
Comparison of Biomolecular Condensate Localization and Protein Phase Separation Predictors

Abstract
:1. Introduction
2. Materials and Methods
- PScore estimates phase separation proclivity based on pi-interaction frequency under the assumption that cation–pi, pi–pi, and charge interactions promote a multivalent behavior. This machine learning model was trained to predict these interactions using over 10,000 structures from the PDB (Protein Data Bank).
- PSPredictor uses evolutionary word2vec sequence encoding with gradient boosting decision tree machine leaning algorithms to generate a prediction model that differentiates phase separating proteins using the LLPSDB dataset.
- Fuzdrop predicts droplet-promoting regions by assuming that the large conformational entropy associated with nonspecific side–chain interactions is the driving force of droplet inclusion. This model was trained on a wide range of data including in vitro and in vivo annotated membraneless organelle proteins.
- catGranule is a linear model trained on 120 granule-forming proteins from the yeast proteome. Protein features in this model largely focus on RNA-binding, protein disorder, and amino acid composition.
- PSAP uses a random forest approach to predict the probability of proteins to undergo phase separation and was trained on 90 high-confidence human phase separating proteins that act as drivers.
- PhaSePred is a meta-predictor that utilizes a XGB16oost tree model to distinguish phase separating proteins and non-phase separating controls. It was trained on 658 experimentally validated phase separating proteins across the PhaSepDB, LLPSDB, and PhaSePro databases. In the current study, we used the eight-feature sum for the generation of our data.
- DeePhase predictor of homotypic liquid–liquid phase separation identifies proteins by combining knowledge-based features with unsupervised embeddings from a pretrained word2vec model using 3-g as words and a context window size of 25.
