
Recent Advances of Degradation Technologies Based on PROTAC Mechanism


Abstract
:1. Introduction
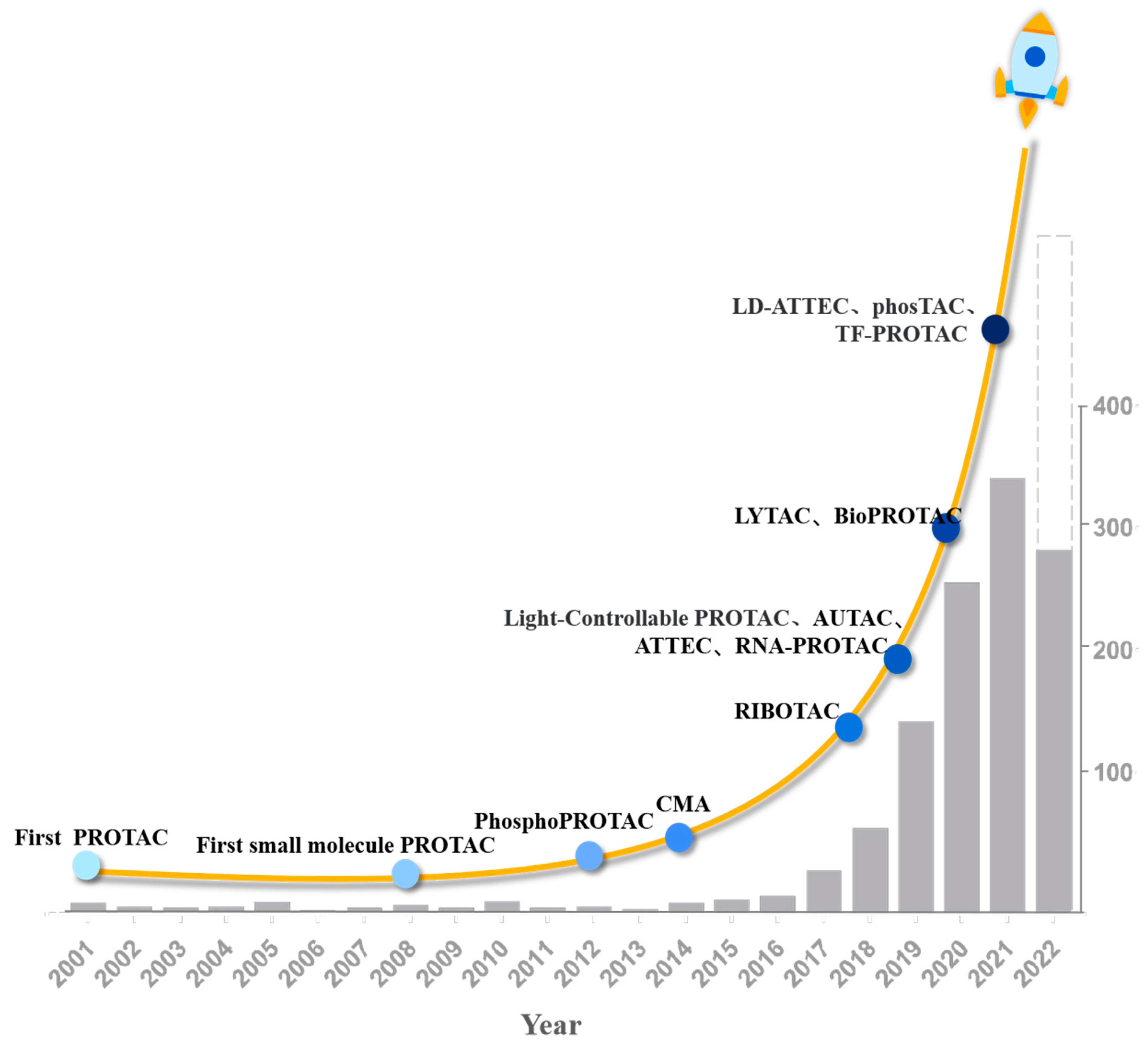
2. Development History of PROTAC
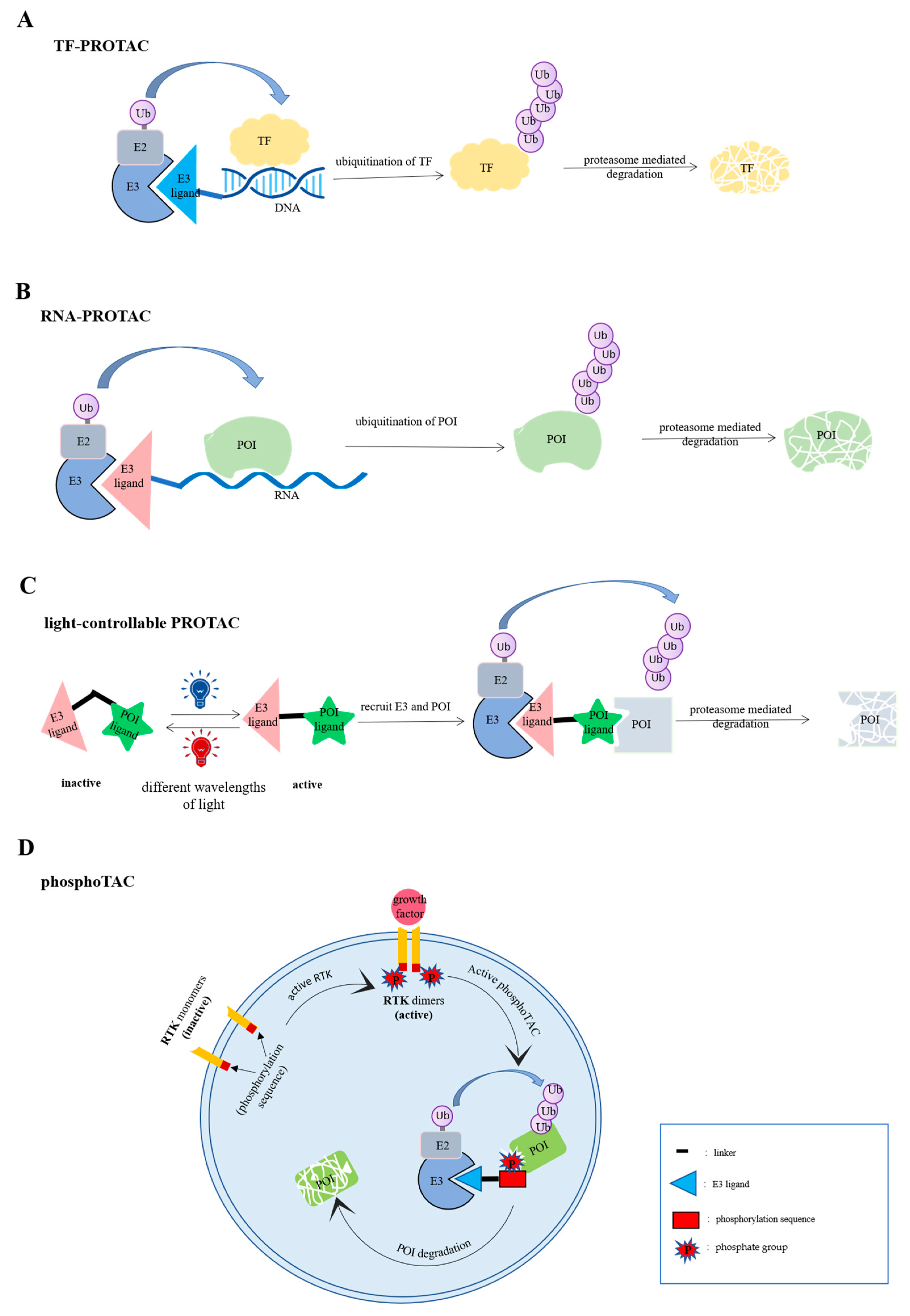
3. TF-PROTACs
4. Light-Controllable PROTAC
5. PhosphoTAC
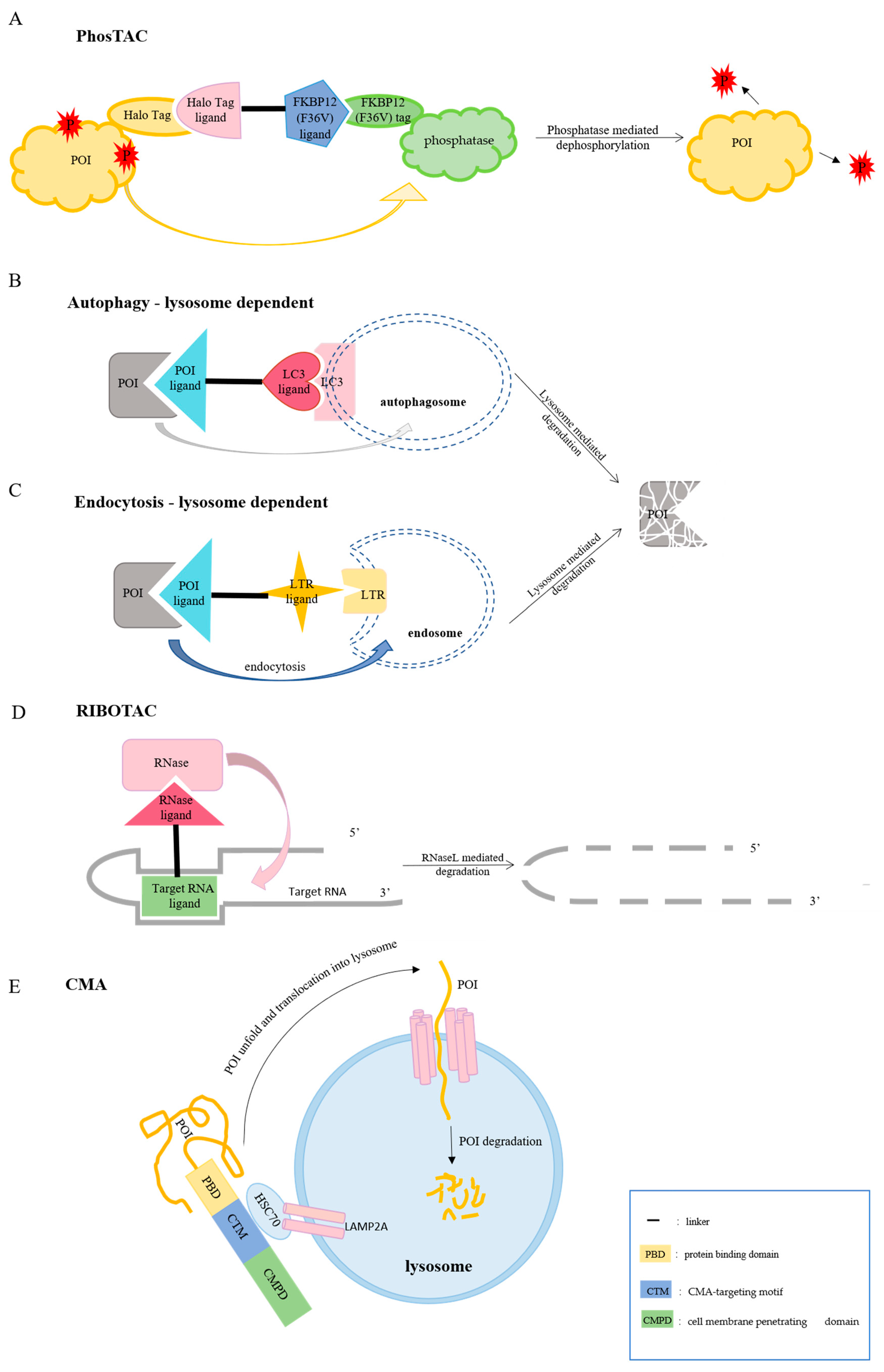
6. PhosTAC
7. LYTAC
8. AUTAC
9. ATTEC
10. CMA
11. RNA-PROTAC
12. RIBOTACs
13. Clinical Progress
14. Current Challenges and Future Prospects of Degradation Technologies Based on PROTAC Mechanism
14.1. More Diversity
14.2. Suitable for More Diseases
14.3. Stronger Druggability
14.4. Based on More Mechanisms
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| ALK | Anaplastic lymphoma kinase |
| AR | androgen receptors |
| ATTEC | autophagosome-tethering compound |
| AUTAC | autophagy-targeting chimera |
| AZO-PROTACs | azobenzene-proteolysis targeting chimeras |
| BRD4 | Bromodomain-containing protein 4 |
| CDK6 | Cyclin-dependent kinase 6 |
| CDK9 | Cyclin-dependent kinase 9 |
| CI-M6PR | Cation-independent mannose-6-phosphate receptor |
| CLL | Chronic lymphatic leukemia |
| CMA | Chaperone-mediated autophagy |
| CMPD | cell membrane penetrating domain |
| COVID | coronavirus disease |
| CRBN | Cereblon |
| CTM | CMA-targeting motif |
| DAPK1 | death-associated protein kinase 1 |
| DCAF16 | DDB1 and CUL4 associated factor 16 |
| DLBCL | Diffuse large B-cell lymphoma |
| E2F | Early 2 factor |
| EGFR | Epidermal Growth Factor Receptor |
| ER | estrogen receptors |
| ERRα | estrogen-related receptor alpha |
| FBDD | fragment-based drug design |
| FEM1B | fem-1 homolog B |
| FKBP12 | FK506 binding protein 12 |
| FL | Follicular lymphoma |
| FOXO3a | Forkhead box class O3a |
| FRS2α | factor receptor substrate 2α |
| HCV | hepatitis C virus |
| HDAC | Histone deacetylase |
| HH8 | human herpesvirus 8 |
| HPV | human papillomavirus |
| HSC70 | heat shock cognate protein of 70KDa |
| HTLV | human T-lymphocyte virus |
| IAP | Inhibitor of Apoptosis |
| ImiDs | immunomodulatory drugs |
| KRAS | Kirsten rat sarcoma |
