
Advances and Challenges in Spatial Transcriptomics for Developmental Biology


Abstract
:1. Introduction
2. scRNA-Seq Techniques and Developmental Biology
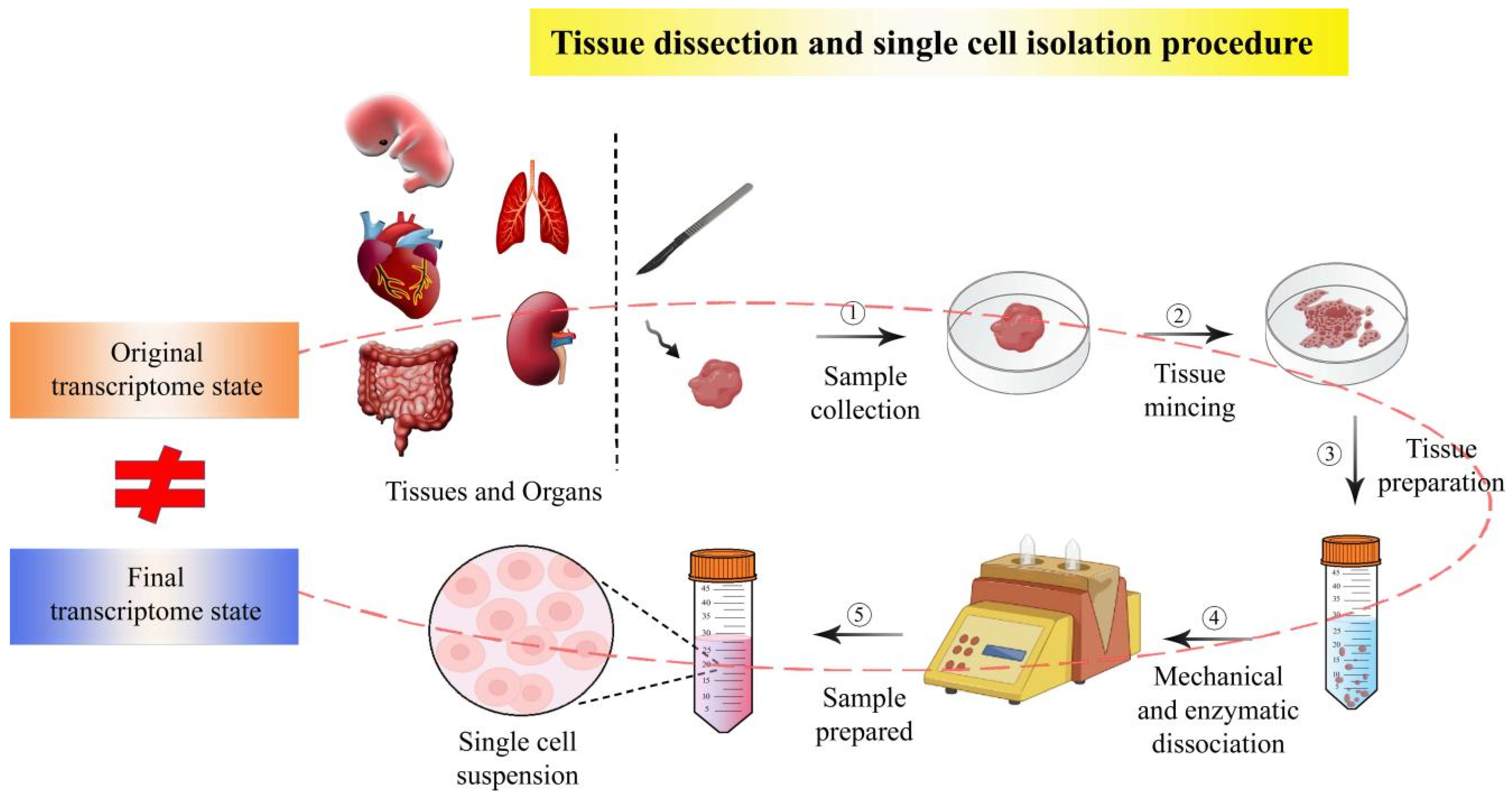
2.1. Single Cell Isolation Techniques Have Enabled scRNA-seq
2.2. Application of scRNA-seq in Developmental Biology Studies
3. ST Techniques and Developmental Biology
3.1. Classification of Ever-Emerged ST Techniques
3.2. General Workflow of the ST Techniques
3.3. Application of ST Techniques in Developmental Biology Studies
4. Limitations of Current Tools and Approaches for Spatial Transcriptomics in Developmental Biology Studies
4.1. Limitations of Current ST Tools
4.2. Limitations in Current Integrative Approaches for Spatial Transcriptomic Studies
5. Integration of Spatial Transcriptomics with Spatial Proteomics or Spatial Metabolomics: Future Prospect
5.1. Spatial Transcriptomics Is Just an Entry for Spatial Omics
5.2. Spatial Multi-Omics Data Would Provide Novel and Comprehensive Insights into Developmental Biology
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ko, M.S.H. Embryogenomics: Developmental biology meets genomics. Trends Biotechnol. 2001, 19, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Catalin Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Barrozo, E.R.; Aagaard, K.M. Human placental biology at single-cell resolution: A contemporaneous review. BJOG 2022, 129, 208–220. [Google Scholar] [CrossRef]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single Cell Isolation and Analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.; Lee, J.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Cai, W.; Sun, Z. Single-cell sequencing technologies: Current and future. J. Genet. Genom. 2014, 41, 513–528. [Google Scholar] [CrossRef]
- Guo, F.; Li, L.; Li, J.; Wu, X.; Hu, B.; Zhu, P.; Wen, L.; Tang, F. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017, 27, 967–988. [Google Scholar] [CrossRef]
- Nakamura, N.; Ruebel, K.; Jin, L.; Qian, X.; Zhang, H.; Lloyd, R.V. Laser capture microdissection for analysis of single cells. Methods Mol. Med. 2007, 132, 11–18. [Google Scholar]
- Espina, V.; Milia, J.; Wu, G.; Cowherd, S.; Liotta, L.A. Laser-capture microdissection. Nat. Protoc. 2006, 1, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Khushalani, D.; Harkare, A.; Agrawal, R. A Review of FACS: Fluorescence Activated Cell Sorting System. Biosci. Biotechnol. Res. Commun. 2020, 13, 436–439. [Google Scholar] [CrossRef]
- Makker, K.; Agarwal, A.; Sharma, R.K. Magnetic activated cell sorting (MACS): Utility in assisted reproduction. Indian J. Exp. Biol. 2008, 46, 491–497. [Google Scholar] [PubMed]
- Miltenyi, S.; Müller, W.; Weichel, W.; Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry 1990, 11, 231–238. [Google Scholar] [CrossRef]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Tam, P.P.L.; Ho, J.W.K. Cellular diversity and lineage trajectory: Insights from mouse single cell transcriptomes. Development 2020, 147, dev179788. [Google Scholar] [CrossRef] [Green Version]