- LLPhyScore was trained on validated phase separating proteins from the PhaSepDB, LLPSDB, and PhaSePro databases and uses 16 weighted features, including pi–pi and charge interactions.
- ParSe estimates phase separation likelihood by combining metrics for the hydrodynamic size of monomeric proteins with protein’s sequence-predicted propensity for β-turns. This model was parameterized using phase separating proteins that were collected from the PhaSePro and DISPROT databases as well as from the literature.
- MaGS and MaGSeq use enriched protein features of granule proteins to generate linear models for protein condensate localization prediction. These models were parameterized using client and scaffold proteins of stress granules listed in the DrLLPS database.
3. Results
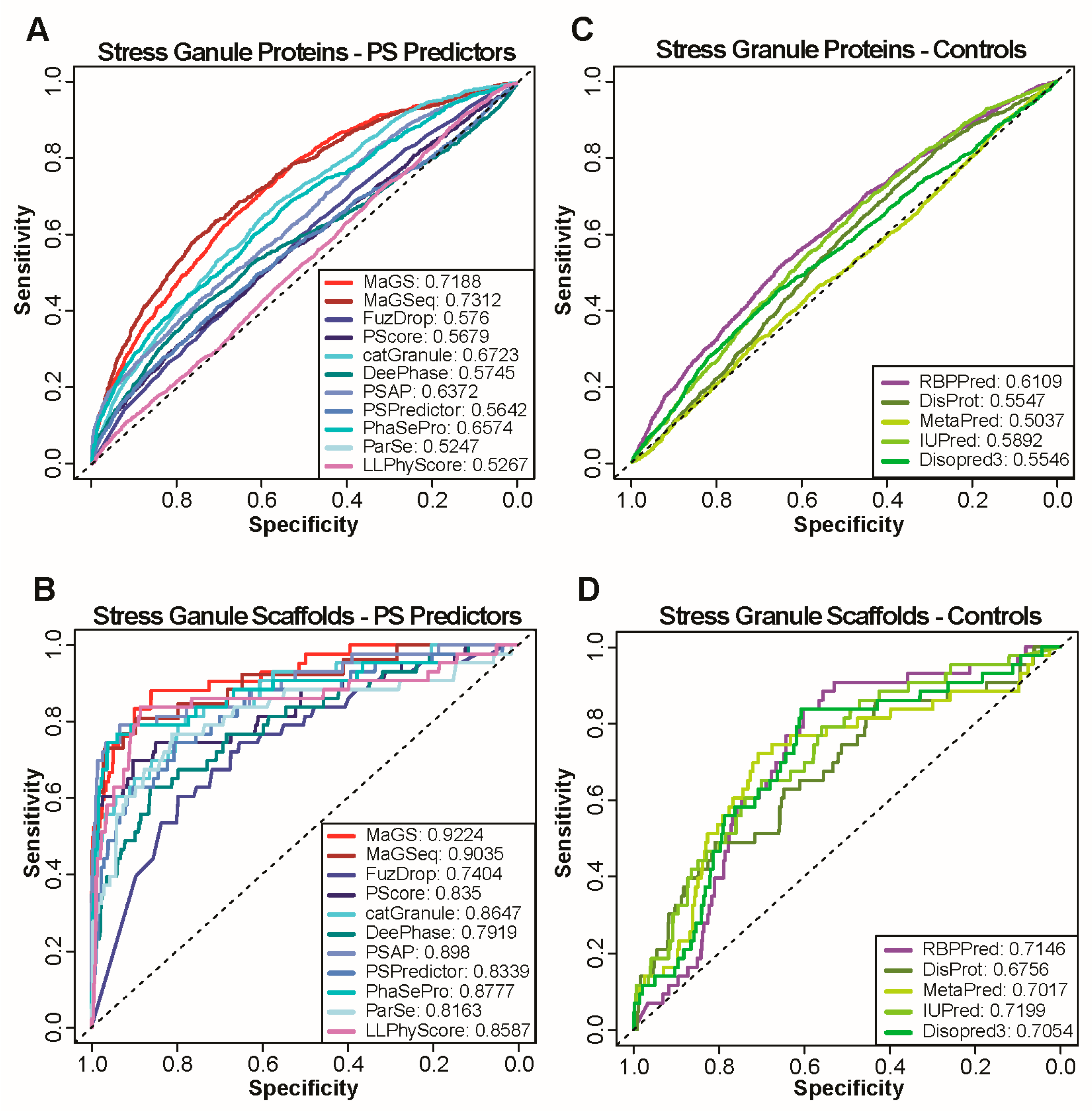
3.1. Prediction of Biomolecular Condensate Localization
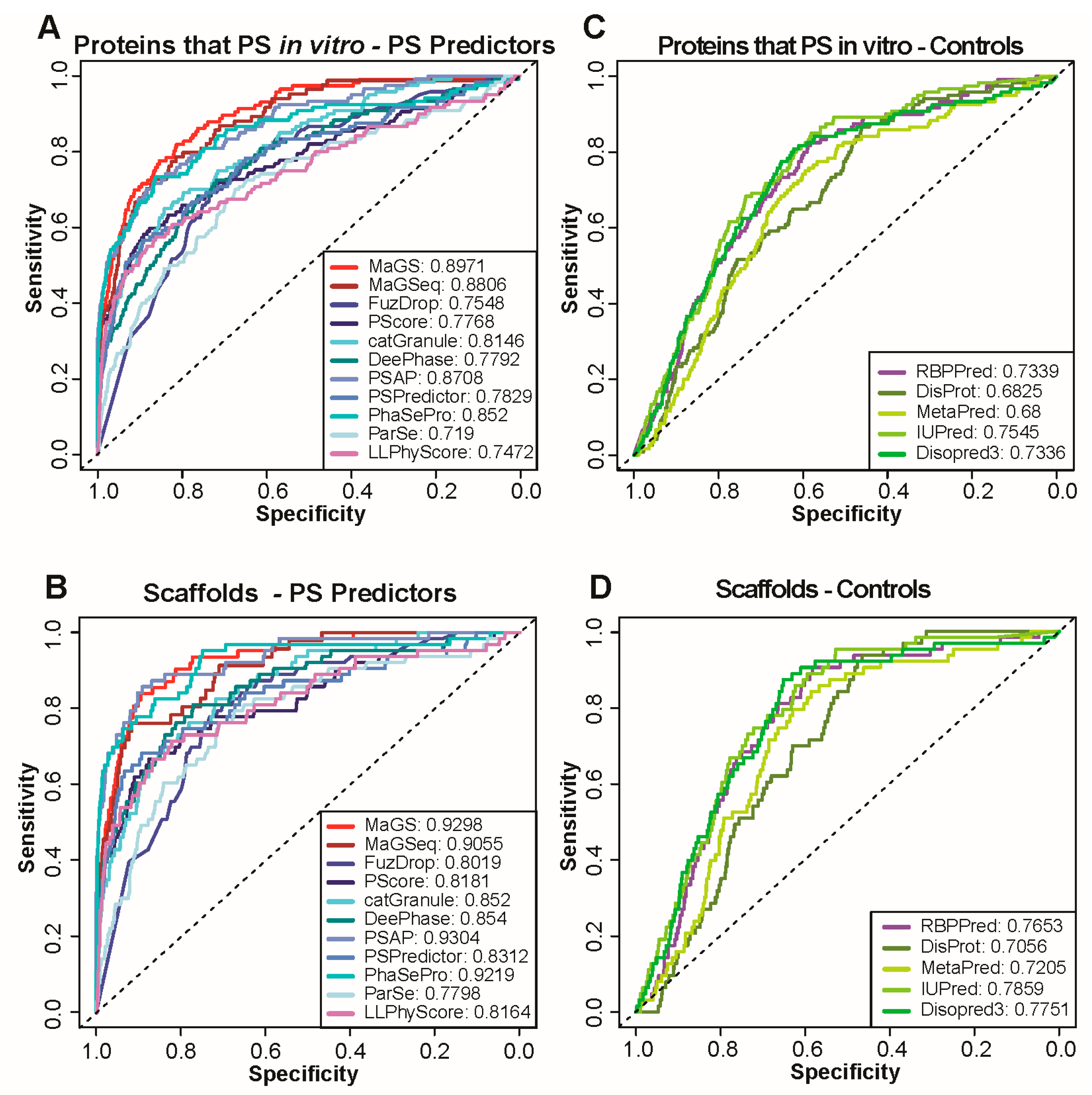
3.2. Prediction of In Vitro Phase Separation
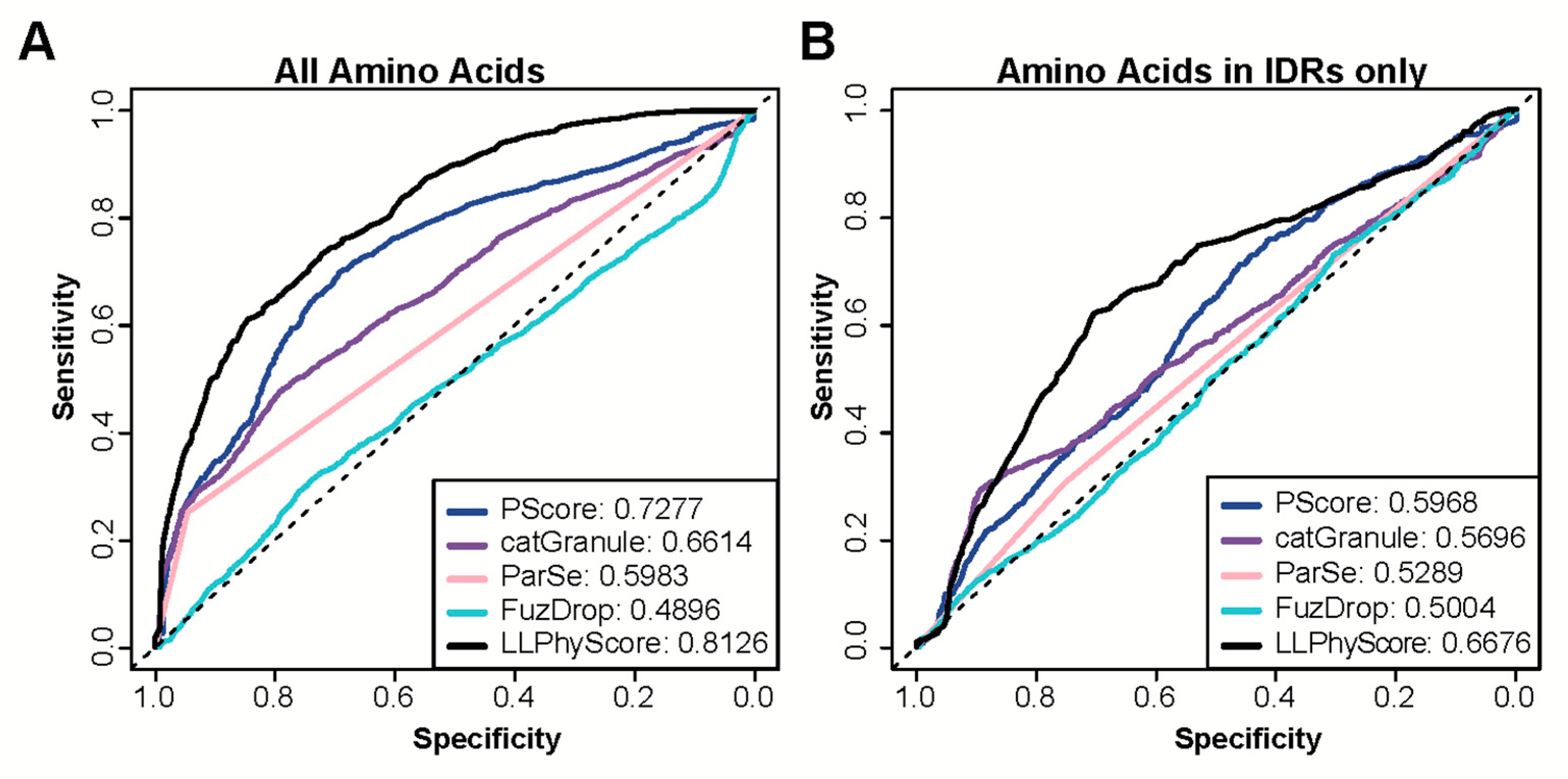
3.3. Prediction Performance of Simple Protein Features
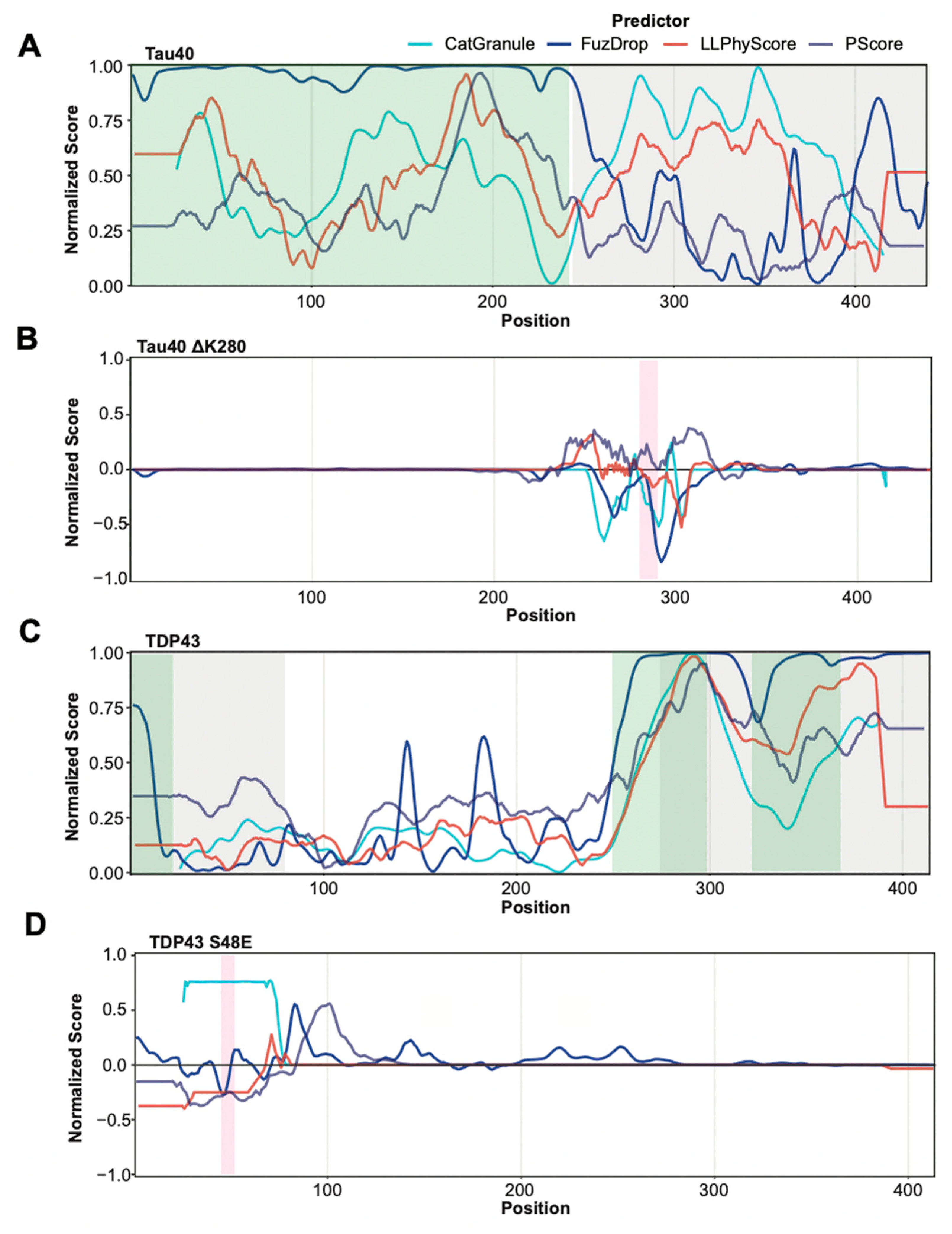
3.4. Prediction of Protein Segments That Are Involved in Phase Separation
3.5. Predicting Mutation Impact on Phase Separation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cusick, M.; Klitgord, N.; Vidal, M.; Hill, D. Interactome: Gateway into systems biology. Hum. Mol. Genet. 2005, 14, R171–R181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinnider, M.; Scott, N.; Prudova, A.; Kerr, C.; Stoynov, N.; Stacey, R.; Chan, Q.; Rattray, D.; Gsponer, J.; Foster, L. An atlas of protein-protein interactions across mouse tissues. Cell 2021, 184, 4073–4089.e17. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Alberti, S.; Fawzi, N.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.; Forman-Kay, J. Liquid-liquid phase separation in cellular signaling systems. Curr. Opin. Struct. Biol. 2016, 41, 180–186. [Google Scholar] [CrossRef]
