
Parietal Epithelial Cell Behavior and Its Modulation by microRNA-193a


Abstract
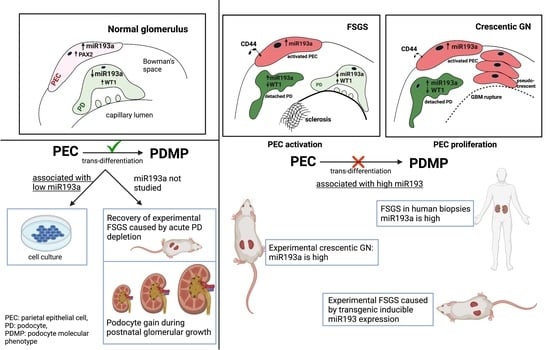
:
1. Introduction
2. PECs and Their Phenotypes
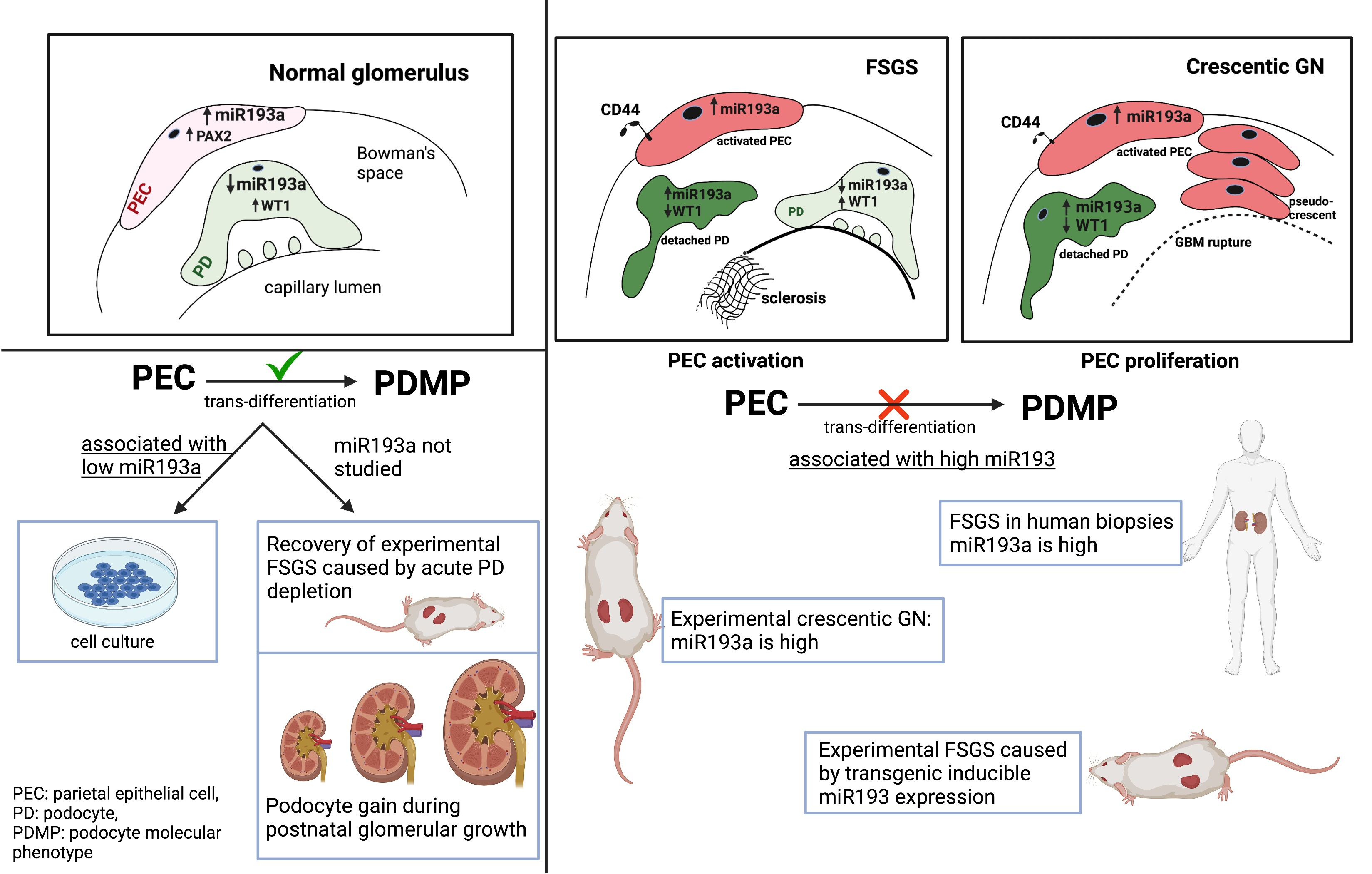
2.1. Types of PECs in Human Kidneys
2.1.1. Based on Location and Expression of Molecular Markers
2.1.2. Based on Morphology
3. Are PECs Reparative?
3.1. Are PECs Progenitors for Podocytes during Postnatal Glomerular Growth?
3.2. Are PECs Progenitors for Podocytes during Aging?
3.3. Are PECs Progenitors for Podocytes during Kidney Disease?
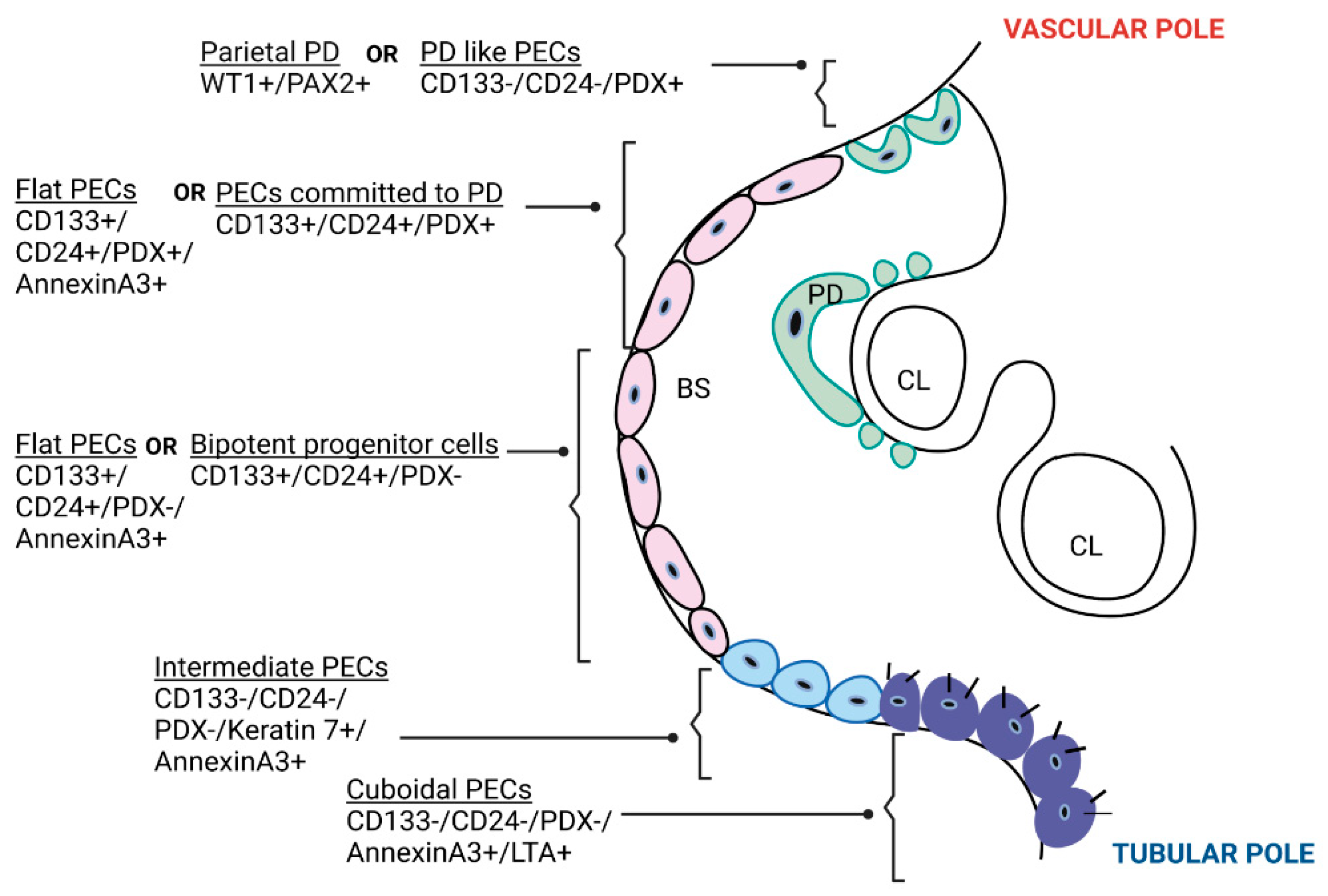
4. Are PECs Pathogenic?
4.1. PEC Activation in Classical FSGS and Collapsing Glomerulopathy
4.2. PEC Activation in Crescentic GN
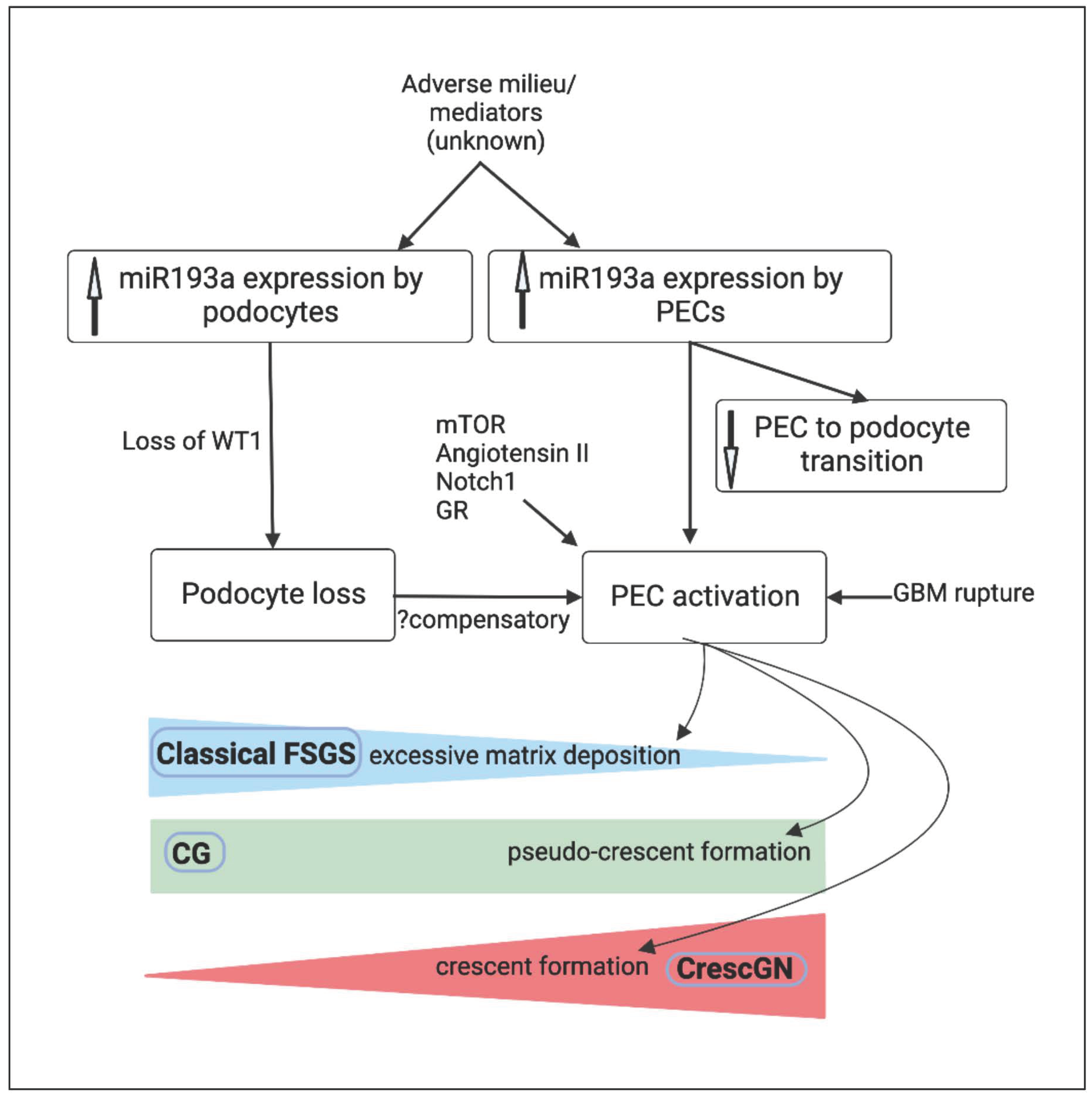
5. MicroRNA-193a and PEC Response
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shankland, S.J.; Smeets, B.; Pippin, J.W.; Moeller, M.J. The emergence of the glomerular parietal epithelial cell. Nat. Rev. Nephrol. 2014, 10, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Ohse, T.; Pippin, J.W.; Chang, A.M.; Krofft, R.D.; Miner, J.H.; Vaughan, M.R.; Shankland, S.J. The enigmatic parietal epithelial cell is finally getting noticed: A review. Kidney Int. 2009, 76, 1225–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronconi, E.; Sagrinati, C.; Angelotti, M.L.; Lazzeri, E.; Mazzinghi, B.; Ballerini, L.; Parente, E.; Becherucci, F.; Gacci, M.; Carini, M.; et al. Regeneration of Glomerular Podocytes by Human Renal Progenitors. J. Am. Soc. Nephrol. 2008, 20, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagrinati, C.; Netti, G.S.; Mazzinghi, B.; Lazzeri, E.; Liotta, F.; Frosali, F.; Ronconi, E.; Meini, C.; Gacci, M.; Squecco, R.; et al. Isolation and Characterization of Multipotent Progenitor Cells from the Bowman’s Capsule of Adult Human Kidneys. J. Am. Soc. Nephrol. 2006, 17, 2443–2456. [Google Scholar] [CrossRef] [Green Version]
- Vogelmann, S.U.; Nelson, W.J.; Myers, B.D.; Lemley, K.V. Urinary excretion of viable podocytes in health and renal disease. Am. J. Physiol. Physiol. 2003, 285, F40–F48. [Google Scholar] [CrossRef] [Green Version]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte Depletion Causes Glomerulosclerosis: Diphtheria Toxin–Induced Podocyte Depletion in Rats Expressing Human Diphtheria Toxin Receptor Transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef] [Green Version]