| LAMP2A | lysosomal-associated membrane protein 2A |
| LC3 | proteolysis-targeting chimeras |
| LD-ATTEC | lipid droplets ATTEC |
| LTRs | lysosome-targeting receptors |
| M6P | mannose-6-phosphate |
| MCL | Mantle cell lymphoma |
| MDM2 | Mouse double minute 2 |
| MetAp-2 | Methionine Aminopeptidase-2 |
| MetAP2 | methionine aminopeptidase 2 |
| mHTT | Mutant huntingtin protein |
| MZL | Marginal zone lymphoma |
| NF-κB | nuclear factor-κB |
| PCNSL | Primary central nervous system lymphoma |
| pc-PROTAC | photo-caged targeting chimera |
| PDCD4 | programmed cell death 4 |
| PD-L1 | Programmed Cell Death-Ligand 1 |
| PDT | protein degradation technology |
| PEG | poly (ethylene glycol) |
| PhosphoTAC | phospho-dependent proteolysis targeting chimeras |
| PhosTACs | phosphorylation targeting chimeras |
| PHOTAC | photochemically targeting chimera |
| photoPROTACs | Photo-switchable proteolysis targeting chimeras |
| PI3K | phosphatidylinositol-3 kinase |
| POI | protein of interest |
| PROTAC | proteolysis-targeting chimera |
| RBPs | RNA binding proteins |
| RIBOTACs | ribonuclease targeting chimera |
| RIPK2 | Receptor-Interacting Serine/Threonine Protein Kinase 2 |
| RKT | receptor tyrosine kinase |
| RNase | ribonuclease |
| RNF114 | ring finger protein 114 |
| RTK | receptor tyrosine kinase |
| SBDD | structure-based drug design |
| SCF | Skp1-cullin-1-F-box protein |
| Sirt2 | Sirtuin 2 |
| SLL | small lymphatic lymphoma |
| TFs | Transcription factors |
| TrkA | tropomyosin receptor kinase A |
| TSPO | Translocator protein |
| UPS | ubiquitin-proteasome system |
| UVA | ultraviolet A |
| VHL | von Hippel-Lindau |
| WM | Waldenstrom macroglobulinemia |
| βTRCP | β-transducin repeat-containing protein |
| ALK | Anaplastic lymphoma kinase |
References
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ma, J.; Liu, Y.; Xia, J.; Li, Y.; Wang, Z.P.; Wei, W. PROTACs: A novel strategy for cancer therapy. Semin. Cancer Biol. 2020, 67, 171–179. [Google Scholar] [CrossRef]
