
The Emerging Role of Deubiquitinases in Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
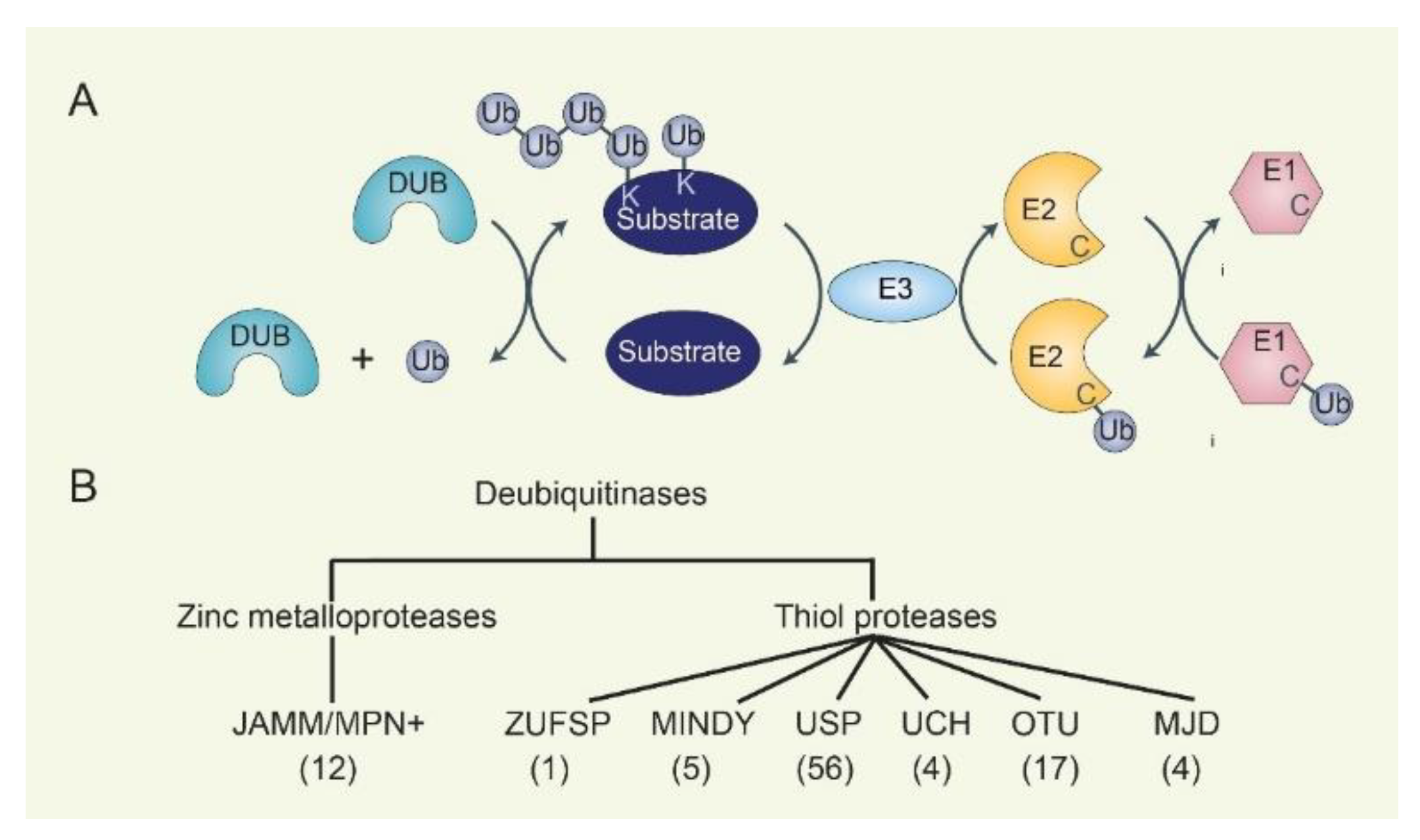
2. The Ubiquitination-Deubiquitination Cycle
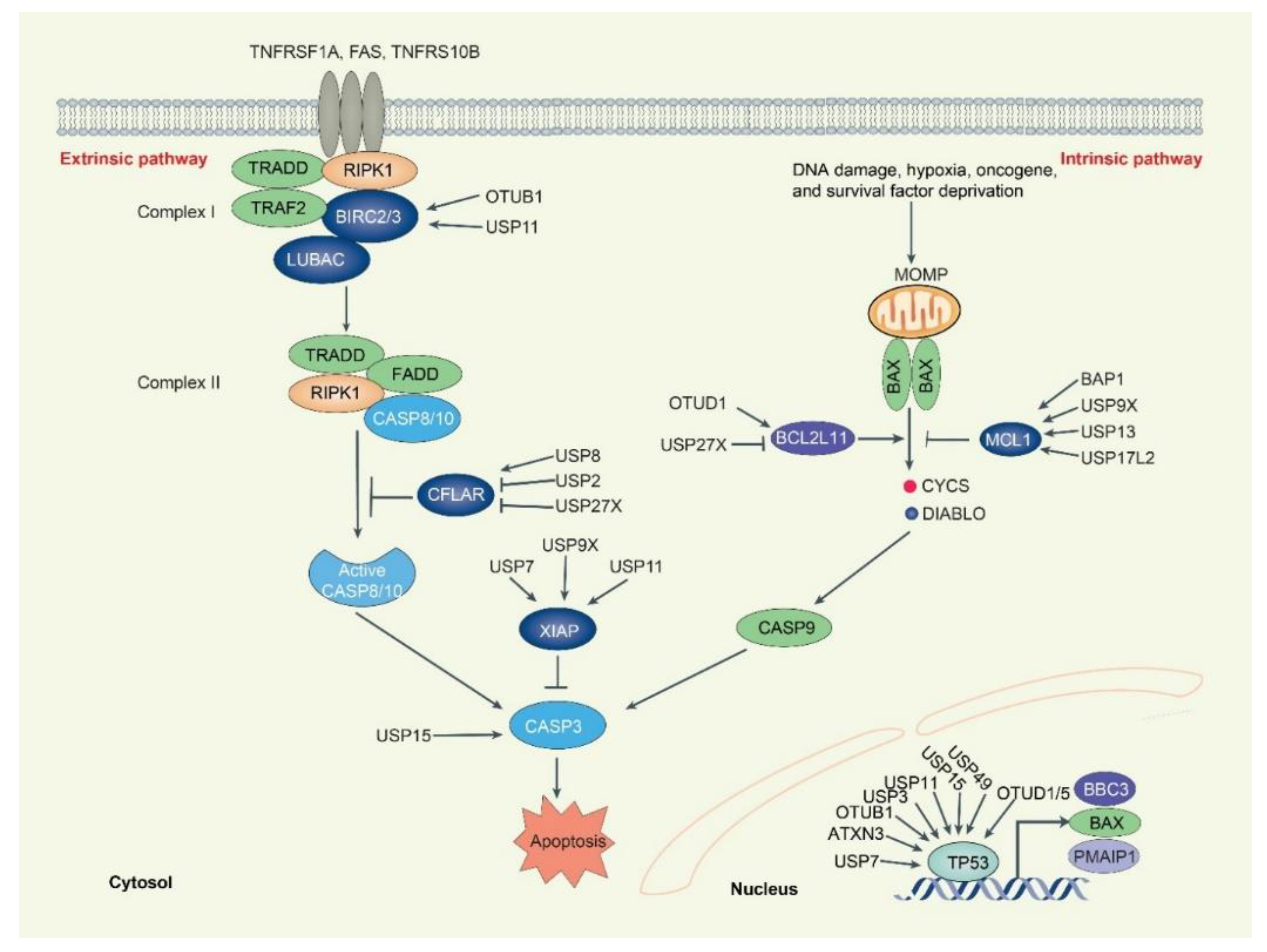
3. DUBs in Apoptosis
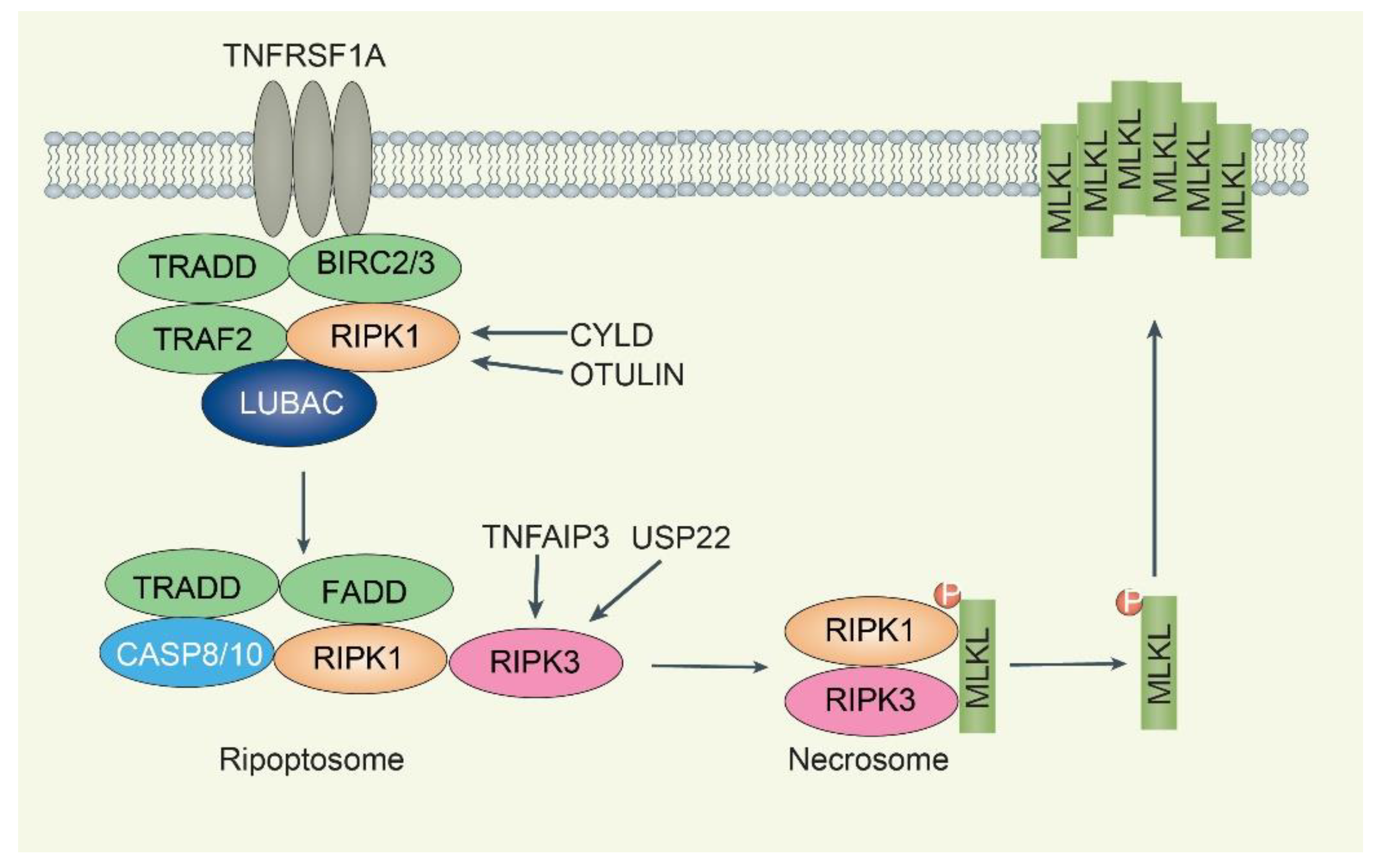
4. DUBs in Necroptosis
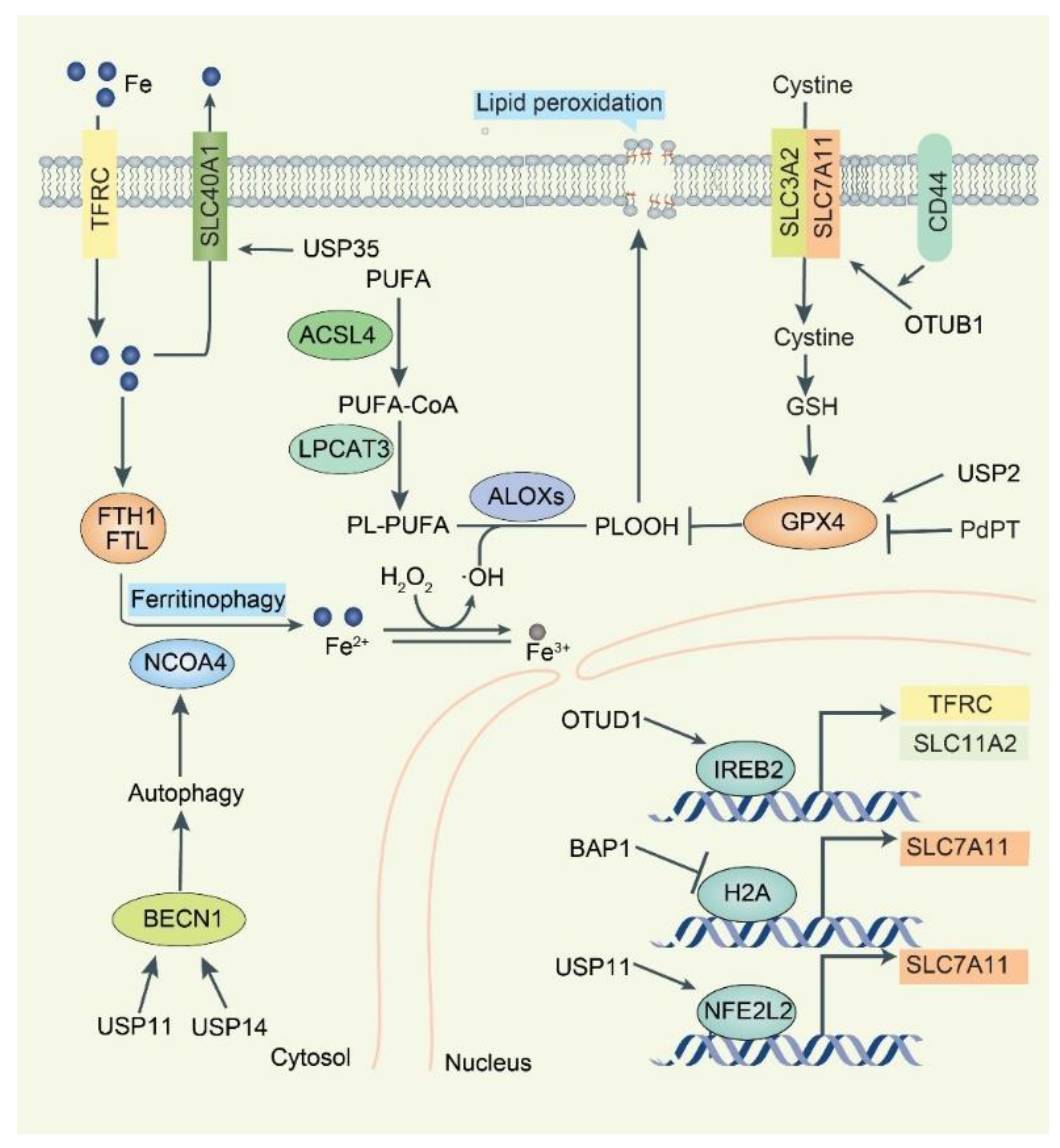
5. DUBs in Ferroptosis
6. DUBs in Pyroptosis
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A. Lessons from the discovery of the ubiquitin system. Trends Biochem. Sci. 1996, 21, 445–449. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164.e156. [Google Scholar] [CrossRef] [Green Version]
- Ronau, J.A.; Beckmann, J.F.; Hochstrasser, M. Substrate specificity of the ubiquitin and Ubl proteases. Cell Res. 2016, 26, 441–456. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 139–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.S.; Mo, S.T.; Hsu, P.N.; Lai, M.Z. c-FLIP is a target of the E3 ligase deltex1 in gastric cancer. Cell Death Dis. 2018, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Jeong, M.; Lee, E.W.; Seong, D.; Seo, J.; Kim, J.H.; Grootjans, S.; Kim, S.Y.; Vandenabeele, P.; Song, J. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability. Oncogene 2017, 36, 458–470. [Google Scholar] [CrossRef]
- Dold, M.N.; Ng, X.; Alber, C.; Gentle, I.E.; Hacker, G.; Weber, A. The deubiquitinase Usp27x as a novel regulator of cFLIPL protein expression and sensitizer to death-receptor-induced apoptosis. Apoptosis 2022, 27, 112–132. [Google Scholar] [CrossRef]
- Haimerl, F.; Erhardt, A.; Sass, G.; Tiegs, G. Down-regulation of the de-ubiquitinating enzyme ubiquitin-specific protease 2 contributes to tumor necrosis factor-alpha-induced hepatocyte survival. J. Biol. Chem. 2009, 284, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Goncharov, T.; Niessen, K.; de Almagro, M.C.; Izrael-Tomasevic, A.; Fedorova, A.V.; Varfolomeev, E.; Arnott, D.; Deshayes, K.; Kirkpatrick, D.S.; Vucic, D. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J. 2013, 32, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.W.; Seong, D.; Seo, J.; Jeong, M.; Lee, H.K.; Song, J. USP11-dependent selective cIAP2 deubiquitylation and stabilization determine sensitivity to Smac mimetics. Cell Death Differ. 