
1-(Arylsulfonyl-isoindol-2-yl)piperazines as 5-HT6R Antagonists: Mechanochemical Synthesis, In Vitro Pharmacological Properties and Glioprotective Activity
 , , , , , , and
, , , , , , and
Abstract
:
1. Introduction
2. Material and Methods
2.1. Chemistry
2.1.1. General Chemical Methods
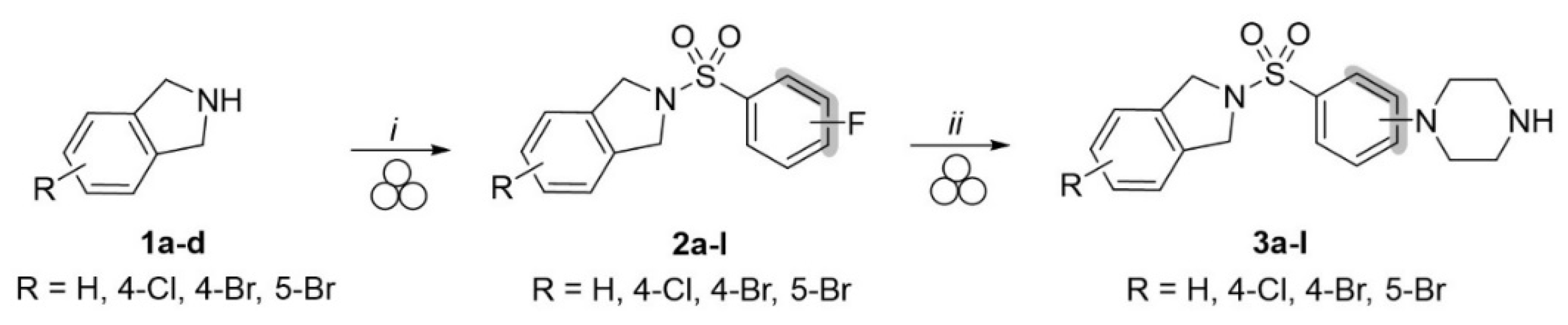
2.1.2. General Procedure A for Sulfonylation of Isoindoline Derivatives to Obtain Intermediates 2a–l
2.1.3. General Procedure B for Aromatic Substitution to Obtain Final Compounds 3a–l
2.1.4. General Procedure C for One-Pot Two-Step Reaction for Obtaining Compounds 3e, 3f, and 3g
2.1.5. Characterization Data for Final Compounds
2-{[4-(Piperazin-1-yl)phenyl]sulfonyl}isoindoline 3a
4-Chloro-2-{[4-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3b
4-Bromo-2-{[4-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3c
5-Bromo-2-{[4-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3d
2-{[3-(Piperazin-1-yl)phenyl]sulfonyl}isoindoline 3e
4-Chloro-2-{[3-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3f
4-Bromo-2-{[3-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3g
5-Bromo-2-{[3-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3h
2-{[2-(Piperazin-1-yl)phenyl]sulfonyl}isoindoline 3i
4-Chloro-2-{[2-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3j
4-Bromo-2-{[2-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3k
5-Bromo-2-{[2-(piperazin-1-yl)phenyl]sulfonyl}isoindoline 3l
2.2. In Vitro Biological Evaluation
2.2.1. Radioligand Binding Assays
2.2.2. Impact of Evaluated Compounds on cAMP Production in 1321N1 Cells
2.2.3. Impact of Evaluated Compounds on cAMP Production Elicited by Constitutively Active 5-HT6R in NG108-15 Cells
2.2.4. Impact of Tested Compounds on Cdk5-Dependent Neurite Growth
2.2.5. In Vitro Assessment of Metabolic Stability
2.2.6. In Vitro Cytotoxicity Evaluation
2.2.7. In Vitro Assessment of Glioprotective Properties
3. Result and Discussion
3.1. Mechanochemical Synthesis of Compounds 3a–3l
3.2. Radioligand Binding Assays in HEK-293 Cells and SAR Studies
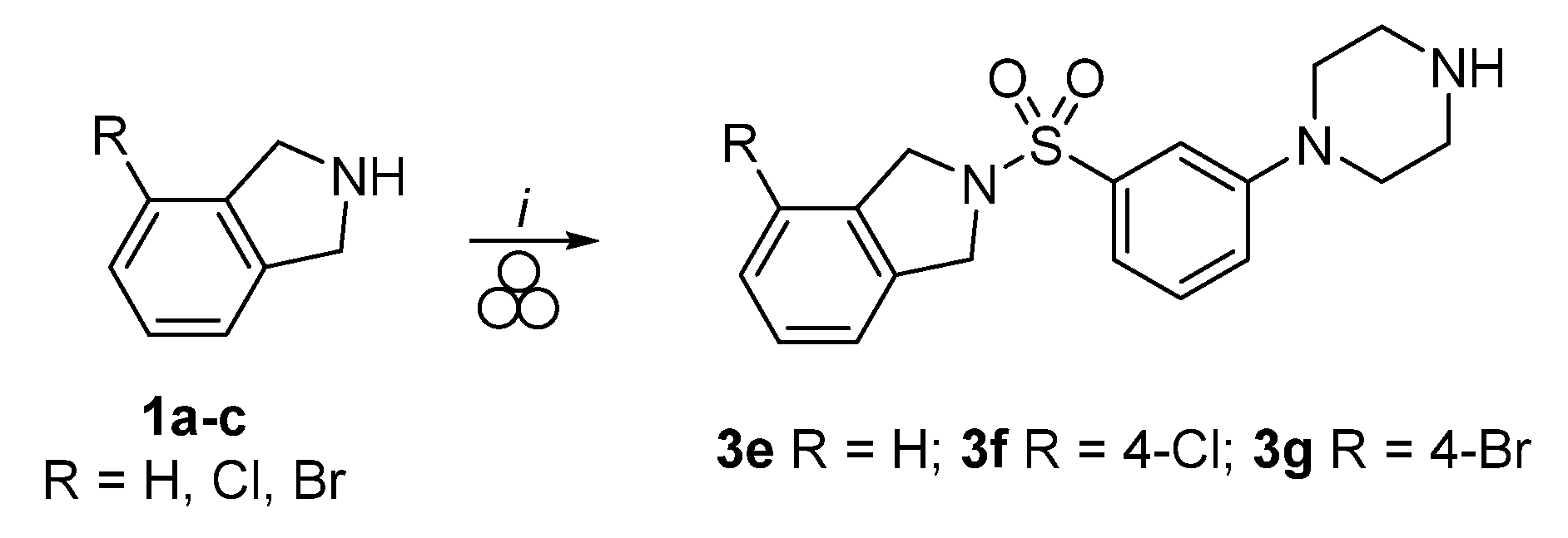
