville, F. Role of mitochondria-associated endoplasmic reticulum membrane (MAMs) interactions and calcium exchange in the development of type 2 diabetes. Int. Rev. Cell. Mol. Biol. 2021, 363, 169–202. [Google Scholar]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- Raghavan, A.; Sheiko, T.; Graham, B.H.; Craigen, W.J. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. Biochim. Biophys. Acta 2012, 1818, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Muhammed, S.J.; Kessler, B.; Salehi, A. Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic beta-cells exposed to high glucose. Islets 2010, 2, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, E.; Al-Amily, I.M.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in beta Cells. Cell. Metab. 2019, 29, 64–77.e6. [Google Scholar] [CrossRef]
- Carvalho, D.S.; de Almeida, A.A.; Borges, A.F.; Campos, D.V. Treatments for diabetes mellitus type II: New perspectives regarding the possible role of calcium and cAMP interaction. Eur. J. Pharmacol. 2018, 830, 9–16. [Google Scholar] [CrossRef]
- Amisten, S.; Salehi, A.; Rorsman, P.; Jones, P.M.; Persaud, S.J. An atlas and functional analysis of G-protein coupled receptors in human islets of Langerhans. Pharmacol. Ther. 2013, 139, 359–391. [Google Scholar] [CrossRef]
- Amisten, S.; Neville, M.; Hawkes, R.; Persaud, S.J.; Karpe, F.; Salehi, A. An atlas of G-protein coupled receptor expression and function in human subcutaneous adipose tissue. Pharmacol. Ther. 2015, 146, 61–93. [Google Scholar] [CrossRef]
- Duner, P.; Al-Amily, I.M.; Soni, A.; Asplund, O.; Safi, F.; Storm, P.; Groop, L.; Amisten, S. Adhesion G Protein-Coupled Receptor G1 (ADGRG1/GPR56) and Pancreatic beta-Cell Function. J. Clin. Endocrinol. Metab. 2016, 101, 4637–4645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaniru, O.E.; Pingitore, A.; Giera, S.; Piao, X.; Gonzalez, R.C.; Jones, P.M.; Persaud, S.J. The adhesion receptor GPR56 is activated by extracellular matrix collagen III to improve beta-cell function. Cell. Mol. Life Sci. 2018, 75, 4007–4019. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Lin, H.H. The role of GPR56/ADGRG1 in health and disease. Biomed. J. 2021, 44, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Guan, Q.; Cheng, H.; Zhu, Z.; Jiang, C.; Guo, P.; Tai, Y.; Sun, H.; Wang, M.; Wei, W.; et al. Functions of G protein-coupled receptor 56 in health and disease. Acta Physiol. 2022, 236, e13866. [Google Scholar] [CrossRef]
- Giera, S.; Deng, Y.; Luo, R.; Ackerman, S.D.; Mogha, A.; Monk, K.R.; Ying, Y.; Jeong, S.J.; Makinodan, M.; Bialas, A.R.; et al. The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat. Commun. 2015, 6, 6121. [Google Scholar] [CrossRef] [Green Version]
- Piao, X.; Hill, R.S.; Bodell, A.; Chang, B.S.; Basel-Vanagaite, L.; Straussberg, R.; Dobyns, W.B.; Qasrawi, B.; Winter, R.M.; Innes, A.M.; et al. G protein-coupled receptor-dependent development of human frontal cortex. Science 2004, 303, 2033–2036. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.P.; Dagar, M.; Dagar, G.; Kumar, S.; Rawal, S.; Sharma, R.D.; Tyagi, R.K.; Bagchi, G. Activation of GPR56, a novel adhesion GPCR, is necessary for nuclear androgen receptor signaling in prostate cells. PLoS ONE 2020, 15, e0226056. [Google Scholar] [CrossRef] [PubMed]
- Olaniru, O.E.; Cheng, J.; Ast, J.; Arvaniti, A.; Atanes, P.; Huang, G.C.; King, A.J.; Jones, P.M.; Broichhagen, J.; Hodson, D.J.; et al. SNAP-tag-enabled super-resolution imaging reveals constitutive and agonist-dependent trafficking of GPR56 in pancreatic beta-cells. Mol. Metab. 2021, 53, 101285. [Google Scholar] [CrossRef] [PubMed]
- Raoux, M.; Vacher, P.; Papin, J.; Picard, A.; Kostrzewa, E.; Devin, A.; Gaitan, J.; Limon, I.; Kas, M.J.; Magnan, C.; et al. Multilevel control of glucose homeostasis by adenylyl cyclase 8. Diabetologia 2015, 58, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Al-Amily, I.M.; Duner, P.; Groop, L.; Salehi, A. The functional impact of G protein-coupled receptor 142 (Gpr142) on pancreatic beta-cell in rodent. Pflugers Arch. 2019, 471, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Amisten, S.; Atanes, P.; Hawkes, R.; Ruz-Maldonado, I.; Liu, B.; Parandeh, F.; Zhao, M.; Huang, G.C.; Salehi, A.; Persaud, S.J. A comparative analysis of human and mouse islet G-protein coupled receptor expression. Sci. Rep. 2017, 7, 46600. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Amisten, S.; Rorsman, P.; Salehi, A. GPRC5B a putative glutamate-receptor candidate is negative modulator of insulin secretion. Biochem. Biophys. Res. Commun. 2013, 441, 643–648. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [PubMed] [Green Version]