2015, 22, 1463–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Galban, S.; Duckett, C.S. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010, 17, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Luo, A.; Shrivastava, I.; He, M.; Huang, Y.; Bahar, I.; Liu, Z.; Wan, Y. Regulation of XIAP Turnover Reveals a Role for USP11 in Promotion of Tumorigenesis. EBioMedicine 2017, 15, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, K.; Rudelius, M.; Slawska, J.; Jacobs, L.; Ahangarian Abhari, B.; Altmann, B.; Kurutz, J.; Rathakrishnan, A.; Fernandez-Saiz, V.; Brunner, A.; et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol. Med. 2016, 8, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Saha, G.; Sarkar, S.; Mohanta, P.S.; Kumar, K.; Chakrabarti, S.; Basu, M.; Ghosh, M.K. USP7 targets XIAP for cancer progression: Establishment of a p53-independent therapeutic avenue for glioma. Oncogene 2022, 41, 5061–5075. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Liu, Z. Ubiquitination and deubiquitination of MCL1 in cancer: Deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis. 2020, 11, 556. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, M.; Jing, Y.; Yin, X.; Ma, P.; Zhang, Z.; Wang, X.; Di, W.; Zhuang, G. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun. 2018, 9, 215. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chaurushiya, M.S.; Webster, J.D.; Kummerfeld, S.; Reja, R.; Chaudhuri, S.; Chen, Y.J.; Modrusan, Z.; Haley, B.; Dugger, D.L.; et al. Intrinsic apoptosis shapes the tumor spectrum.m linked to inactivation of the deubiquitinase BAP1. Science 2019, 364, 283–285. [Google Scholar] [CrossRef]
- Dehan, E.; Bassermann, F.; Guardavaccaro, D.; Vasiliver-Shamis, G.; Cohen, M.; Lowes, K.N.; Dustin, M.; Huang, D.C.; Taunton, J.; Pagano, M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol. Cell 2009, 33, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Wang, Q.; Srinivasan, B.; Xu, J.; Padilla, M.T.; Li, Z.; Wang, X.; Liu, Y.; Gou, X.; Shen, H.M.; et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 2014, 33, 3004–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.; Heinlein, M.; Dengjel, J.; Alber, C.; Singh, P.K.; Hacker, G. The deubiquitinase Usp27x stabilizes the BH3-only protein Bim and enhances apoptosis. EMBO Rep. 2016, 17, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.M.; Seo, S.U.; Min, K.J.; Kwon, T.K. Melatonin induces apoptotic cell death through Bim stabilization by Sp1-mediated OTUD1 upregulation. J. Pineal Res. 2022, 72, e12781. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Gallegos, J.R.; Gu, Q.; Huang, Y.; Li, J.; Jin, Y.; Lu, H.; Sun, Y. SAG/ROC-SCF beta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia 2006, 8, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef]
- Xu, M.; Takanashi, M.; Oikawa, K.; Tanaka, M.; Nishi, H.; Isaka, K.; Kudo, M.; Kuroda, M. USP15 plays an essential role for caspase-3 activation during Paclitaxel-induced apoptosis. Biochem. Biophys. Res. Commun. 2009, 388, 366–371. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Li, X.; Ning, G.; Zhu, S.; Ma, X.; Liu, X.; Liu, C.; Huang, M.; Schmitt, I.; Wullner, U.; et al. The Machado-Joseph Disease Deubiquitinase Ataxin-3 Regulates the Stability and Apoptotic Function of p53. PLoS Biol. 2016, 14, e2000733. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef]
- Sun, X.X.; Challagundla, K.B.; Dai, M.S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J. 2012, 31, 576–592. [Google Scholar] [CrossRef]
- Ke, J.Y.; Dai, C.J.; Wu, W.L.; Gao, J.H.; Xia, A.J.; Liu, G.P.; Lv, K.S.; Wu, C.L. USP11 regulates p53 stability by deubiquitinating p53. J. Zhejiang Univ. Sci. B 2014, 15, 1032–1038. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.T.; Huang, K.Y.; Lu, M.C.; Huang, H.L.; Chen, C.Y.; Cheng, Y.L.; Yu, H.C.; Liu, S.Q.; Lai, N.S.; Huang, H.B. TGF-beta upregulates the translation of USP15 via the PI3K/AKT pathway to promote p53 stability. Oncogene 2017, 36, 2715–2723. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Candelaria, N.; Wong, K.K.; Nikolai, B.C.; Lonard, D.M.; O’Malley, B.W.; Richards, J.S. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat. Commun. 2018, 9, 1270. [Google Scholar] [CrossRef] [Green Version]
- Tu, R.; Kang, W.; Yang, X.; Zhang, Q.; Xie, X.; Liu, W.; Zhang, J.; Zhang, X.D.; Wang, H.; Du, R.L. USP49 participates in the DNA damage response by forming a positive feedback loop with p53. Cell Death Dis. 2018, 9, 553. [Google Scholar] [CrossRef]