3.3. One-Pot Two-Step Mechanochemical Synthesis of Compounds 3e, 3f, and 3g
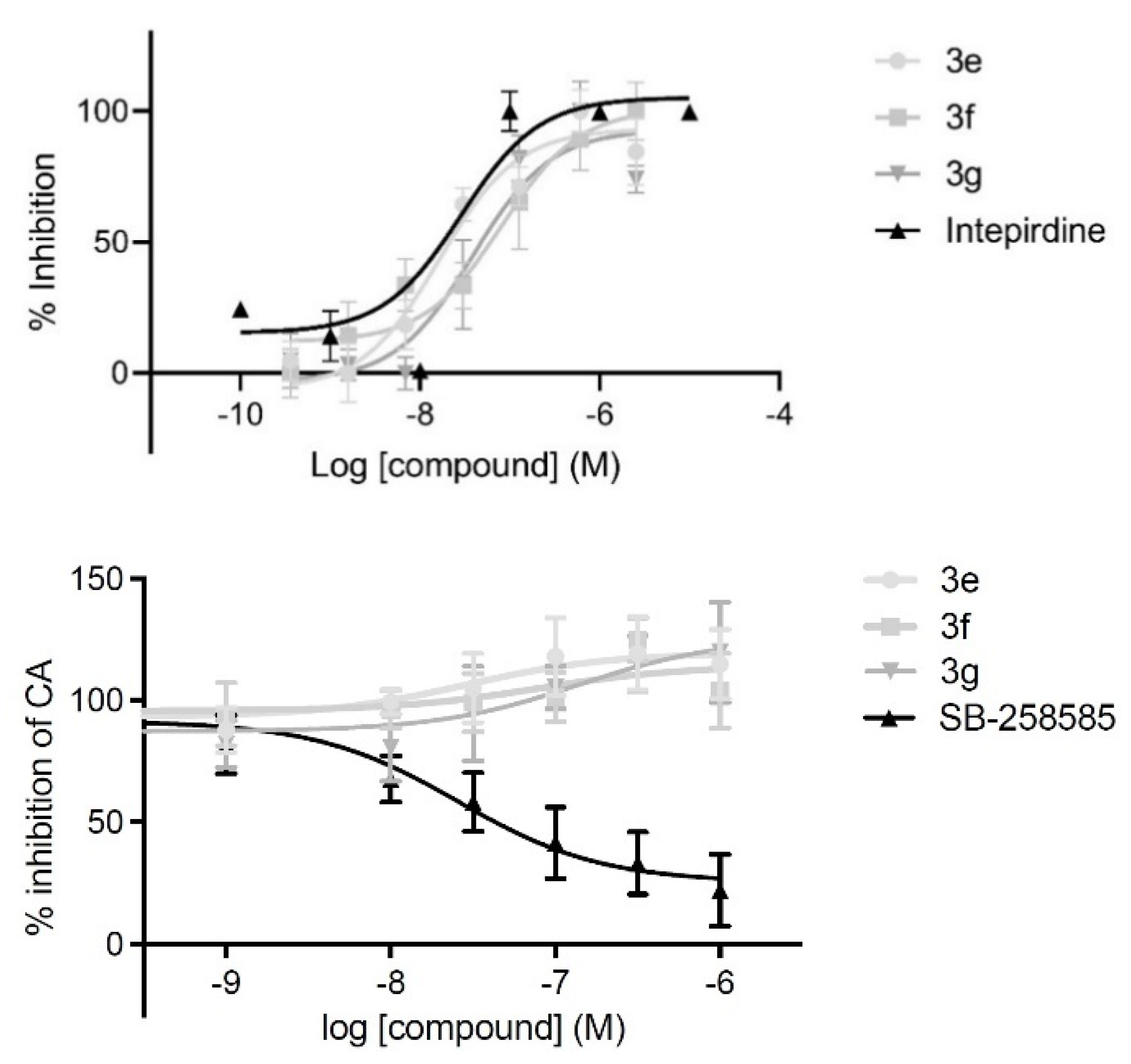
3.4. Antagonist Properties of Selected Compounds at 5-HT6R-Operated Gs Signaling
3.5. In Vitro Metabolic Stability and Preliminary Safety Assessment for 3e, 3f, and 3g
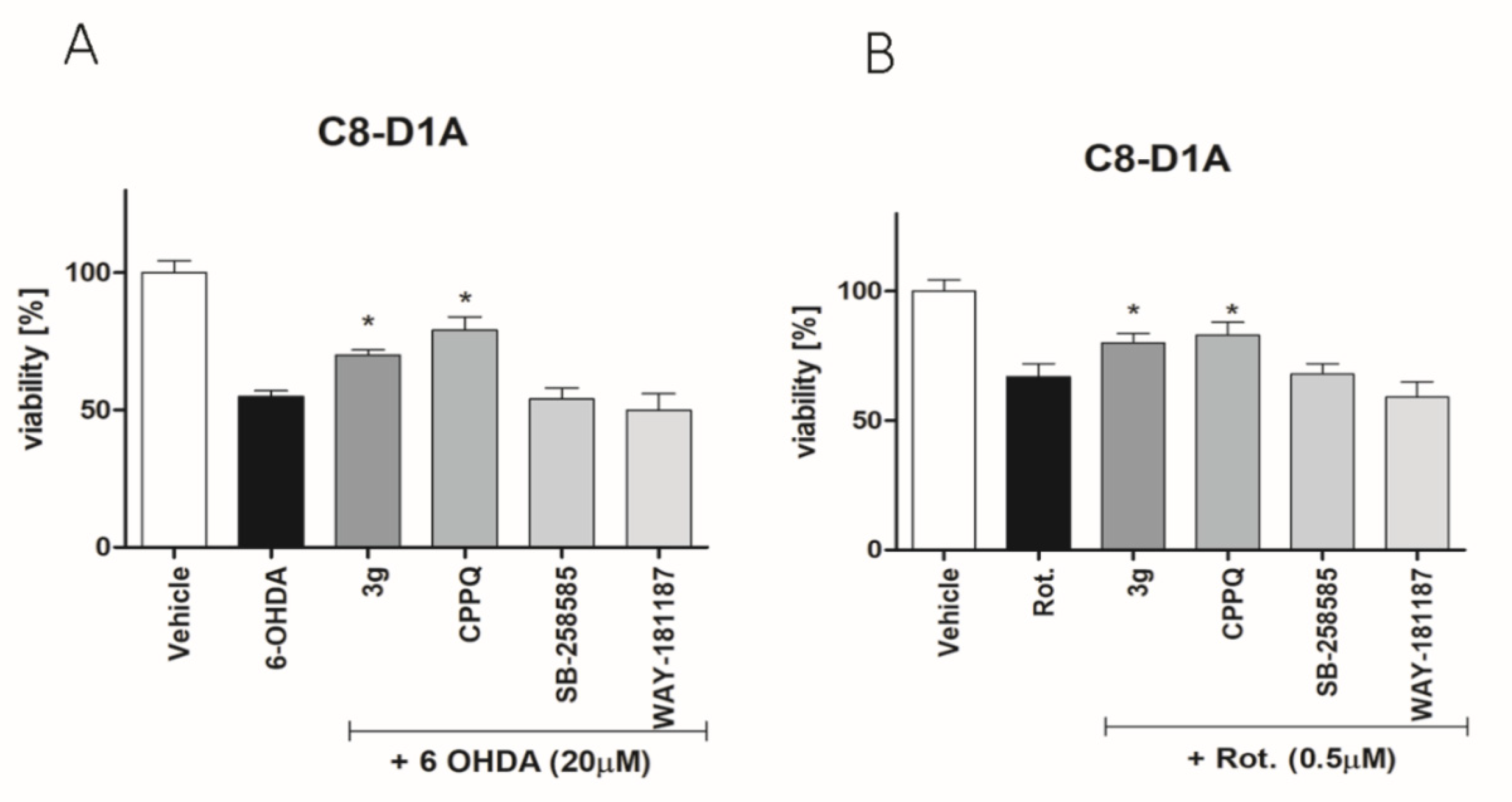
3.6. Glioprotective Properties of Compound 3g
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- Barnes, N.M.; Ahern, G.P.; Becamel, C.; Bockaert, J.; Camilleri, M.; Chaumont-Dubel, S.; Claeysen, S.; Cunningham, K.A.; Fone, K.C.; Gershon, M.; et al. International Union of Basic and Clinical Pharmacology. CX. Classification of Receptors for 5-Hydroxytryptamine; Pharmacology and Function. Pharmacol. Rev. 2021, 73, 310–520. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, P.; Marciniec, K.; Satała, G.; Canale, V.; Kos, T.; Partyka, A.; Jastrzębska-Więsek, M.; Wesołowska, A.; Basińska-Ziobroń, A.; Wójcikowski, J.; et al. N1-Azinylsulfonyl-1H-indoles: 5-HT6 Receptor Antagonists With Procognitive and Antidepressant-Like Properties. ACS Med. Chem. Lett. 2016, 7, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogendorf, A.S.; Hogendorf, A.; Kurczab, R.; Kalinowska-Tłuścik, J.; Popik, P.; Nikiforuk, A.; Krawczyk, M.; Satała, G.; Lenda, T.; Knutelska, J.; et al. 2-Aminoimidazole-Based Antagonists of the 5-HT(6) Receptor—A New Concept in Aminergic GPCR Ligand Design. Eur. J. Med. Chem. 2019, 179, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Abraham, R.; Benade, V.; Medapati, R.B.; Jayarajan, P.; Bhyrapuneni, G.; Muddana, N.; Mekala, V.R.; Subramanian, R.; Shinde, A.; et al. SUVN-502, a Novel, Potent, Pure, and Orally Active 5-HT6 Receptor Antagonist: Pharmacological, Behavioral, and Neurochemical Characterization. Behav. Pharmacol. 2019, 30, 16–35. [Google Scholar] [CrossRef]
- Sudoł, S.; Cios, A.; Jastrzębska-Więsek, M.; Honkisz-Orzechowska, E.; Mordyl, B.; Wilczyńska-Zawal, N.; Satała, G.; Kucwaj-Brysz, K.; Partyka, A.; Latacz, G.; et al. The Phenoxyalkyltriazine Antagonists for 5-HT(6) Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases. Int. J. Mol. Sci. 2021, 22, 10773. [Google Scholar] [CrossRef]