- Smeets, B.; Uhlig, S.; Fuss, A.; Mooren, F.; Wetzels, J.F.; Floege, J.; Moeller, M.J. Tracing the Origin of Glomerular Extracapillary Lesions from Parietal Epithelial Cells. J. Am. Soc. Nephrol. 2009, 20, 2604–2615. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-H.; Guo, X.-Y.; Quan, X.-Y.; Yang, C.; Liu, Z.-J.; Su, H.-Y.; An, N.; Liu, H.-F. The Role of Parietal Epithelial Cells in the Pathogenesis of Podocytopathy. Front. Physiol. 2022, 13, 342. [Google Scholar] [CrossRef]
- Moeller, M.J.; Smeets, B. Role of Parietal Epithelial Cells in Kidney Injury: The Case of Rapidly Progressing Glomerulonephritis and Focal and Segmental Glomerulosclerosis. Nephron Exp. Nephrol. 2014, 126, 97–100. [Google Scholar] [CrossRef]
- Wong, M.N.; Tharaux, P.-L.; Grahammer, F.; Puelles, V.G. Parietal epithelial cell dysfunction in crescentic glomerulonephritis. Cell Tissue Res. 2021, 385, 345–354. [Google Scholar] [CrossRef]
- Rizzo, P.; Perico, N.; Gagliardini, E.; Novelli, R.; Alison, M.R.; Remuzzi, G.; Benigni, A. Nature and Mediators of Parietal Epithelial Cell Activation in Glomerulonephritides of Human and Rat. Am. J. Pathol. 2013, 183, 1769–1778. [Google Scholar] [CrossRef]
- Smeets, B.; Kuppe, C.; Sicking, E.-M.; Fuss, A.; Jirak, P.; van Kuppevelt, T.H.; Endlich, K.; Wetzels, J.F.; Gröne, H.-J.; Floege, J.; et al. Parietal Epithelial Cells Participate in the Formation of Sclerotic Lesions in Focal Segmental Glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 1262–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [Green Version]
- Gebeshuber, C.A.; Kornauth, C.; Dong, L.; Sierig, R.; Seibler, J.; Reiss, M.; Tauber, S.; Bilban, M.; Wang, S.; Kain, R.; et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat. Med. 2013, 19, 481–487. [Google Scholar] [CrossRef]