- Banani, S.; Lee, H.; Hyman, A.; Rosen, M. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Hyman, A.; Weber, C.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [Green Version]
- Riback, J.; Katanski, C.; Kear-Scott, J.; Pilipenko, E.V.; Rojek, A.; Sosnick, T.; Drummond, D. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e19. [Google Scholar] [CrossRef] [Green Version]
- Turoverov, K.; Kuznetsova, I.; Fonin, A.V.; Darling, A.; Zaslavsky, B.; Uversky, V. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem. Sci. 2019, 44, 716–728. [Google Scholar] [CrossRef]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.; King, D.; Banani, S.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Brangwynne, C.; Eckmann, C.; Courson, D.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Weber, S. Sequence-encoded material properties dictate the structure and function of nuclear bodies. Curr. Opin. Cell Biol. 2017, 46, 62–71. [Google Scholar] [CrossRef]
- Zhu, M.; Kuechler, E.; Zhang, J.; Matalon, O.; Dubreuil, B.; Hofmann, A.; Loewen, C.; Levy, E.; Gsponer, J.; Mayor, T. Proteomic analysis reveals the direct recruitment of intrinsically disordered regions to stress granules. J. Cell Sci. 2020, 133, 13. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Dunham, W.; Hong, S.; Knight, J.; Bashkurov, M.; Chen, G.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.; Mak, R.; Jin, W.; Fang, M.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Wheeler, J.; Walters, R.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-Y.; Dyakov, B.; Zhang, J.; Knight, J.; Vernon, R.; Forman-Kay, J.; Gingras, A.-C. Properties of Stress Granule and P-Body Proteomes. Mol. Cell 2019, 76, 286–294. [Google Scholar] [CrossRef]
- Wheeler, J.; Jain, S.; Khong, A.; Parker, R. Isolation of yeast and mammalian stress granule cores. Methods 2017, 126, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Ning, W.; Guo, Y.; Lin, S.; Mei, B.; Wu, Y.; Jiang, P.; Tan, X.; Zhang, W.; Chen, G.; Peng, D.; et al. DrLLPS: A data resource of liquid-liquid phase separation in eukaryotes. Nucleic Acids Res. 2020, 48, D288–D295. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Szabó, B.; Schád, É.; Tantos, Á.; Abukhairan, R.; Horváth, T.; Murvai, N.; Kovács, O.; Kovács, M.; et al. PhaSePro: The database of proteins driving liquid–liquid phase separation. Nucleic Acids Res. 2019, 48, D360–D367. [Google Scholar] [CrossRef]
- You, K.; Huang, Q.; Yu, C.; Shen, B.; Sevilla, C.; Shi, M.; Hermjakob, H.; Chen, Y.; Li, T. PhaSepDB: A database of liquid-liquid phase separation related proteins. Nucleic Acids Res. 2020, 48, D354–D359. [Google Scholar] [CrossRef]
- Hou, C.; Wang, X.; Xie, H.; Chen, T.; Zhu, P.; Xu, X.; You, K.; Li, T. PhaSepDB in 2022: Annotating phase separation-related proteins with droplet states, co-phase separation partners and other experimental information. Nucleic Acids Res. 2023, 51, D460–D465. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, S.; Rosen, M. Using quantitative reconstitution to investigate multicomponent condensates. RNA 2022, 28, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.; Rice, A.; Peeples, W.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Brangwynne, C.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Ginell, G.; Holehouse, A. An Introduction to the Stickers-and-Spacers Framework as Applied to Biomolecular Condensates. Methods Mol. Biol. 2023, 2563, 95–116. [Google Scholar] [CrossRef]
- Cumberworth, A.; Lamour, G.; Babu, M.; Gsponer, J. Promiscuity as a functional trait: Intrinsically disordered regions as central players of interactomes. Biochem. J. 2013, 454, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Vernon, R.; Chong, P.; Tsang, B.; Kim, T.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7, 1–48. [Google Scholar] [CrossRef]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of liquid–liquid phase separating proteins using machine learning. BMC Bioinform. 2022, 23, 72. [Google Scholar] [CrossRef]