- Bond, M.J.; Crews, C.M. Proteolysis targeting chimeras (PROTACs) come of age: Entering the third decade of targeted protein degradation. RSC Chem. Biol. 2021, 2, 725–742. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, J.; Hu, J.; Qi, H.; Xu, H. Research Progress of Proteolysis Targeting Chimeria in NSCLC Therapy. Zhongguo Fei Ai Za Zhi 2022, 25, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.; Crews, C.M. Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef]
- Bargagna-Mohan, P.; Baek, S.H.; Lee, H.; Kim, K.; Mohan, R. Use of PROTACS as molecular probes of angiogenesis. Bioorganic Med. Chem. Lett. 2005, 15, 2724–2727. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 2020, 27, 19–31.e16. [Google Scholar] [CrossRef]
- You, I.; Erickson, E.C.; Donovan, K.A.; Eleuteri, N.A.; Fischer, E.S.; Gray, N.S.; Toker, A. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem. Biol. 2020, 27, 66–73.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef]
- Peng, L.; Zhang, Z.; Lei, C.; Li, S.; Zhang, Z.; Ren, X.; Chang, Y.; Zhang, Y.; Xu, Y.; Ding, K. Identification of New Small-Molecule Inducers of Estrogen-related Receptor α (ERRα) Degradation. ACS Med. Chem. Lett. 2019, 10, 767–772. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e63. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Spradlin, J.N.; Boike, L.; Tong, B.; Brittain, S.M.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol. 2021, 28, 559–566.e515. [Google Scholar] [CrossRef]
- Tong, B.; Spradlin, J.N.; Novaes, L.F.T.; Zhang, E.; Hu, X.; Moeller, M.; Brittain, S.M.; McGregor, L.M.; McKenna, J.M.; Tallarico, J.A.; et al. A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL. ACS Chem. Biol. 2020, 15, 1788–1794. [Google Scholar] [CrossRef]
- Miah, A.H.; Smith, I.E.D.; Rackham, M.; Mares, A.; Thawani, A.R.; Nagilla, R.; Haile, P.A.; Votta, B.J.; Gordon, L.J.; Watt, G.; et al. Optimization of a Series of RIPK2 PROTACs. J. Med. Chem. 2021, 64, 12978–13003. [Google Scholar] [CrossRef] [PubMed]