- Allison, S.J. Glomerular disease: MicroRNA-193a—A new pathogenic player and target in focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 2013, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, L.; Guhr, S.S.; Meyer, T.N.; Ni, L.; Sachs, M.; Panzer, U.; Stahl, R.A.; Saleem, M.A.; Kerjaschki, D.; Gebeshuber, C.A.; et al. MicroRNA-193a Regulates the Transdifferentiation of Human Parietal Epithelial Cells toward a Podocyte Phenotype. J. Am. Soc. Nephrol. 2015, 26, 1389–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Ayasolla, K.; Kumar, V.; Lan, X.; Vashistha, H.; Aslam, R.; Hussain, A.; Chowdhary, S.; Shoshtari, S.M.; Paliwal, N.; et al. Modulation of apolipoprotein L1-microRNA-193a axis prevents podocyte dedifferentiation in high-glucose milieu. Am. J. Physiol. Physiol. 2018, 314, F832–F843. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Vashistha, H.; Lan, X.; Chandel, N.; Ayasolla, K.; Shoshtari, S.S.M.; Aslam, R.; Paliwal, N.; Abbruscato, F.; Mikulak, J.; et al. Role of Apolipoprotein L1 in Human Parietal Epithelial Cell Transition. Am. J. Pathol. 2018, 188, 2508–2528. [Google Scholar] [CrossRef] [Green Version]
- Pabst, R.; Sterzel, R.B. Cell renewal of glomerular cell types in normal rats. An autoradiographic analysis. Kidney Int. 1983, 24, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Bariety, J.; Mandet, C.; Hill, G.S.; Bruneval, P. Parietal Podocytes in Normal Human Glomeruli. J. Am. Soc. Nephrol. 2006, 17, 2770–2780. [Google Scholar] [CrossRef]
- Kuppe, C.; Leuchtle, K.; Wagner, A.; Kabgani, N.; Saritas, T.; Puelles, V.G.; Smeets, B.; Hakroush, S.; van der Vlag, J.; Boor, P.; et al. Novel parietal epithelial cell subpopulations contribute to focal segmental glomerulosclerosis and glomerular tip lesions. Kidney Int. 2019, 96, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fioretto, P.; Steffes, M.W.; Sutherland, D.E.; Goetz, F.C.; Mauer, M. Reversal of Lesions of Diabetic Nephropathy after Pancreas Transplantation. N. Engl. J. Med. 1998, 339, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Pichaiwong, W.; Hudkins, K.L.; Wietecha, T.; Nguyen, T.Q.; Tachaudomdach, C.; Li, W.; Askari, B.; Kobayashi, T.; O’Brien, K.D.; Pippin, J.W.; et al. Reversibility of Structural and Functional Damage in a Model of Advanced Diabetic Nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1088–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudkins, K.L.; Li, X.; Holland, A.L.; Swaminathan, S.; E Alpers, C. Regression of diabetic nephropathy by treatment with empagliflozin in BTBR ob/ob mice. Nephrol. Dial. Transplant. 2022, 37, 847–859. [Google Scholar] [CrossRef]