- Kuechler, E.; Jacobson, M.; Mayor, T.; Gsponer, J. GraPES: The Granule Protein Enrichment Server for prediction of biological condensate constituents. Nucleic Acids Res. 2022, 50, W384–W391. [Google Scholar] [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Bolognesi, B.; Gotor, N.L.; Dhar, R.; Cirillo, D.; Baldrighi, M.; Tartaglia, G.; Lehner, B. A Concentration-Dependent Liquid Phase Separation Can Cause Toxicity upon Increased Protein Expression. Cell Rep. 2016, 16, 222–231. [Google Scholar] [CrossRef] [Green Version]
- van Mierlo, G.; Jansen, J.; Wang, J.; Poser, I.; van Heeringen, S.; Vermeulen, M. Predicting protein condensate formation using machine learning. Cell Rep. 2021, 34, 108705. [Google Scholar] [CrossRef]
- Chen, Z.; Hou, C.; Wang, L.; Yu, C.; Chen, T.; Shen, B.; Hou, Y.; Li, P.; Li, T. Screening membraneless organelle participants with machine-learning models that integrate multimodal features. Proc. Natl. Acad. Sci. USA 2022, 119, e2115369119. [Google Scholar] [CrossRef]
- Saar, K.; Morgunov, A.; Qi, R.; Arter, W.; Krainer, G.; Lee, A.; Knowles, T. Learning the molecular grammar of protein condensates from sequence determinants and embeddings. Proc. Natl. Acad. Sci. USA 2021, 118, e2019053118. [Google Scholar] [CrossRef]
- Cai, H.; Vernon, R.; Forman-Kay, J. An Interpretable Machine-Learning Algorithm to Predict Disordered Protein Phase Separation Based on Biophysical Interactions. Biomolecules 2022, 12, 1131. [Google Scholar] [CrossRef]
- Badasyan, A.; Mamasakhlisov, Y.; Podgornik, R.; Parsegian, V. Solvent effects in the helix-coil transition model can explain the unusual biophysics of intrinsically disordered proteins. J. Chem. Phys. 2015, 143, 014102. [Google Scholar] [CrossRef] [Green Version]
- Kuechler, E.; Budzyńska, P.; Bernardini, J.; Gsponer, J.; Mayor, T. Distinct Features of Stress Granule Proteins Predict Localization in Membraneless Organelles. J. Mol. Biol. 2020, 432, 2349–2368. [Google Scholar] [CrossRef]