- Piao, S.; Pei, H.Z.; Huang, B.; Baek, S.H. Ovarian tumor domain-containing protein 1 deubiquitinates and stabilizes p53. Cell Signal. 2017, 33, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lu, Z.; Lu, X.; Chen, L.; Cao, J.; Zhang, S.; Ling, Y.; Zhou, X. OTUD5 regulates p53 stability by deubiquitinating p53. PLoS ONE 2013, 8, e77682. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.; Reiley, W.W.; Chang, M.; Jin, W.; Lee, A.J.; Zhang, M.; Sun, S.C. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev. Cell 2007, 13, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; He, W.; Sun, L. Necrosome core machinery: MLKL. Cell Mol. Life Sci. 2016, 73, 2153–2163. [Google Scholar] [CrossRef]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461. [Google Scholar] [CrossRef]
- De Almagro, M.C.; Goncharov, T.; Izrael-Tomasevic, A.; Duttler, S.; Kist, M.; Varfolomeev, E.; Wu, X.; Lee, W.P.; Murray, J.; Webster, J.D.; et al. Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ. 2017, 24, 26–37. [Google Scholar] [CrossRef]
- Douglas, T.; Saleh, M. Post-translational Modification of OTULIN Regulates Ubiquitin Dynamics and Cell Death. Cell Rep. 2019, 29, 3652–3663.e3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, P.R.; Leske, D.; Hrdinka, M.; Bagola, K.; Fiil, B.K.; McLaughlin, S.H.; Wagstaff, J.; Volkmar, N.; Christianson, J.C.; Kessler, B.M.; et al. SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol. Cell 2016, 63, 990–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, S.L.; Chen, T.T.; Lawrence, D.A.; Marsters, S.A.; Gonzalvez, F.; Ashkenazi, A. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 2015, 22, 1846–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziedzic, S.A.; Su, Z.; Jean Barrett, V.; Najafov, A.; Mookhtiar, A.K.; Amin, P.; Pan, H.; Sun, L.; Zhu, H.; Ma, A.; et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 2018, 20, 58–68. [Google Scholar] [CrossRef]
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302. [Google Scholar] [CrossRef]
- Wang, H.; Meng, H.; Li, X.; Zhu, K.; Dong, K.; Mookhtiar, A.K.; Wei, H.; Li, Y.; Sun, S.C.; Yuan, J. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc. Natl. Acad. Sci. USA 2017, 114, 11944–11949. [Google Scholar] [CrossRef] [Green Version]