- Grouls, S.; Iglesias, D.M.; Wentzensen, N.; Moeller, M.J.; Bouchard, M.; Kemler, R.; Goodyer, P.; Niggli, F.; Gröne, H.J.; Kriz, W.; et al. Lineage specification of parietal epithelial cells requires β-catenin/Wnt signaling. J. Am. Soc. Nephrol. 2012, 23, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Puelles, V.G.; Douglas-Denton, R.N.; Cullen-McEwen, L.A.; Li, J.; Hughson, M.D.; Hoy, W.E.; Kerr, P.G.; Bertram, J.F. Podocyte Number in Children and Adults: Associations with Glomerular Size and Numbers of Other Glomerular Resident Cells. J. Am. Soc. Nephrol. 2015, 26, 2277–2288. [Google Scholar] [CrossRef] [Green Version]
- Appel, D.; Kershaw, D.B.; Smeets, B.; Yuan, G.; Fuss, A.; Frye, B.; Elger, M.; Kriz, W.; Floege, J.; Moeller, M.J. Recruitment of Podocytes from Glomerular Parietal Epithelial Cells. J. Am. Soc. Nephrol. 2008, 20, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Berger, K.; Schulte, K.; Boor, P.; Kuppe, C.; van Kuppevelt, T.H.; Floege, J.; Smeets, B.; Moeller, M.J. The Regenerative Potential of Parietal Epithelial Cells in Adult Mice. J. Am. Soc. Nephrol. 2014, 25, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Wanner, N.; Hartleben, B.; Herbach, N.; Goedel, M.; Stickel, N.; Zeiser, R.; Walz, G.; Moeller, M.J.; Grahammer, F.; Huber, T.B. Unraveling the Role of Podocyte Turnover in Glomerular Aging and Injury. J. Am. Soc. Nephrol. 2014, 25, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Kaverina, N.V.; Eng, D.G.; Miner, J.H.; Pippin, J.W.; Shankland, S.J. Parietal epithelial cell differentiation to a podocyte fate in the aged mouse kidney. Aging 2020, 12, 17601–17624. [Google Scholar] [CrossRef] [PubMed]
- Eng, D.G.; Sunseri, M.W.; Kaverina, N.V.; Roeder, S.S.; Pippin, J.W.; Shankland, S.J. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 999–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaverina, N.V.; Eng, D.G.; Freedman, B.S.; Kutz, J.N.; Chozinski, T.J.; Vaughan, J.C.; Miner, J.H.; Pippin, J.W.; Shankland, S.J. Dual lineage tracing shows that glomerular parietal epithelial cells can transdifferentiate toward the adult podocyte fate. Kidney Int. 2019, 96, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, L.; Angelotti, M.L.; Ronconi, E.; Lombardi, D.; Nardi, S.; Peired, A.; Becherucci, F.; Mazzinghi, B.; Sisti, A.; Romoli, S.; et al. Podocyte Regeneration Driven by Renal Progenitors Determines Glomerular Disease Remission and Can Be Pharmacologically Enhanced. Stem Cell Rep. 2015, 5, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, M.J.; Tharaux, P.-L. Cellular regeneration of podocytes from parietal cells: The debate is still open. Kidney Int. 2019, 96, 542–544. [Google Scholar] [CrossRef]
- Hakroush, S.; Cebulla, A.; Schaldecker, T.; Behr, D.; Mundel, P.; Weins, A. Extensive Podocyte Loss Triggers a Rapid Parietal Epithelial Cell Response. J. Am. Soc. Nephrol. 2013, 25, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Miesen, L.; Steenbergen, E.; Smeets, B. Parietal cells—New perspectives in glomerular disease. Cell Tissue Res. 2017, 369, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Froes, B.P.; de Almeida Araújo, S.; Bambirra, E.A.; Oliveira, E.A.; Simões ESilva, A.C.; Pinheiro, S.V.B. Is CD44 in glomerular parietal epithelial cells a pathological marker of renal function deterioration in primary focal segmental glomerulosclerosis? Pediatr. Nephrol. 2017, 32, 2165–2169. [Google Scholar] [CrossRef]
- Benigni, A.; Morigi, M.; Rizzo, P.; Gagliardini, E.; Rota, C.; Abbate, M.; Ghezzi, S.; Remuzzi, A.; Remuzzi, G. Inhibiting Angiotensin-Converting Enzyme Promotes Renal Repair by Limiting Progenitor Cell Proliferation and Restoring the Glomerular Architecture. Am. J. Pathol. 2011, 179, 628–638. [Google Scholar] [CrossRef]
- Albaqumi, M.; Barisoni, L. Current Views on Collapsing Glomerulopathy. J. Am. Soc. Nephrol. 2008, 19, 1276–1281. [Google Scholar] [CrossRef] [Green Version]
- Meyrier, A.Y. Collapsing glomerulopathy: Expanding interest in a shrinking tuft. Am. J. Kidney Dis. 1999, 33, 801–803. [Google Scholar] [CrossRef]