- Farahi, N.; Lazar, T.; Wodak, S.; Tompa, P.; Pancsa, R. Integration of Data from Liquid–Liquid Phase Separation Databases Highlights Concentration and Dosage Sensitivity of LLPS Drivers. Int. J. Mol. Sci. 2021, 22, 3017. [Google Scholar] [CrossRef]
- Jones, D.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emenecker, R.; Griffith, D.; Holehouse, A. Metapredict: A fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys. J. 2021, 120, 4312–4319. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K. Predicting intrinsic disorder from amino acid sequence. Proteins 2003, 53, 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, S. RBPPred: Predicting RNA-binding proteins from sequence using SVM. Bioinformatics 2017, 33, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Core Team, R. R: A Language and Environment for Statistical Computing. 2017. Available online: https://www.r-project.org/ (accessed on 1 January 2022).
- Wall, L.; Christiansen, T.; Orwant, J. Programming Perl, 3rd ed.; O’Reilly Media: Newton, MA, USA, 2000. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Lafontaine, D.; Riback, J.; Bascetin, R.; Brangwynne, C. The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol. 2021, 22, 165–182. [Google Scholar] [CrossRef]
- Ilık, İ.; Aktaş, T. Nuclear speckles: Dynamic hubs of gene expression regulation. FEBS J. 2022, 289, 7234–7245. [Google Scholar] [CrossRef]
- Nott, T.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.; Bazett-Jones, D.; Pawson, T.; Forman-Kay, J.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Brady, J.; Farber, P.; Sekhar, A.; Lin, Y.; Huang, R.; Bah, A.; Nott, T.; Chan, H.; Baldwin, A.; Forman-Kay, J.; et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. USA 2017, 114, E8194–E8203. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Choi, J.-M.; Holehouse, A.; Lee, H.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; Holehouse, A.; Peran, I.; Farag, M.; Incicco, J.; Bremer, A.; Grace, C.; Soranno, A.; Pappu, R.