- Mei, P.; Xie, F.; Pan, J.; Wang, S.; Gao, W.; Ge, R.; Gao, B.; Gao, S.; Chen, X.; Wang, Y.; et al. E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis. Cell Death Differ. 2021, 28, 2888–2899. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Han, S.A.; Fan, Y.; Guo, L.S.; Aziz, K.; Nowsheen, S.; Kim, S.S.; Park, S.Y.; Luo, Q.; et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat. Cell Biol. 2019, 21, 940–951. [Google Scholar] [CrossRef]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Roedig, J.; Kowald, L.; Juretschke, T.; Karlowitz, R.; Ahangarian Abhari, B.; Roedig, H.; Fulda, S.; Beli, P.; van Wijk, S.J. USP22 controls necroptosis by regulating receptor-interacting protein kinase 3 ubiquitination. EMBO Rep. 2021, 22, e50163. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Liu, J.; Zou, B.; Li, C.; Zeh, H.J.; Kang, R.; Kroemer, G.; Huang, J.; Tang, D. Trypsin-Mediated Sensitization to Ferroptosis Increases the Severity of Pancreatitis in Mice. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 483–500. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Tang, Z.; Jiang, W.; Mao, M.; Zhao, J.; Chen, J.; Cheng, N. Deubiquitinase USP35 modulates ferroptosis in lung cancer via targeting ferroportin. Clin. Transl. Med. 2021, 11, e390. [Google Scholar] [CrossRef]
- Zumbrennen, K.B.; Wallander, M.L.; Romney, S.J.; Leibold, E.A. Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Mol. Cell Biol. 2009, 29, 2219–2229. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Liu, T.; Yin, Y.; Zhao, W.; Lin, Z.; Yin, Y.; Lu, D.; You, F. The deubiquitinase OTUD1 enhances iron transport and potentiates host antitumor immunity. EMBO Rep. 2021, 22, e51162. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Chen, X.; Li, J.; Comish, P.; Kang, R.; Tang, D. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020, 27, 645–656. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef]
- Zhang, Y.; Koppula, P.; Gan, B. Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1. Cell Cycle 2019, 18, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Ma, S.; Sun, L.; Wu, W.; Wu, J.; Sun, Z.; Ren, J. USP22 Protects Against Myocardial Ischemia-Reperfusion Injury via the SIRT1-p53/SLC7A11-Dependent Inhibition of Ferroptosis-Induced Cardiomyocyte Death. Front. Physiol. 2020, 11, 551318. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef]