- Detwiler, R.K.; Falk, R.J.; Hogan, S.L.; Jennette, J.C. Collapsing glomerulopathy: A clinically and pathologically distinct variant of focal segmental glomerulosclerosis. Kidney Int. 1994, 45, 1416–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Knob, A.L.U.; et al. Association of Trypanolytic ApoL1 Variants with Kidney Disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruggeman, L.A.; Wu, Z.; Luo, L.; Madhavan, S.; Drawz, P.E.; Thomas, D.B.; Barisoni, L.; O'Toole, J.F.; Sedor, J.R. APOL1-G0 protects podocytes in a mouse model of HIV-associated nephropathy. PLoS ONE 2019, 14, e0224408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behar, D.M.; Kedem, E.; Rosset, S.; Haileselassie, Y.; Tzur, S.; Kra-Oz, Z.; Wasser, W.G.; Shenhar, Y.; Shahar, E.; Hassoun, G.; et al. Absence of APOL1 Risk Variants Protects against HIV-Associated Nephropathy in the Ethiopian Population. Am. J. Nephrol. 2011, 34, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.; Jog, P.; Lee, J.; Blackler, D.; Wilmot, M.; D’Agati, V.; Markowitz, G.; Kopp, J.; Alper, S.; Pollak, M.; et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015, 87, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barisoni, L.; Bruggeman, L.A.; Mundel, P.; D’Agati, V.D.; Klotman, P.E. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int. 2000, 58, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.D.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; A Hu, C.-A.; et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Lazareth, H.; Henique, C.; Lenoir, O.; Puelles, V.G.; Flamant, M.; Bollée, G.; Fligny, C.; Camus, M.; Guyonnet, L.; Millien, C.; et al. The tetraspanin CD9 controls migration and proliferation of parietal epithelial cells and glomerular disease progression. Nat. Commun. 2019, 10, 3303. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, P.; Novelli, R.; Rota, C.; Gagliardini, E.; Ruggiero, B.; Rottoli, D.; Benigni, A.; Remuzzi, G. The Role of Angiotensin II in Parietal Epithelial Cell Proliferation and Crescent Formation in Glomerular Diseases. Am. J. Pathol. 2017, 187, 2441–2450. [Google Scholar] [CrossRef] [Green Version]
- Wilkening, A.; Krappe, J.; Mühe, A.M.; Lindenmeyer, M.T.; Eltrich, N.; Luckow, B.; Vielhauer, V. C-C chemokine receptor type 2 mediates glomerular injury and interstitial fibrosis in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2020, 35, 227–239. [Google Scholar] [CrossRef]