- Scialdone, A.; Tanaka, Y.; Jawaid, W.; Moignard, V.; Wilson, N.K.; Macaulay, I.C.; Marioni, J.C.; Göttgens, B. Resolving early mesoderm diversification through single-cell expression profiling. Nature 2016, 535, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann1, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.E.; Weinreb, C.; Collins, Z.M.; Briggs, J.A.; Megason, S.G.; Klein, A.M. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 2018, 360, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, J.A.; Wang, Y.; Riesenfeld, S.J.; Shekhar, K.; Regev, A.; Schier, A.F. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 2018, 360, eaar3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, J.A.; Weinreb, C.; Wagner, D.E.; Megason, S.; Peshkin, L.; Kirschner, M.W.; Klein, A.M. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 2018, 360, eaar5780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Sun, N.; Li, C.; Lei, Y.; Huang, Z.; Wu, J.; Si, C.; Dai, X.; Liu, C.; Wei, J.; et al. Dissecting primate early post-implantation development using long-term in vitro embryo culture. Science 2019, 366, eaaw5754. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Reyes, A.P.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Wang, R.; Yuan, P.; Ren, Y.; Mao, Y.; Li, R.; Lian, Y.; Li, J.; Wen, L.; Yan, L.; et al. Reconstituting the transcriptome and DNA methylome landscapes of human implantation. Nature 2019, 572, 660–664. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, S.; Fan, X.; Wu, Q.; Yan, L.; Dong, J.; Zhang, H.; Li, L.; Sun, L.; Pan, N.; et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018, 555, 524–528. [Google Scholar] [CrossRef]
- Gao, S.; Yan, L.; Wang, R.; Li, J.; Yong, J.; Zhou, X.; Wei, Y.; Wu, X.; Wang, X.; Fan, X.; et al. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat. Cell Biol. 2018, 20, 721–734. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, F.; Shangguan, Y.; Yang, Y.; Shi, W.; Hu, W.; Zeng, Z.; Hu, N.; Zhang, X.; Hocher, B.; et al. Integrating spatial transcriptomics with single-cell transcriptomics reveals a spatiotemporal gene landscape of the human developing kidney. Cell Biosci. 2022, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 2020, 42, e1900221. [Google Scholar] [CrossRef] [PubMed]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef]
- Simone, N.I.; Bonner, R.F.; Gillespie, J.W.; Emmert-Buck, M.R.; Liotta, L.A. Laser-capture microdissection: Opening the microscopic frontier to molecular analysis. Trends Genet. 1998, 14, 272–276. [Google Scholar] [CrossRef]
- Combs, P.A.; Eisen, M.B. Sequencing mRNA from cryo-sliced Drosophila embryos to determine genome-wide spatial patterns of gene expression. PLoS ONE 2013, 8, e71820. [Google Scholar] [CrossRef]
- Lovatt, D.; Ruble, B.K.; Lee, J.; Dueck, H.; Kim, T.K.; Fisher, S.; Francis, C.; Spaethling, J.M.; Wolf, J.A.; Grady, M.S.; et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat. Methods 2014, 11, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Junker, J.P.; Noël, E.S.; Guryev, V.; Peterson, K.A.; Shah, G.; Huisken, J.; McMahon, A.P.; Berezikov, E.; Bakkers, J.; Oudenaarden, A. Genome-wide RNA Tomography in the zebrafish embryo. Cell 2014, 159, 662–675. [Google Scholar] [CrossRef] [Green Version]