- Steinebach, C.; Ng, Y.L.D.; Sosič, I.; Lee, C.S.; Chen, S.; Lindner, S.; Vu, L.P.; Bricelj, A.; Haschemi, R.; Monschke, M.; et al. Systematic exploration of different E3 ubiquitin ligases: An approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11, 3474–3486. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Kaniskan, H.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef]
- Reynders, M.; Trauner, D. PHOTACs Enable Optical Control of Protein Degradation. Methods Mol. Biol. 2021, 2365, 315–329. [Google Scholar] [CrossRef]
- Jin, Y.H.; Lu, M.C.; Wang, Y.; Shan, W.X.; Wang, X.Y.; You, Q.D.; Jiang, Z.Y. Azo-PROTAC: Novel Light-Controlled Small-Molecule Tool for Protein Knockdown. J. Med. Chem. 2020, 63, 4644–4654. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell. Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lin, H.; Wu, G.; Zhu, M.; Li, M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 760971. [Google Scholar] [CrossRef]
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.H.; Hu, Z.; An, E.; Okeke, I.; Zheng, S.; Luo, X.; Gong, A.; Jaime-Figueroa, S.; Crews, C.M. Modulation of Phosphoprotein Activity by Phosphorylation Targeting Chimeras (PhosTACs). ACS Chem. Biol. 2021, 16, 2808–2815. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Prata, M.J.; Alves, S. A shortcut to the lysosome: The mannose-6-phosphate-independent pathway. Mol. Genet. Metab. 2012, 107, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10. [Google Scholar] [CrossRef]
- Li, Z.; Wang, C.; Wang, Z.; Zhu, C.; Li, J.; Sha, T.; Ma, L.; Gao, C.; Yang, Y.; Sun, Y.; et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575, 203–209. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, N.; Wang, Z.; Luo, S.; Ding, Y.; Lu, B. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 2021, 31, 965–979. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, A.; Cléry, A.; Halloy, F.; Allain, F.H.T.; Hall, J. RNA-PROTACs: Degraders of RNA-Binding Proteins. Angew. Chem. Int. Ed. Engl. 2021, 60, 3163–3169. [Google Scholar] [CrossRef]