- Zhang, J.; Qiu, Q.; Wang, H.; Chen, C.; Luo, D. TRIM46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating GPX4 ubiquitination. Exp. Cell Res. 2021, 407, 112800. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Wei, R.; Jin, T.; Zhang, M.; Shen, J.; Xiang, H.; Shan, B.; Yuan, J.; Li, Y. HOIP modulates the stability of GPx4 by linear ubiquitination. Proc. Natl. Acad. Sci. USA 2022, 119, e2214227119. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, X.; Yang, Q.; Chen, J.; Huang, Q.; Yao, L.; Yan, D.; Wu, J.; Zhang, P.; Tang, D.; et al. Broad Spectrum Deubiquitinase Inhibition Induces Both Apoptosis and Ferroptosis in Cancer Cells. Front. Oncol. 2020, 10, 949. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, C.; Zhan, J.; Chen, D.; Shao, G.; Zhang, H.; Gu, W.; Luo, J. The deubiquitinase USP11 regulates cell proliferation and ferroptotic cell death via stabilization of NRF2 USP11 deubiquitinates and stabilizes NRF2. Oncogene 2021, 40, 1706–1720. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, J.; Zhang, R.; Yu, C.; Zhou, Z.; Liu, J.; Liao, S.; Klionsky, D.J.; Kroemer, G.; et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy 2022, 1–21. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Wang, Y.; Li, C.; Xie, Y.; Klionsky, D.J.; Kang, R.; Tang, D. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy 2022, 1–12. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr. Biol. 2018, 28, 2388–2399.e2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zhu, S.; Zeh, H.J.; Klionsky, D.J.; Tang, D. BECN1 is a new driver of ferroptosis. Autophagy 2018, 14, 2173–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.; Xia, C.; Sun, Z. The Inhibitory Effect of 6-Gingerol on Ubiquitin-Specific Peptidase 14 Enhances Autophagy-Dependent Ferroptosis and Anti-Tumor in vivo and in vitro. Front. Pharmacol. 2020, 11, 598555. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Fan, J.; Ji, C.; Wang, Z.; Ge, X.; Wang, J.; Ye, W.; Yin, G.; Cai, W.; Liu, W. USP11 regulates autophagy-dependent ferroptosis after spinal cord ischemia-reperfusion injury by deubiquitinating Beclin 1. Cell Death Differ. 2022, 29, 1164–1175. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. Autophagy and Ferroptosis—What’s the Connection? Curr. Pathobiol. Rep. 2017, 5, 153–159. [Google Scholar] [CrossRef]