- Kruse, F.; Junker, J.P.; Oudenaarden, A.V.; Bakkers, J. Tomo-seq: A method to obtain genome-wide expression data with spatial resolution. Methods Cell Biol. 2016, 135, 299–307. [Google Scholar]
- Nichterwitz, S.; Chen, G.; Benitez, J.A.; Yilmaz, M.; Storvall, H.; Cao, M.; Sandberg, R.; Deng, Q.; Hedlund, E. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 2016, 7, 12139. [Google Scholar] [CrossRef] [Green Version]
- Moor, A.E.; Harnik, Y.; Ben-Moshe, S.; Massasa, E.E.; Rozenberg, M.; Eilam, R.; Halpern, K.B.; Itzkovitz, S. Spatial Reconstruction of Single Enterocytes Uncovers Broad Zonation along the Intestinal Villus Axis. Cell 2018, 175, 1156–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Suo, S.; Tam, P.P.; Han, J.D.; Peng, G.; Jing, N. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protoc. 2017, 12, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Medaglia, C.; Giladi, A.; Stoler-Barak, L.; Giovanni, M.D.; Salame, T.M.; Biram, A.; David, E.; Li, H.; Iannacone, M.; Shulman, Z.; et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 2017, 358, 1622–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Sun, L.; Chen, J.Y.; Dong, R.; Lin, Y.; Palmiter, R.D.; Lin, S.; Gu, L. Continuous Polony Gels for Tissue Mapping with High Resolution and RNA Capture Efficiency. bioRxiv 2021. [Google Scholar]
- Boisset, J.C.; Vivié1, J.; Grün, D.; Muraro, M.J.; Lyubimova, A.; Oudenaarden, A. Mapping the physical network of cellular interactions. Nat. Methods 2018, 15, 547–553. [Google Scholar] [CrossRef]
- Femino, A.M.; Fay, F.S.; Fogarty, K.; Singer, R.H. Visualization of single RNA transcripts in situ. Science 1998, 280, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; Bogaard, P.; Rifkin, S.A.; Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.T.; Ma, X.J.; Luo, Y. RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012, 14, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Lubeck, E.; Zhou, W.; Cai, L. In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron 2016, 92, 342–357. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nat. Methods 2021, 18, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Lubeck, E.; Schwarzkopf, M.; He, T.F.; Greenbaum, A.; Sohn, C.H.; Lignell, A.; Choi, H.M.T.; Gradinaru, V.; Pierce, N.A.; et al. Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development 2016, 143, 2862–2867. [Google Scholar] [CrossRef] [Green Version]
- Codeluppi, S.; Borm, L.E.; Zeisel, A.; Manno, G.L.; Lunteren, J.A.; Svensson, C.I.; Linnarsson, S. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 2018, 15, 932–935. [Google Scholar] [CrossRef]
- Eng, C.H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Weinstein, J.A.; Regev, A.; Zhang, F. DNA Microscopy: Optics-free Spatio-genetic Imaging by a Stand-Alone Chemical Reaction. Cell 2019, 178, 229–241.e16. [Google Scholar] [CrossRef]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Ferrante, T.C.; Terry, R.; Turczyk, B.M.; Yang, J.L.; Lee, H.S.; Aach, J.; et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 2015, 10, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Sun, Y.C.; Church, G.M.; Lee, J.H.; Zador, A.M. Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 2018, 46, e22. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Punthambaker, S.; Iyer, E.P.R.; Ferrante, T.; Goodwin, D.; Fürth, D.; Pawlowski, A.C.; Jindal, K.; Tam, J.M.; Mifflin, L.; et al. Barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses. Nucleic Acids Res. 2021, 49, e58. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; Goodwin, D.R.; Sinha, A.; Wassie, A.T.; Chen, F.; Daugharthy, E.R.; Bando, Y.; Kajita, A.; Xue, A.G.; Marrett, K.; et al. Expansion sequencing: Spatially precise in situ transcriptomics in intact biological systems. Science 2021, 371, eaax2656. [Google Scholar] [CrossRef]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Faza, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490.e26. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e18. [Google Scholar] [CrossRef]
- Srivatsan, S.R.; Regier, M.C.; Barkan, E.; Franks, J.M.; Packer, J.S.; Grosjean, P.; Duran1, M.; Saxton, S.; Ladd, J.J.; Spielmann, M.; et al. Embryo-scale, single-cell spatial transcriptomics. Science 2021, 373, 111–117. [Google Scholar] [CrossRef]
- Stenbeck, L.; Taborsak-Lines, F.; Giacomello, S. Enabling automated and reproducible spatially resolved transcriptomics at scale. Heliyon 2022, 8, e09651. [Google Scholar] [CrossRef]
- Cho, C.S.; Xi, J.; Si, Y.; Park, S.R.; Hsu, J.E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572.e22. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Yang, J.; Li, W.; Xu, J.; Hao, S.; et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. bioRxiv 2021. [Google Scholar]
- Zollinger, D.R.; Lingle, S.E.; Sorg, K.; Beechem, J.M.; Merritt, C.R. GeoMx™ RNA Assay: High Multiplex, Digital, Spatial Analysis of RNA in FFPE Tissue. Methods Mol. Biol. 2020, 2148, 331–345. [Google Scholar] [PubMed]
- Honda, M.; Oki, S.; Kimura, R.; Harada, A.; Maehara, K.; Tanaka, K.; Meno, C.; Ohkawa, Y. High-depth spatial transcriptome analysis by photo-isolation chemistry. Nat. Commun. 2021, 12, 4416. [Google Scholar] [CrossRef] [PubMed]
- Jansova, D.; Tetkova, A.; Koncicka, M.; Kubelka, M.; Susor, A. Localization of RNA and translation in the mammalian oocyte and embryo. PLoS ONE 2018, 13, e0192544. [Google Scholar] [CrossRef] [Green Version]
- Burkhard, S.B.; Bakkers, J. Spatially resolved RNA-sequencing of the embryonic heart identifies a role for Wnt/β-catenin signaling in autonomic control of heart rate. Elife 2018, 7, e31515. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Peng, G.; Suo, S.; Cui, G.; Yu, F.; Wang, R.; Chen, J.; Chen, S.; Liu, Z.; Chen, G.; Qian, Y.; et al. Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature 2019, 572, 528–532. [Google Scholar] [CrossRef]
- Brink, S.C.; Alemany, A.; Batenburg, V.; Moris, N.; Blotenburg, M.; Vivié, J.; Baillie-Johnson, P.; Nichols, J.; Sonnen, K.F.; Arias, A.M.; et al. Single-cell and spatial transcriptomics reveal somitogenesis in gastruloids. Nature 2020, 582, 405–409. [Google Scholar] [CrossRef]