- Wang, E.T.; Taliaferro, J.M.; Lee, J.A.; Sudhakaran, I.P.; Rossoll, W.; Gross, C.; Moss, K.R.; Bassell, G.J. Dysregulation of mRNA Localization and Translation in Genetic Disease. J. Neurosci. 2016, 36, 11418–11426. [Google Scholar] [CrossRef]
- Costales, M.G.; Matsumoto, Y.; Velagapudi, S.P.; Disney, M.D. Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 2018, 140, 6741–6744. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, X.; Abegg, D.; Tanaka, T.; Tong, Y.; Benhamou, R.I.; Baisden, J.; Crynen, G.; Meyer, S.M.; Cameron, M.D.; et al. Reprogramming of Protein-Targeted Small-Molecule Medicines to RNA by Ribonuclease Recruitment. J. Am. Chem. Soc. 2021, 143, 13044–13055. [Google Scholar] [CrossRef]
- Su, X.; Ma, W.; Feng, D.; Cheng, B.; Wang, Q.; Guo, Z.; Zhou, D.; Tang, X. Efficient Inhibition of SARS-CoV-2 Using Chimeric Antisense Oligonucleotides through RNase L Activation*. Angew. Chem. Int. Ed. Engl. 2021, 60, 21662–21667. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Q.; Jiang, T.; Li, S.; Ye, J.; Zheng, J.; Wang, X.; Liu, Y.; Deng, M.; Ke, D.; et al. A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics 2021, 11, 5279–5295. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Liu, J.; Wei, W.; Rezaeian, A.H. PROTACs technology for treatment of Alzheimer’s disease: Advances and perspectives. Acta Mater. Med. 2022, 1, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021, 20, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Rozenbaum, R.T.; Andrén, O.C.J.; van der Mei, H.C.; Woudstra, W.; Busscher, H.J.; Malkoch, M.; Sharma, P.K. Penetration and Accumulation of Dendrons with Different Peripheral Composition in Pseudomonas aeruginosa Biofilms. Nano Lett. 2019, 19, 4327–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Technology | Degradation Mechanism | Advantage | Disadvantage |
|---|---|---|---|
| TF-PROTACs | ubiquitin-proteasome system | TFs without active sites or allosteric regulatory pockets can be degraded by TF-PROTAC. | It is difficult to design TF-PROTAC with an unknown DNA-binding sequence. |