- Sicking, E.-M.; Fuss, A.; Uhlig, S.; Jirak, P.; Dijkman, H.; Wetzels, J.; Engel, D.R.; Urzynicok, T.; Heidenreich, S.; Kriz, W.; et al. Subtotal ablation of parietal epithelial cells induces crescent formation. J. Am. Soc. Nephrol. 2012, 23, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell. 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ma, L.; Yu, H.; Yao, Y.; Xu, Z.; Lin, W.; Wang, L.; Wang, X.; Yang, H. MicroRNAs as Potential Biomarkers for the Diagnosis of Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Front. Med. 2022, 8, 782561. [Google Scholar] [CrossRef] [PubMed]
- Mahtal, N.; Lenoir, O.; Tinel, C.; Anglicheau, D.; Tharaux, P.-L. MicroRNAs in kidney injury and disease. Nat. Rev. Nephrol. 2022, 18, 643–662. [Google Scholar]
- Chou, N.H.; Tsai, C.Y.; Tu, Y.T.; Wang, K.C.; Kang, C.H.; Chang, P.M.; Li, G.C.; Lam, H.C.; Liu, S.I.; Tsai, K.W. MiR-193a-5p and -3p Play a Distinct Role in Gastric Cancer: miR-193a-3p Suppresses Gastric Cancer Cell Growth by Targeting ETS1 and CCND1. Anticancer Res. 2018, 38, 3309–3318. [Google Scholar] [CrossRef]
- Takahashi, H.; Takahashi, M.; Ohnuma, S.; Unno, M.; Yoshino, Y.; Ouchi, K.; Takahashi, S.; Yamada, Y.; Shimodaira, H.; Ishioka, C. microRNA-193a-3p is specifically down-regulated and acts as a tumor suppressor in BRAF-mutated colorectal cancer. BMC Cancer 2017, 17, 723. [Google Scholar] [CrossRef]
- Azar, M.R.M.H.; Aghazadeh, H.; Mohammed, H.N.; Sara, M.R.S.; Hosseini, A.; Shomali, N.; Tamjidifar, R.; Tarzi, S.; Mansouri, M.; Sarand, S.P.; et al. miR-193a-5p as a promising therapeutic candidate in colorectal cancer by reducing 5-FU and Oxaliplatin chemoresistance by targeting CXCR4. Int. Immunopharmacol. 2021, 92, 107355. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers. 2019, 5, 11. [Google Scholar]
- Zhang, S.; Liu, J.; He, J.; Yi, N. MicroRNA-193a-5p exerts a tumor suppressive role in epithelial ovarian cancer by modulating RBBP6. Mol. Med. Rep. 2021, 24, 582. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, J.-S.; Wen, J.-K.; Gao, H.-T.; Zheng, B.; Qu, C.-B.; Liu, K.-L.; Zhang, M.-L.; Gu, J.-F.; Li, J.-D.; et al. Silencing of miR-193a-5p increases the chemosensitivity of prostate cancer cells to docetaxel. J. Exp. Clin. Cancer Res. 2017, 36, 178. [Google Scholar] [CrossRef] [Green Version]