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Rao, B.; Parker, R. Numerous interactions act redundantly to assemble a tunable size of P bodies in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, E9569–E9578. [Google Scholar] [CrossRef] [Green Version]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 2015, 4, e06807. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.; Gleixner, A.; Mauna, J.; Gomes, E.; DeChellis-Marks, M.; Needham, P.; Copley, K.; Hurtle, B.; Portz, B.; Pyles, N.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Conicella, A.; Schmidt, H.; Martin, E.; Rhoads, S.; Reeb, A.; Nourse, A.; Montero, D.R.; Ryan, V.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.; Bennett, R.; Dujardin, S.; Laskowski, P.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Hyman, Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, 1–21. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C. Liquid phase condensation in cell physiology and disease. Science 2017, 357, 1253. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Lin, Y.-H.; Vernon, R.; Forman-Kay, J.; Chan, H. Comparative roles of charge, π, and hydrophobic interactions in sequence-dependent phase separation of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 28795–28805. [Google Scholar] [CrossRef]
- Wei, S.; Wang, Y.; Yang, G. Liquid–Liquid Phase Separation Prediction of Proteins in Salt Solution by Deep Neural Network. Biomolecules 2023, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Holehouse, A. Intrinsically disordered protein regions and phase separation: Sequence determinants of assembly or lack thereof. Emerg. Top. Life Sci. 2020, 4, 307–329. [Google Scholar] [CrossRef]
- Marzahn, M.; Marada, S.; Lee, J.; Nourse, A.; Kenrick, S.; Zhao, H.; Ben-Nissan, G.; Kolaitis, R.-M.; Peters, J.; Pounds, S.; et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016, 35, 1254–1275. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Ditlev, J.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.; Taunton, J.; Rosen, M.; Vale, R. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-M.; Dar, F.; Pappu, R.V. LASSI: A lattice model for simulating phase transitions of multivalent proteins. PLoS Comput. Biol. 2019, 15, e1007028. [Google Scholar] [CrossRef] [Green Version]