- Nowotschin, S.; Setty, M.; Kuo, Y.Y.; Liu, V.; Garg, V.; Sharma, R.; Simon, C.S.; Saiz, N.; Gardner, R.; Boutet, S.C.; et al. The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature 2019, 569, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Anlas, K.; Brink, S.C.; Alemany, A.; Schröder, J.; Ghimire, S.; Balayo, T.; Oudenaarden, A.; Arias, A.M. An in vitro model of early anteroposterior organization during human development. Nature 2020, 582, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Negretti, N.M.; Plosa, E.J.; Benjamin, J.T.; Schuler, B.A.; Habermann, A.C.; Jetter, C.S.; Gulleman, P.; Bunn, C.; Hackett, A.N.; Ransom, M.; et al. A single-cell atlas of mouse lung development. Development 2021, 148, dev199512. [Google Scholar] [CrossRef]
- Lohoff, T.; Ghazanfar, S.; Missarova, A.; Koulena, N.; Pierson, N.; Griffiths, J.A.; Bardot, E.S.; Eng, C.H.L.; Tyser, R.C.V.; Argelaguet, R.; et al. Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis. Nat. Biotechnol. 2022, 40, 74–85. [Google Scholar] [CrossRef]
- Bella, D.J.D.; Habibi, E.; Stickels, R.R.; Scalia, G.; Brown, J.; Yadollahpour, P.; Yang, S.M.; Abbate, C.; Biancalani, T.; Macosko, E.J.; et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 2021, 595, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.X.; Saillour, Y.; Cwetsch, A.W.; Pierani, A.; Causeret, F. Single-cell transcriptomics of the early developing mouse cerebral cortex disentangle the spatial and temporal components of neuronal fate acquisition. Development 2021, 148, dev197962. [Google Scholar] [CrossRef]
- Zhao, L.; Song, W.; Chen, Y.G. Mesenchymal-epithelial interaction regulates gastrointestinal tract development in mouse embryos. Cell Rep. 2022, 40, 111053. [Google Scholar] [CrossRef]
- Hou, X.; Yang, Y.; Li, P.; Zeng, Z.; Hu, W.; Zhe, R.; Liu, X.; Tang, D.; Ou, M.; Dai, Y. Integrating Spatial Transcriptomics and Single-Cell RNA-seq Reveals the Gene Expression Profling of the Human Embryonic Liver. Front. Cell Dev. Biol. 2021, 9, 652408. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, G.; Zhao, G.; Guo, C.; Zeng, Y.; Xue, Y.; Zeng, F. Spatial transcriptomic profiling to identify mesoderm progenitors with precision genomic screening and functional confirmation. Cell Prolif. 2022, 55, e13298. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, S.; Deng, D.; Wang, W.; Xu, X.; Liu, X.; Zhao, S.; Yu, M. Integrating LCM-Based Spatio-Temporal Transcriptomics Uncovers Conceptus and Endometrial Luminal Epithelium Communication that Coordinates the Conceptus Attachment in Pigs. Int. J. Mol. Sci. 2021, 22, 1248. [Google Scholar] [CrossRef]
- Wang, M.; Hu, Q.; Lv, T.; Wang, Y.; Lan, Q.; Xiang, R.; Tu, Z.; Wei, Y.; Han, K.; Shi, C.; et al. High-resolution 3D spatiotemporal transcriptomic maps of developing Drosophila embryos and larvae. Dev. Cell 2022, 57, 1271–1283.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, R.; Li, Y.; Lin, X.; Zhao, K.; Liu, Q.; Wang, S.; Yang, X.; Shi, X.; Ma, Y.; et al. Spatiotemporal mapping of gene expression landscapes and developmental trajectories during zebrafish embryogenesis. Dev. Cell 2022, 57, 1284–1298.e5. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso1, L.; Lorenzi, V.; Mazzeo, C.I.; Alves-Lopes, J.P.; Roberts, K.; Sancho-Serra, C.; Engelbert, J.; Marečková, M.; Gruhn, W.H.; Botting, R.A.; et al. Single-cell roadmap of human gonadal development. Nature 2022, 607, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Gerós, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E.; et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 2021, 184, 810–826.e23. [Google Scholar] [CrossRef]