| Light-controllable PROTAC | The activity can be controlled bydifferent lights. | The PROTAC may be not effective for deep tissue that light cannot penetrate. | |
| PhosphoTAC | The activity of PROTAC is dependent on the phosphorylation of the signal pathway. | Mutations of phosphorylation sites may affect the activity of PhosphoTAC. | |
| RNA-PROTAC | RNA-PROTAC specifically degrades RNA-binding proteins. | RNA-PROTAC may be easily degraded since RNA is unstable. | |
| PhosTACs | phosphatase | Compared with degrading the target protein, PhosTAC induces the dephosphorylation of the target protein, which is a more precise way of regulating the protein. | Protein dephosphorylation induced by PhosTAC is only applicable to diseases caused by abnormal phosphorylation. |
| CMA | CMA-lysosome | The peptide of CMA is easy to design. | CMA is chimeric polypeptides, so it has poor transmembrane ability and low stability. |
| LYTAC | Endocytosis-lysosome | LYTAC can induce targeted degradation of secreted and cell membrane proteins | LYTAC is not stable enough in vivo. In addition, the LTR ligands of LYTACs are chemically synthesized sugar, which may produce strong immunogenicity in the body. |
| AUTAC | autophagy-lysosome | induce the degradation of proteins and organelles by lysosomes. | The degradation process is complicated and there are many influencing factors. |
| ATTEC | ATTEC can degrade not only proteins but also lipid droplets. In addition, ATTEC molecules are small, so it is easy to penetrate cell membranes. | Whether the ATTEC will affect the overall autophagy activity and how to avoid the non-specific degradation of autophagy-related proteins remains to be further explored. | |
| RIBOTAC | RNaseL | RIBOTAC selectively degrades target RNA. | It is difficult to develop target RNA ligands. |
| Drug | Company | Targeted Protein | Indication | Stage of Clinical Trial |
|---|---|---|---|---|
| ARV-110 | Arvinas | Androgen receptor (AR) | Metastatic castrate resistant prostate cancer | Phase II |