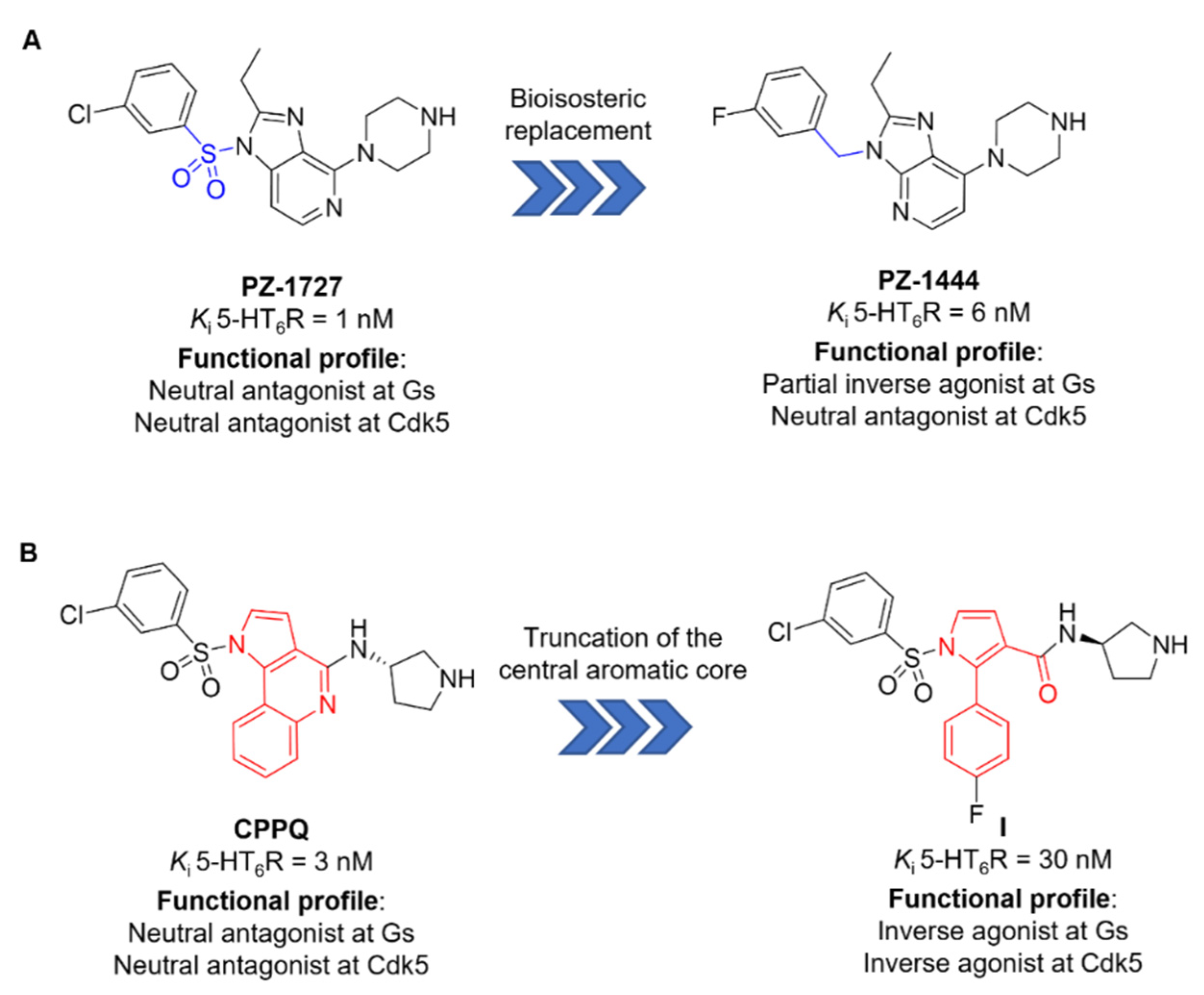
- Zajdel, P.; Grychowska, K.; Mogilski, S.; Kurczab, R.; Satała, G.; Bugno, R.; Kos, T.; Gołębiowska, J.; Malikowska-Racia, N.; Nikiforuk, A.; et al. Structure-Based Design and Optimization of FPPQ, a Dual-Acting 5-HT(3) and 5-HT(6) Receptor Antagonist with Antipsychotic and Procognitive Properties. J. Med. Chem. 2021, 64, 13279–13298. [Google Scholar] [CrossRef]
- Dayer, A.G.; Jacobshagen, M.; Chaumont-Dubel, S.; Marin, P. 5-HT6 Receptor: A New Player Controlling the Development of Neural Circuits. ACS Chem. Neurosci. 2015, 6, 951–960. [Google Scholar] [CrossRef]
- Pujol, C.N.; Dupuy, V.; Séveno, M.; Runtz, L.; Bockaert, J.; Marin, P.; Chaumont-Dubel, S. Dynamic Interactions of the 5-HT6 Receptor with Protein Partners Control Dendritic Tree Morphogenesis. Sci. Signal. 2020, 13, eaax9520. [Google Scholar] [CrossRef]
- Sheu, S.-H.; Upadhyayula, S.; Dupuy, V.; Pang, S.; Deng, F.; Wan, J.; Walpita, D.; Pasolli, H.A.; Houser, J.; Sanchez-Martinez, S.; et al. A Serotonergic Axon-Cilium Synapse Drives Nuclear Signaling to Alter Chromatin Accessibility. Cell 2022, 185, 3390–3407. [Google Scholar] [CrossRef]
- De Deurwaerdère, P.; Bharatiya, R.; Chagraoui, A.; Di Giovanni, G. Constitutive Activity of 5-HT Receptors: Factual Analysis. Neuropharmacology 2020, 168, 107967. [Google Scholar] [CrossRef]
- Berthoux, C.; Hamieh, A.M.; Rogliardo, A.; Doucet, E.L.; Coudert, C.; Ango, F.; Grychowska, K.; Chaumont-Dubel, S.; Zajdel, P.; Maldonado, R.; et al. Early 5-HT(6) Receptor Blockade Prevents Symptom Onset in a Model of Adolescent Cannabis Abuse. EMBO Mol. Med. 2020, 12, e10605. [Google Scholar] [CrossRef] [PubMed]
- Doucet, E.; Grychowska, K.; Zajdel, P.; Bockaert, J.; Marin, P.; Bécamel, C. Blockade of Serotonin 5-HT(6) Receptor Constitutive Activity Alleviates Cognitive Deficits in a Preclinical Model of Neurofibromatosis Type 1. Int. J. Mol. Sci. 2021, 22, 10178. [Google Scholar] [CrossRef] [PubMed]