- Zou, J.; Zheng, Y.; Huang, Y.; Tang, D.; Kang, R.; Chen, R. The Versatile Gasdermin Family: Their Function and Roles in Diseases. Front. Immunol. 2021, 12, 751533. [Google Scholar] [CrossRef]
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Wei, X.; Xie, F.; Zhou, X.; Wu, Y.; Yan, H.; Liu, T.; Huang, J.; Wang, F.; Zhou, F.; Zhang, L. Role of pyroptosis in inflammation and cancer. Cell Mol. Immunol. 2022, 19, 971–992. [Google Scholar] [CrossRef]
- Tang, D.; Wang, H.; Billiar, T.R.; Kroemer, G.; Kang, R. Emerging mechanisms of immunocoagulation in sepsis and septic shock. Trends Immunol. 2021, 42, 508–522. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 2018, 24, 97–108.e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zeng, L.; Xie, M.; Liu, J.; Zhou, B.; Wu, R.; Cao, L.; Kroemer, G.; Wang, H.; Billiar, T.R.; et al. TMEM173 Drives Lethal Coagulation in Bacterial Infections. Cell Host Microbe 2020, 27, 556–570.E6. [Google Scholar] [CrossRef]
- Wu, R.; Wang, N.; Comish, P.B.; Tang, D.; Kang, R. Inflammasome-Dependent Coagulation Activation in Sepsis. Front. Immunol. 2021, 12, 641750. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, L.; Zhu, S.; Liu, J.; Zeh, H.J.; Kroemer, G.; Wang, H.; Billiar, T.R.; Jiang, J.; Tang, D.; et al. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv. 2019, 5, eaav5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
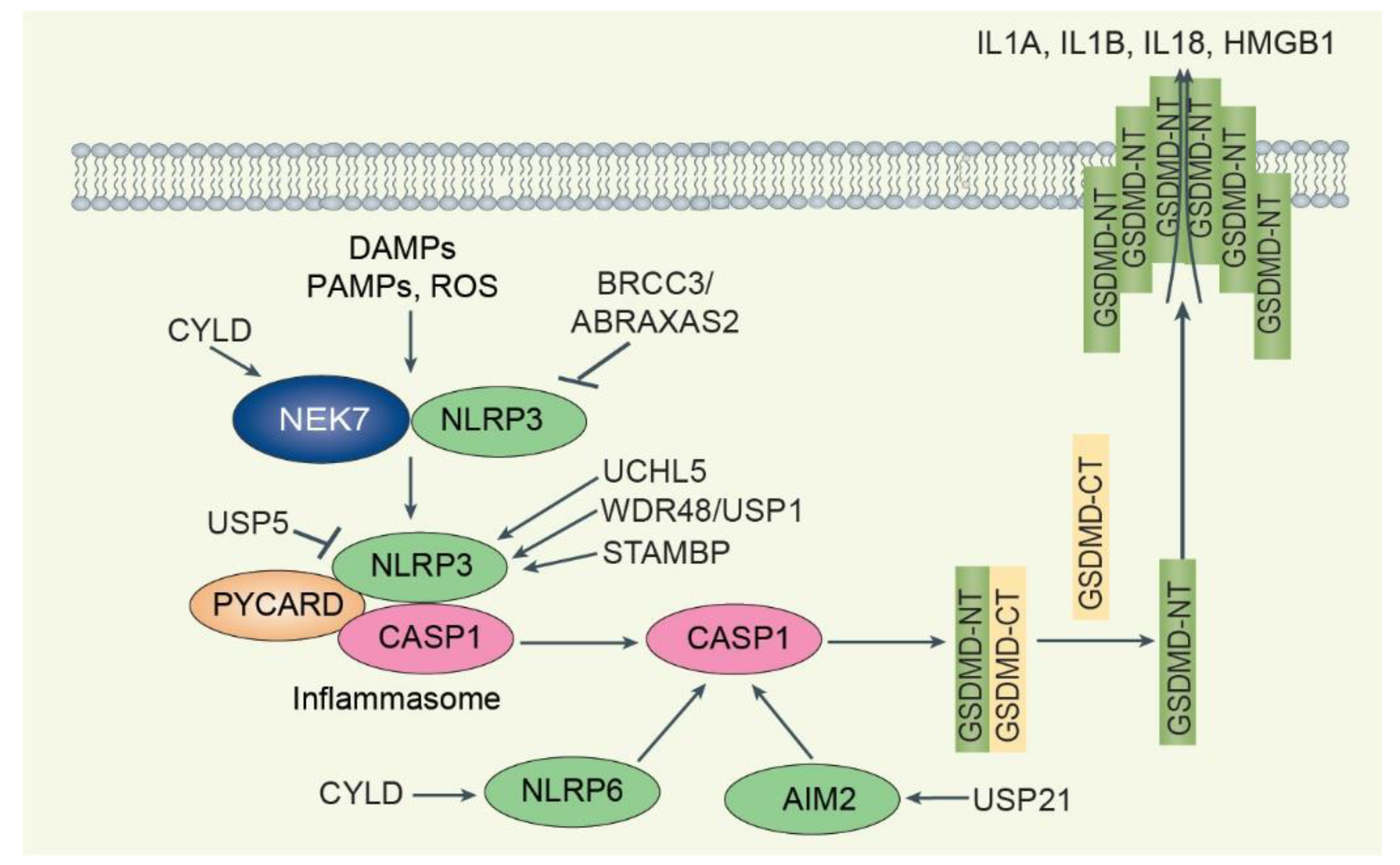
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akther, M.; Haque, M.E.; Park, J.; Kang, T.B.; Lee, K.H. NLRP3 Ubiquitination-A New Approach to Target NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2021, 22, 8780. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tu, S.; Lin, G.; Guo, H.; Yan, C.; Liu, Q.; Huang, L.; Tang, N.; Xiao, Y.; Pope, R.M.; et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. 2020, 217, e20182091. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Ren, G.M.; Li, J.; Zhang, X.C.; Wang, Y.; Xiao, Y.; Zhang, X.Y.; Liu, X.; Zhang, W.; Ma, W.B.; Zhang, J.; et al. Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3-associated inflammatory diseases. Sci. Immunol. 2021, 6, eabe2933. [Google Scholar] [CrossRef]