- Shankland, S.J.; Pippin, J.W.; Duffield, J.S. Progenitor Cells and Podocyte Regeneration. Semin. Nephrol. 2014, 34, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, J.; Wang, Z.; Zhou, J.; Zhang, Y. Higher Urine Exosomal miR-193a Is Associated With a Higher Probability of Primary Focal Segmental Glomerulosclerosis and an Increased Risk of Poor Prognosis Among Children With Nephrotic Syndrome. Front. Cell Dev. Biol. 2021, 9, 2872. [Google Scholar] [CrossRef] [PubMed]
- Jessee, J.; Kopp, J.B. APOL1-miR-193 Axis as a Bifunctional Regulator of the Glomerular Parietal Epithelium: Maintaining Parietal Cell Phenotype versus Promoting Podocyte Differentiation. Am. J. Pathol. 2018, 188, 2461–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Paliwal, N.; Ayasolla, K.; Vashistha, H.; Jha, A.; Chandel, N.; Chowdhary, S.; Saleem, M.A.; Malhotra, A.; Chander, P.N.; et al. Disruption of APOL1-miR193a Axis Induces Disorganization of Podocyte Actin Cytoskeleton. Sci. Rep. 2019, 9, 3582. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Future Research Perspectives |
|---|
| Evaluation of PEC characteristics after miR193a inhibition in transgenic mice with miR193a-induced FSGS |
| Analysis of miR193a profile of the PECs and podocytes in animal models of crescentic GN during active disease and after resolution |
| Validation of miR193a profile in primary FSGS by other investigators |
| Comparison of miR193a profile in animals with transgenic expression of APOL1G0 and those without APOL1G0 |
| Long-term tumorigenesis effects of miR193a inhibition in animal models |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bharati, J.; Chander, P.N.; Singhal, P.C. Parietal Epithelial Cell Behavior and Its Modulation by microRNA-193a. Biomolecules 2023, 13, 266. https://doi.org/10.3390/biom13020266
Bharati J, Chander PN, Singhal PC. Parietal Epithelial Cell Behavior and Its Modulation by microRNA-193a. Biomolecules. 2023; 13(2):266. https://doi.org/10.3390/biom13020266
Chicago/Turabian StyleBharati, Joyita, Praveen N. Chander, and Pravin C. Singhal. 2023. "Parietal Epithelial Cell Behavior and Its Modulation by microRNA-193a" Biomolecules 13, no. 2: 266. https://doi.org/10.3390/biom13020266