- Deraredj Nadim, W.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.-L.; Zajdel, P.; Bénédetti, H.; Marin, P.; Morisset-Lopez, S. Physical Interaction Between Neurofibromin and Serotonin 5-HT6 Receptor Promotes Receptor Constitutive Activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.-Y.; Doly, S.; Hamieh, A.M.; Chapuy, E.; Canale, V.; Drop, M.; Chaumont-Dubel, S.; Bantreil, X.; Lamaty, F.; Bojarski, A.J.; et al. mTOR Activation by Constitutively Active Serotonin6 Receptors as New Paradigm in Neuropathic Pain and its Treatment. Prog. Neurobiol. 2020, 193, 101846. [Google Scholar] [CrossRef] [PubMed]
- Drop, M.; Jacquot, F.; Canale, V.; Chaumont-Dubel, S.; Walczak, M.; Satała, G.; Nosalska, K.; Mahoro, G.U.; Słoczyńska, K.; Piska, K.; et al. Neuropathic Pain-Alleviating Activity of Novel 5-HT(6) Receptor Inverse Agonists derived from 2-aryl-1H-pyrrole-3-carboxamide. Bioorg. Chem. 2021, 115, 105218. [Google Scholar] [CrossRef] [PubMed]
- Vanda, D.; Soural, M.; Canale, V.; Chaumont-Dubel, S.; Satała, G.; Kos, T.; Funk, P.; Fülöpová, V.; Lemrová, B.; Koczurkiewicz, P.; et al. Novel Non-Sulfonamide 5-HT(6) Receptor Partial Inverse Agonist in a Group of Imidazo[4,5-b]pyridines With Cognition Enhancing Properties. Eur. J. Med. Chem. 2018, 144, 716–729. [Google Scholar] [CrossRef]
- Vanda, D.; Canale, V.; Chaumont-Dubel, S.; Kurczab, R.; Satała, G.; Koczurkiewicz-Adamczyk, P.; Krawczyk, M.; Pietruś, W.; Blicharz, K.; Pękala, E.; et al. Imidazopyridine-Based 5-HT(6) Receptor Neutral Antagonists: Impact of N(1)-Benzyl and N(1)-Phenylsulfonyl Fragments on Different Receptor Conformational States. J. Med. Chem. 2021, 64, 1180–1196. [Google Scholar] [CrossRef] [PubMed]
- Grychowska, K.; Satała, G.; Kos, T.; Partyka, A.; Colacino, E.; Chaumont-Dubel, S.; Bantreil, X.; Wesołowska, A.; Pawłowski, M.; Martinez, J.; et al. Novel 1H-Pyrrolo[3,2-c]quinoline Based 5-HT6 Receptor Antagonists with Potential Application for the Treatment of Cognitive Disorders Associated with Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 972–983. [Google Scholar] [CrossRef]