- Yang, X.D.; Li, W.; Zhang, S.; Wu, D.; Jiang, X.; Tan, R.; Niu, X.; Wang, Q.; Wu, X.; Liu, Z.; et al. PLK4 deubiquitination by Spata2-CYLD suppresses NEK7-mediated NLRP3 inflammasome activation at the centrosome. EMBO J. 2020, 39, e102201. [Google Scholar] [CrossRef]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Zhou, J.; Zhou, Y.; Xie, Y.; Jiang, Y.; Wu, J.; Luo, Z.; Liu, G.; Yin, L.; Zhang, X.L. Mycobacterial EST12 activates a RACK1-NLRP3-gasdermin D pyroptosis-IL-1beta immune pathway. Sci. Adv. 2020, 6, eaba4733. [Google Scholar] [CrossRef]
- Palazon-Riquelme, P.; Worboys, J.D.; Green, J.; Valera, A.; Martin-Sanchez, F.; Pellegrini, C.; Brough, D.; Lopez-Castejon, G. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018, 19, e44766. [Google Scholar] [CrossRef]
- Song, H.; Zhao, C.; Yu, Z.; Li, Q.; Yan, R.; Qin, Y.; Jia, M.; Zhao, W. UAF1 deubiquitinase complexes facilitate NLRP3 inflammasome activation by promoting NLRP3 expression. Nat. Commun. 2020, 11, 6042. [Google Scholar] [CrossRef] [PubMed]
- Bednash, J.S.; Johns, F.; Patel, N.; Smail, T.R.; Londino, J.D.; Mallampalli, R.K. The deubiquitinase STAMBP modulates cytokine secretion through the NLRP3 inflammasome. Cell Signal. 2021, 79, 109859. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Zhao, J.; Zhang, Y.; Liu, Y.; Ma, C.; Yi, F.; Zheng, Y.; Zhang, L.; Chen, T.; Liu, H.; et al. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy 2022, 18, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kumar, R.; Tsakem Lenou, E.; Basrur, V.; Kontoyiannis, D.L.; Ioakeimidis, F.; Mosialos, G.; Theiss, A.L.; Flavell, R.A.; Venuprasad, K. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat. Immunol. 2020, 21, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Lee, S.O.; Oh, C.; Kang, K.; Ryoo, J.; Kim, D.; Ahn, K. USP21 Deubiquitinase Regulates AIM2 Inflammasome Activation. J. Immunol. 2021, 207, 1926–1936. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Song, X.; Kang, R.; Tang, D. The Emerging Role of Deubiquitinases in Cell Death. Biomolecules 2022, 12, 1825. https://doi.org/10.3390/biom12121825
Zhou Z, Song X, Kang R, Tang D. The Emerging Role of Deubiquitinases in Cell Death. Biomolecules. 2022; 12(12):1825. https://doi.org/10.3390/biom12121825
Chicago/Turabian StyleZhou, Zhuan, Xinxin Song, Rui Kang, and Daolin Tang. 2022. "The Emerging Role of Deubiquitinases in Cell Death" Biomolecules 12, no. 12: 1825. https://doi.org/10.3390/biom12121825