- Bergmann, S.; Penfold, C.A.; Slatery, E.; Siriwardena, D.; Drummer, C.; Clark, S.; Strawbridge, S.E.; Kishimoto, K.; Vickers, A.; Tewary, M.; et al. Spatial profiling of early primate gastrulation in utero. Nature 2022, 609, 136–143. [Google Scholar] [CrossRef]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef]
- Li, H.; Huang, Q.; Liu, Y.; Garmire, L.X. Single cell transcriptome research in human placenta. Reproduction 2020, 160, R155–R167. [Google Scholar] [CrossRef]
- Ahmed, R.; Zaman, T.; Chowdhury, F.; Mraiche, F.; Tariq, M.; Ahmad, I.S.; Hasan, A. Single-Cell RNA Sequencing with Spatial Transcriptomics of Cancer Tissues. Int. J. Mol. Sci. 2022, 23, 3042. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef]
- Fox, B.W.; Schroeder, F.C. Toward spatially resolved metabolomics. Nat. Chem. Biol. 2020, 16, 1039–1040. [Google Scholar] [CrossRef]
- Padrón, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA Labeling by APEX-Seq Reveals the Organization of Translation Initiation Complexes and Repressive RNA Granules. Mol. Cell 2019, 75, 875–887.e5. [Google Scholar] [CrossRef] [PubMed]
- Gouin III, K.H.; Ing, N.; Plummer, J.T.; Rosser, C.J.; Cheikh, B.B.; Oh, C.; Chen, S.S.; Chan, K.S.; Furuya, H.; Tourtellotte, W.G.; et al. An N-Cadherin 2 expressing epithelial cell subpopulation predicts response to surgery, chemotherapy and immunotherapy in bladder cancer. Nat. Commun. 2021, 12, 4906. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhou, Q.; Cai, L.; Pan, L.; Sun, W.; Qumu, S.; Yu, S.; Feng, J.; Zhao, H.; Zheng, Y.; et al. SEAM is a spatial single nuclear metabolomics method for dissecting tissue microenvironment. Nat. Methods 2021, 18, 1223–1232. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Organism | Sequenced Samples | Sample Preparation | Sequencing Techniques | How to Obtain Single Cell- and Spatial-Resolution Transcriptome Data | References |
|---|---|---|---|---|---|
| human | heart tissues at 4.5~9 PCW single cells | tissue dissection tissue dissociation cell suspension | 10× Visium scRNA-seq ISS | 1. Generation of spatial transcriptome data at spot (~equal to size of 30 cells) resolution by 10× Visium 2. Deconvolution of spot transcriptome data into single-cell-resolution with scRNA-seq data 3. Validation of overall spatial transcriptome data by ISS | [78] |
| intestine tissues at 8~22 PCW single cells | full tissue digestion tissue block sectioning | scRNA-seq 10× Visium | 1. Generation of single-cell-resolution transcriptome data by scRNA-seq 2. Generation of spatial transcriptome data by 10× Visium and spatial mapping of scRNA-seq data with 10× Visium data | [94] | |
| gastruloids | chiron pre-treatment cryo-sectioning | Tomo-seq | Generation of spatial transcriptome data at each section (20-μm sections) by Tomo-seq | [82] | |