- Drop, M.; Canale, V.; Chaumont-Dubel, S.; Kurczab, R.; Satała, G.; Bantreil, X.; Walczak, M.; Koczurkiewicz-Adamczyk, P.; Latacz, G.; Gwizdak, A.; et al. 2-Phenyl-1H-pyrrole-3-carboxamide as a New Scaffold for Developing 5-HT(6) Receptor Inverse Agonists with Cognition Enhancing Activity. ACS Chem. Neurosci. 2021, 12, 1228–1240. [Google Scholar] [CrossRef]
- Hirst, W.D.; Minton, J.A.; Bromidge, S.M.; Moss, S.F.; Latter, A.J.; Riley, G.; Routledge, C.; Middlemiss, D.N.; Price, G.W. Characterization of [(125)I]-SB-258585 Binding to human Recombinant and Native 5-HT(6) Receptors in Rat, Pig and Human Brain Tissue. Br. J. Pharmacol. 2000, 130, 1597–1605. [Google Scholar] [CrossRef]
- Duhr, F.; Déléris, P.; Raynaud, F.; Séveno, M.; Morisset-Lopez, S.; Mannoury la Cour, C.; Millan, M.J.; Bockaert, J.; Marin, P.; Chaumont-Dubel, S. Cdk5 Induces Constitutive Activation of 5-HT6 Receptors to Promote Neurite Growth. Nat. Chem. Biol. 2014, 10, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partyka, A.; Kurczab, R.; Canale, V.; Satała, G.; Marciniec, K.; Pasierb, A.; Jastrzębska-Więsek, M.; Pawłowski, M.; Wesołowska, A.; Bojarski, A.J.; et al. The Impact of The Halogen Bonding on D(2) and 5-HT(1A)/5-HT(7) Receptor Activity of Azinesulfonamides of 4-[(2-ethyl)piperidinyl-1-yl]phenylpiperazines With Antipsychotic and Antidepressant Properties. Bioorg. Med. Chem. 2017, 25, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R.; Canale, V.; Satała, G.; Zajdel, P.; Bojarski, A.J. Amino Acid Hot Spots of Halogen Bonding: A Combined Theoretical and Experimental Case Study of the 5-HT(7) Receptor. J. Med. Chem. 2018, 61, 8717–8733. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, P.; Kos, T.; Marciniec, K.; Satała, G.; Canale, V.; Kamiński, K.; Hołuj, M.; Lenda, T.; Koralewski, R.; Bednarski, M.; et al. Novel Multi-Target Azinesulfonamides of Cyclic Amine Derivatives as Potential Antipsychotics with Pro-Social and Pro-Cognitive Effects. Eur. J. Med. Chem. 2018, 145, 790–804. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship Between the Inhibition Constant (K1) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.