| ARV-471 | Arvinas | Estrogen Receptor-α(ER-α) | ER+/HER2-Breast cancer | Phase II |
| ARV-766 | Arvinas | Androgen receptor (AR) | Metastatic castrate resistant prostate cancer | Phase I |
| DT2216 | Dialectic | BCL-XL | Liquid and solid tumors | Phase I |
| KT-474 | Kymera/Sanofi | Interleukin 1 receptor associated kinase4(IRAK4) | Autoimmune diseases | Phase I |
| NX-2127 | Nurix | Bruton tyrosine kinase (BTK) | B-cell Malignancies, including CLL, SLL, WM, MCL, MZL, FL, DLBCL | Phase I |
| NX-5948 | Nurix | Bruton tyrosine kinase (BTK) | B-cell Malignancies, including CLL, SLL, DLBCL, FL, MCL, MZL, WM, PCNSL | Phase I |
| FHD-609 | Foghorn | Bromodomain containing9(BRD9) | Synovial Sarcoma | Phase I |
| HSK29116 | Haisco | Bruton tyrosine kinase (BTK) | B-cell Malignancies | Phase I |
| BGB-16673 | BeiGene | Bruton tyrosine kinase (BTK) | B-cell Malignancies | Phase I |
| AR-LDD | Bristol Myers Squibb | Androgen receptor (AR) | Prostate Cancer | Phase I |
| KT-413 | Kymera | Interleukin 1 receptor associated kinase4(IRAK4) | MYD88-mutant Diffuse Large B-Cell Lymphoma | Phase I |
| KT333 | Kymera | signal transducer and activator of transcription 3 | Liquid and solid tumors | Phase I |
| GT-00029 | Kintor | Androgen receptor (AR) | Androgenetic alopecia and acne | Phase I |
| AC0682 | Accutar | Estrogen Receptor (ER) | Breast cancer | Phase I |
| AC0176 | Accutar | Androgen receptor (AR) | Prostate cancer | Phase I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Zhao, J.; Wang, Q.; Liu, J.; Ma, L. Recent Advances of Degradation Technologies Based on PROTAC Mechanism. Biomolecules 2022, 12, 1257. https://doi.org/10.3390/biom12091257
Xiao M, Zhao J, Wang Q, Liu J, Ma L. Recent Advances of Degradation Technologies Based on PROTAC Mechanism. Biomolecules. 2022; 12(9):1257. https://doi.org/10.3390/biom12091257
Chicago/Turabian StyleXiao, Mingchao, Jiaojiao Zhao, Qiang Wang, Jia Liu, and Leina Ma. 2022. "Recent Advances of Degradation Technologies Based on PROTAC Mechanism" Biomolecules 12, no. 9: 1257. https://doi.org/10.3390/biom12091257