- Majtnerova, P.; Capek, J.; Petira, F.; Handl, J.; Rousar, T. Quantitative spectrofluorometric assay detecting nuclear condensation and fragmentation in intact cells. Sci. Rep. 2021, 11, 11921. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Henry, T.A.N.; Pronin, A.N.; Jang, G.F.; Lubaczeuski, C.; Crabb, J.W.; Bernal-Mizrachi, E.; Slepak, V.Z. The regulatory G protein signaling complex, Gbeta5-R7, promotes glucose- and extracellular signal-stimulated insulin secretion. J. Biol. Chem. 2020, 295, 7213–7223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Nwe, P.K.; Yang, Y.; Rosen, C.E.; Bielecka, A.A.; Kuchroo, M.; Cline, G.W.; Kruse, A.C.; Ring, A.M.; Crawford, J.M.; et al. A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell. 2019, 177, 1217–1231.e18. [Google Scholar] [CrossRef]
- Lin, H.V.; Efanov, A.M.; Fang, X.; Beavers, L.S.; Wang, X.; Wang, J.; Valcarcel, I.C.G.; Ma, T. GPR142 Controls Tryptophan-Induced Insulin and Incretin Hormone Secretion to Improve Glucose Metabolism. PLoS ONE 2016, 11, e0157298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, K.D.; Hemler, M.E.; Stipp, C.S. Dynamic regulation of a GPCR-tetraspanin-G protein complex on intact cells: Central role of CD81 in facilitating GPR56-Galpha q/11 association. Mol. Biol. Cell. 2004, 15, 2375–2387. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Chang, G.W.; Hsiao, C.C.; Peng, Y.M.; Braga, F.A.V.; Kragten, N.A.; Remmerswaal, E.B.; van de Garde, M.D.; Straussberg, R.; König, G.M.; Kostenis, E.; et al. The Adhesion G Protein-Coupled Receptor GPR56/ADGRG1 Is an Inhibitory Receptor on Human NK Cells. Cell. Rep. 2016, 15, 1757–1770. [Google Scholar] [CrossRef] [Green Version]
- Banes-Berceli, A.K.; Ketsawatsomkron, P.; Ogbi, S.; Patel, B.; Pollock, D.M.; Marrero, M.B. Angiotensin II and endothelin-1 augment the vascular complications of diabetes via JAK2 activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1291–H1299. [Google Scholar] [CrossRef] [Green Version]
- Shalev, A. Minireview: Thioredoxin-interacting protein: Regulation and function in the pancreatic beta-cell. Mol. Endocrinol. 2014, 28, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Poungvarin, N.; Lee, J.K.; Yechoor, V.K.; Li, M.V.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.J.; Saha, P.K.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Yu, Z.; Fantus, I.G.; Jin, T. Cyclic AMP signaling stimulates proteasome degradation of thioredoxin interacting protein (TxNIP) in pancreatic beta-cells. Cell. Signal. 2010, 22, 1240–1246. [Google Scholar] [CrossRef]
- Takahashi, N.; Kadowaki, T.; Yazaki, Y.; Ellis-Davies, G.C.; Miyashita, Y.; Kasai, H. Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc. Natl. Acad. Sci. USA 1999, 96, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pinto, V.; Messina, A.; Lane, D.J.; Lawen, A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 2010, 584, 1793–1799. [Google Scholar]
- Akanda, N.; Tofighi, R.; Brask, J.; Tamm, C.; Elinder, F.; Ceccatelli, S. Voltage-dependent anion channels (VDAC) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell. Cycle 2008, 7, 3225–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, S.F.; O’Neal, W.K.; Huang, P.; Nicholas, R.A.; Ostrowski, L.E.; Craigen, W.J.; Lazarowski, E.R.; Boucher, R.C. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 2004, 124, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. Int. J. Mol. Sci. 2022, 23, 7722. [Google Scholar] [CrossRef]
- Verma, A.; Shteinfer-Kuzmine, A.; Kamenetsky, N.; Pittala, S.; Paul, A.; Nahon Crystal, E.; Ouro, A.; Chalifa-Caspi, V.; Pandey, S.K.; Monsengo, A.; et al. Targeting the overexpressed mitochondrial protein VDAC1 in a mouse model of Alzheimer’s disease protects against mitochondrial dysfunction and mitigates brain pathology. Transl. Neurodegener. 2022, 11, 58. [Google Scholar] [CrossRef]
- Midha, A.; Pan, H.; Abarca, C.; Andle, J.; Carapeto, P.; Bonner-Weir, S.; Aguayo-Mazzucato, C. Unique Human and Mouse beta-Cell Senescence-Associated Secretory Phenotype (SASP) Reveal Conserved Signaling Pathways and Heterogeneous Factors. Diabetes 2021, 70, 1098–1116. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad Al-Amily, I.; Sjögren, M.; Duner, P.; Tariq, M.; Wollheim, C.B.; Salehi, A. Ablation of GPR56 Causes β-Cell Dysfunction by ATP Loss through Mistargeting of Mitochondrial VDAC1 to the Plasma Membrane. Biomolecules 2023, 13, 557. https://doi.org/10.3390/biom13030557
Mohammad Al-Amily I, Sjögren M, Duner P, Tariq M, Wollheim CB, Salehi A. Ablation of GPR56 Causes β-Cell Dysfunction by ATP Loss through Mistargeting of Mitochondrial VDAC1 to the Plasma Membrane. Biomolecules. 2023; 13(3):557. https://doi.org/10.3390/biom13030557
Chicago/Turabian StyleMohammad Al-Amily, Israa, Marie Sjögren, Pontus Duner, Mohammad Tariq, Claes B. Wollheim, and Albert Salehi. 2023. "Ablation of GPR56 Causes β-Cell Dysfunction by ATP Loss through Mistargeting of Mitochondrial VDAC1 to the Plasma Membrane" Biomolecules 13, no. 3: 557. https://doi.org/10.3390/biom13030557