-M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET Sensor to Characterize a Novel Regulation of cAMP by the Sphingosine 1-Phosphate/G13 Pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Solanki, A.; Shirsath, V.S. Comparative in-vitro Intrinsic Clearance of Imipramine in Multiple Species Liver Microsomes: Human, Rat, Mouse and Dog. J. Drug Metab. Toxicol. 2012, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Canale, V.; Kotańska, M.; Dziubina, A.; Stefaniak, M.; Siwek, A.; Starowicz, G.; Marciniec, K.; Kasza, P.; Satała, G.; Duszyńska, B.; et al. Design, Sustainable Synthesis and Biological Evaluation of a Novel Dual α2A/5-HT7 Receptor Antagonist with Antidepressant-Like Properties. Molecules 2021, 26, 3228. [Google Scholar] [CrossRef]
- Koczurkiewicz-Adamczyk, P.; Gąsiorkiewicz, B.; Piska, K.; Gunia-Krzyżak, A.; Jamrozik, M.; Bucki, A.; Słoczyńska, K.; Bojdo, P.; Wójcik-Pszczoła, K.; Władyka, B.; et al. Cinnamamide Derivatives With 4-Hydroxypiperidine Moiety Enhance Effect of Doxorubicin to Cancer Cells and Protect Cardiomyocytes Against Drug-Induced Toxicity Through CBR1 Inhibition Mechanism. Life Sci. 2022, 305, 120777. [Google Scholar] [CrossRef]
- Colacino, E.; Porcheddu, A.; Charnay, C.; Delogu, F. From Enabling Technologies to Medicinal Mechanochemistry: An Eco-Friendly Access to Hydantoin-Based Active Pharmaceutical Ingredients. React. Chem. Eng. 2019, 4, 1179–1188. [Google Scholar] [CrossRef]
- Canale, V.; Frisi, V.; Bantreil, X.; Lamaty, F.; Zajdel, P. Sustainable Synthesis of a Potent and Selective 5-HT(7) Receptor Antagonist Using a Mechanochemical Approach. J. Org. Chem. 2020, 85, 10958–10965. [Google Scholar] [CrossRef]
- Bento, O.; Luttringer, F.; Mohy El Dine, T.; Pétry, N.; Bantreil, X.; Lamaty, F. Sustainable Mechanosynthesis of Biologically Active Molecules. European J. Org. Chem. 2022, e202101516. [Google Scholar] [CrossRef]
- Andersen, J.; Brunemann, J.; Mack, J. Exploring Stable, Sub-ambient Temperatures in Mechanochemistry Via a Diverse Set of Enantioselective Reactions. React. Chem. Eng. 2019, 4, 1229–1236. [Google Scholar] [CrossRef]