| gonadal tissue at 6~21 PCW single cells | tissue dissection tissue dissociation cell suspension cryo-sectioning paraformaldehyde fixing | scRNA-seq smFISH 10× Visium | 1. Generation of single-cell-resolution transcriptome data by scRNA-seq 2. Providing spatial information for partial genes by probe-dependent smFISH 3. Generation of spatial transcriptome data by 10× Visium and spatial mapping of scRNA-seq data with 10× Visium data | [93] | |
| liver tissue at 8~17 PCW | cryo-sectioning tissue dissection | 10× Visium | 1. Generation of spatial transcriptome data at spot (100μm) resolution by 10× Visium 2. Deconvolution of spot data into single-cell resolution with previous scRNA-seq data | [88] | |
| kidney tissue at 9~18 PCW single cells | cryo-sectioning cell suspension | 10× Visium scRNA-seq | 1. Generation of spatial transcriptome data at spot (100μm) resolution by 10× Visium 2. Deconvolution of spot transcriptome data into single-cell-resolution with scRNA-seq data | [32] | |
| mouse | lung tissues at E12~P14 single cells | tissue dissection tissue dissociation cell suspension | scRNA-seq RNAScope | 1. Generation of single-cell resolution transcriptome data by scRNA-seq 2. Providing spatial information for partial genes of scRNA-seq data by probe-dependent RNA Scope | [83] |
| embryo tissues at E2.5~E7.5 single cells | 1. cryo-sectioning tissue dissection tissue dissociation 2. manually cell picking | Geo-seq scRNAseq | 1. Generation of spatial transcriptome data at capture area (5–40 cells) by Geo-seq 2. Deconvolution of the Geo-seq data into single-cell resolution using scRNA-seq data | [79] | |
| somatosensory cortex tissues at E10.5~E18.5 and P1~P4 single cells | tissue dissection tissue dissociation cell suspension tissue block sectioning | scRNA-seq Slide-seq | 1. Generation of single-cell resolution transcriptome data by scRNA-seq 2. Spatial mapping of scRNA-seq data onto Slide-seq data with Tangram | [85] | |
| oocyte and 2-cell embryo | IBMX treatment oocytes picking paraformaldehyde fixing permeabilizing in Triton X-100 | smRNA FISH RCA FISH | 1. Detecting RNA localization by smRNA FISH 2. Visualizing the whole cellular transcriptome by RCA FISH | [76] | |
| Embryo tissues at E14.0 | cryo-sectioning | sci-Space | Generation of spatial transcriptome data at spot (200 μm) resolution by sci-Space | [69] | |
| embryo tissues at E14.5 | cryo-sectioning | PIC RNA-seq | Generation of spatial transcriptome data at regions of interest by PIC RNA-seq | [75] | |
| embryo tissues at E8.5~E8.75 single cells | tissue dissection paraformaldehyde fixing cryo-sectioning | seqFISH scRNA-seq | 1. Obtaining spatial information for partial genes by probe-dependent seqFISH 2. Spatial mapping of scRNA-seq data onto seqFISH data | [84] | |
| gut endoderm tissue at E3.5~E8.75 single cells | tissue dissection tissue dissociation cell suspension paraformaldehyde fixing | scRNA-seq ISH | 1. Generation of single-cell resolution transcriptome data by scRNA-seq 2. Validation of cell cluster- and position-specific expression profile data by ISH | [81] | |