- Dignon, G.; Zheng, W.; Best, R.; Kim, Y.; Mittal, J. Relation between single-molecule properties and phase behavior of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 9929–9934. [Google Scholar] [CrossRef] [Green Version]
- Dignon, G.; Zheng, W.; Mittal, J. Simulation methods for liquid–liquid phase separation of disordered proteins. Curr. Opin. Chem. Eng. 2019, 23, 92–98. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification of Mutants Using Confidence Intervals | ||||
|---|---|---|---|---|
| catGranule | FuzDrop | LLPhyScore | PScore | |
| PS-reducing mutations | 4/6 (4/11) | 1/6 (1/11) | 5/6 (6/11) | 3/6 (7/11) |
| PS-promoting mutations | 2/7 (2/12) | 1/7 (1/12) | 2/7 (3/12) | 5/7 (6/12) |
| Classification of Mutants Based on Net Prediction Change | ||||
| catGranule | FuzDrop | LLPhyScore | PScore | |
| PS-reducing mutations | 4/6 (4/11) | 2/6 (5/11) | 5/6 (6/11) | 3/6 (7/11) |
| PS-promoting mutations | 2/7 (2/12) | 3/7 (3/12) | 2/7 (3/12) | 5/7 (6/12) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuechler, E.R.; Huang, A.; Bui, J.M.; Mayor, T.; Gsponer, J. Comparison of Biomolecular Condensate Localization and Protein Phase Separation Predictors. Biomolecules 2023, 13, 527. https://doi.org/10.3390/biom13030527
Kuechler ER, Huang A, Bui JM, Mayor T, Gsponer J. Comparison of Biomolecular Condensate Localization and Protein Phase Separation Predictors. Biomolecules. 2023; 13(3):527. https://doi.org/10.3390/biom13030527
Chicago/Turabian StyleKuechler, Erich R., Alex Huang, Jennifer M. Bui, Thibault Mayor, and Jörg Gsponer. 2023. "Comparison of Biomolecular Condensate Localization and Protein Phase Separation Predictors" Biomolecules 13, no. 3: 527. https://doi.org/10.3390/biom13030527