- Cindro, N.; Tireli, M.; Karadeniz, B.; Mrla, T.; Užarević, K. Investigations of Thermally Controlled Mechanochemical Milling Reactions. ACS Sustain. Chem. Eng. 2019, 7, 16301–16309. [Google Scholar] [CrossRef]
- Kubota, K.; Baba, E.; Seo, T.; Ishiyama, T.; Ito, H. Palladium-Catalyzed Solid-State Borylation of Aryl Halides Using Mechanochemistry. Beilstein J. Org. Chem. 2022, 18, 855–862. [Google Scholar] [CrossRef]
- Reynes, J.F.; Garcia, F. Temperature-Controlled Mechanochemistry Unlocks The Nickel-Catalyzed Suzuki-Miyaura-Type Coupling of Aryl Sulfamates at Different Scales. Angew. Chemie Int. Ed. 2022. [Google Scholar] [CrossRef]
- Chaumont-Dubel, S.; Dupuy, V.; Bockaert, J.; Bécamel, C.; Marin, P. The 5-HT(6) Receptor Interactome: New Insight in Receptor Signaling and Its Impact on Brain Physiology and Pathologies. Neuropharmacology 2020, 172, 107839. [Google Scholar] [CrossRef]
- Allen, N.J.; Lyons, D.A. Glia as Architects of Central Nervous System Formation and Function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Grychowska, K.; Chaumont-Dubel, S.; Kurczab, R.; Koczurkiewicz, P.; Deville, C.; Krawczyk, M.; Pietruś, W.; Satała, G.; Buda, S.; Piska, K.; et al. Dual 5-HT(6) and D(3) Receptor Antagonists in a Group of 1H-Pyrrolo[3,2-c]quinolines with Neuroprotective and Procognitive Activity. ACS Chem. Neurosci. 2019, 10, 3183–3196. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| ID | R | R1 | Ki [nM] ± SEM a |
| 3a | H | 4-piperazinyl | 6 ± 1 |
| 3b | 4-Cl | 7 ± 1 | |
| 3c | 4-Br | 10 ± 2 | |
| 3d | 5-Br | 87 ± 12 | |
| 3e | H | 3-piperazinyl | 2 ± 0.8 |
| 3f | 4-Cl | 1 ± 0.2 | |
| 3g | 4-Br | 1 ± 0.3 | |
| 3h | 5-Br | 4 ± 0.5 | |
| 3i | H | 2-piperazinyl | 977 ± 43 |
| 3j | 4-Cl | 918 ± 55 | |
| 3k | 4-Br | 32 ± 5 | |
| 3l | 5-Br | 1175 ± 99 | |
| SB-258585 b | - | - | 8.9 |
 | |||||||
|---|---|---|---|---|---|---|---|
| ID | 5-HT6R | Ki [nM] a | |||||
| Ki [nM] a | Kb [nM] b | Functional Profileat Gs Signalingc | 5-HT1AR | 5-HT2AR | 5-HT7R | D2 | |
| 3e | 2 ± 0.2 | 0.7 ± 0.2 | Neutral antagonist | 951 ± 120 | 128 ± 37 | 4529 ± 327 | 3424 ± 415 |