| gastruloids tissue at 120 h after aggregation Embryo tissues at E8.5~E9.5 | cryo-sectioning | tomo-seq | Generation of transcriptome data at each section (8-μm and 20-μm sections) by Tomo-seq | [80] | |
| cerebral cortex tissue at E12.5 single cells | tissue dissociation cell suspension paraformaldehyde fixing | scRNA-seq ISH | 1. Generation of single-cell resolution transcriptome data by scRNA-seq 2. Providing spatial information for partial genes of scRNA-seq data by probe-dependent ISH | [86] | |
| stomach and intestine tissues at E9.5~E15.5 single cells | tissue dissection tissue dissociation cell suspension cryo-sectioning | scRNA-seq 10× Visium, | 1. Generation of single-cell resolution transcriptome data by scRNA-seq 2. Mapping spatial distributions of scRNA-seq data by 10× Visium | [87] | |
| embryo tissues at E9.5~E16.5 single cells | cryo-sectioning | Stereo-seq ISH scRNA-seq | 1. Generation of spatial transcriptome data at spot (220 nm) resolution by Stereo-seq 2. Validation of spatial transcriptome data by ISH 3. Spatial alignment of scRNA-seq data with Stereo-seq by Tangram | [72] | |
| embryo tissues at E7.5 | cryo-sectioning | LCM-seq | Generation of spatial transcriptome data at capture area (50–300 cells) by LCM-seq | [89] | |
| pig | uterine tissue at G12~G15 | formalin fixing cryo-sectioning | LCM-seq | Generation of spatial transcriptome data at capture area by LCM-seq | [90] |
| Marm-oset | preimplantation and postimplantation embryos (E15~E25) uterine tissue | cryo-sectioning cell picking by LCM | LCM-seq | Generation of spatial transcriptome data at capture area (1–3 cells) by LCM-seq | [95] |
| Zebra-fish | heart tissue at 2 dpf | cryo-sectioning | Tomo-seq | Generation of transcriptome data at each section (10-μm sections) by Tomo-seq | [77] |
| embryo tissues at 3.3~24 hpf single cells | cryo-sectioning paraformaldehyde fixing tissue dissociation cell suspension | Stereo-seq ISH scRNA-seq | 1. Generation of spatial transcriptome data at spot (220 nm) resolution by Stereo-seq 2. Obtaining spatial information for partial genes by probe-dependent ISH 3. Construction of single-cell- and spatial-resolution developmental trajectory by integrating scRNA-seq and Stereo-seq data | [92] | |
| Droso-phila | embryo tissues at 14–16 ELh, 14–18 E, and 1–3 L | cryo-sectioning | Stereo-seq ISH | 1. Generation of spatial transcriptome data at spot (220 nm) resolution by Stereo-seq 2. Validation of spatial transcriptome data by ISH | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, K.; Pak, U.; Pang, Y.; Hao, W.; Yang, X. Advances and Challenges in Spatial Transcriptomics for Developmental Biology. Biomolecules 2023, 13, 156. https://doi.org/10.3390/biom13010156
Choe K, Pak U, Pang Y, Hao W, Yang X. Advances and Challenges in Spatial Transcriptomics for Developmental Biology. Biomolecules. 2023; 13(1):156. https://doi.org/10.3390/biom13010156
Chicago/Turabian StyleChoe, Kyongho, Unil Pak, Yu Pang, Wanjun Hao, and Xiuqin Yang. 2023. "Advances and Challenges in Spatial Transcriptomics for Developmental Biology" Biomolecules 13, no. 1: 156. https://doi.org/10.3390/biom13010156