| 3f | 1 ± 0.2 | 3.5 ± 0.8 | Neutral antagonist | 385 ± 71 | 170 ± 32 | 10660 ± 1025 | 2026 ± 299 |
| 3g | 1 ± 0.4 | 1.6 ± 0.3 | Neutral antagonist | 680 ± 33 | 159 ± 25 | 8798 ± 854 | 2440 ± 324 |
| SB-258585 d | 8.9 | NT | Inverse agonist | 645 | 1023 | 3388 | 3802 |
| ID | Metabolic Stability a | Cytotoxicity b | ||||
|---|---|---|---|---|---|---|
| t1/2 [min] | Clint [µL/min/mg] | Major Metabolite | HepG2 | SH-SY5Y | C8-D1A | |
| 3e | 367 | 3.78 | not detected | IC50 > 50 µM | IC50 = 42 µM | IC50 > 50 µM |
| 3f | 76.95 | 18.01 | hydroxylated | IC50 > 50 µM | IC50 = 38 µM | IC50 > 50 µM |
| 3g | 94.41 | 14.68 | hydroxylated | IC50 > 50 µM | IC50 = 48 µM | IC50 > 50 µM |
| Donepezil | 379 | 3.65 | not detected | NT | NT | NT |
| Imipramine | 11.98 | 115.65 | Des-methylated | NT | NT | NT |
| Doxorubicin | NT | NT | NT | IC50 = 10.8 µM | IC50 = 4.4 µM | IC50 = 12.3 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canale, V.; Trybała, W.; Chaumont-Dubel, S.; Koczurkiewicz-Adamczyk, P.; Satała, G.; Bento, O.; Blicharz-Futera, K.; Bantreil, X.; Pękala, E.; Bojarski, A.J.; et al. 1-(Arylsulfonyl-isoindol-2-yl)piperazines as 5-HT6R Antagonists: Mechanochemical Synthesis, In Vitro Pharmacological Properties and Glioprotective Activity. Biomolecules 2023, 13, 12. https://doi.org/10.3390/biom13010012
Canale V, Trybała W, Chaumont-Dubel S, Koczurkiewicz-Adamczyk P, Satała G, Bento O, Blicharz-Futera K, Bantreil X, Pękala E, Bojarski AJ, et al. 1-(Arylsulfonyl-isoindol-2-yl)piperazines as 5-HT6R Antagonists: Mechanochemical Synthesis, In Vitro Pharmacological Properties and Glioprotective Activity. Biomolecules. 2023; 13(1):12. https://doi.org/10.3390/biom13010012
Chicago/Turabian StyleCanale, Vittorio, Wojciech Trybała, Séverine Chaumont-Dubel, Paulina Koczurkiewicz-Adamczyk, Grzegorz Satała, Ophélie Bento, Klaudia Blicharz-Futera, Xavier Bantreil, Elżbieta Pękala, Andrzej J. Bojarski, and et al. 2023. "1-(Arylsulfonyl-isoindol-2-yl)piperazines as 5-HT6R Antagonists: Mechanochemical Synthesis, In Vitro Pharmacological Properties and Glioprotective Activity" Biomolecules 13, no. 1: 12. https://doi.org/10.3390/biom13010012