
Advances in Gene Therapy Techniques to Treat LRRK2 Gene Mutation
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
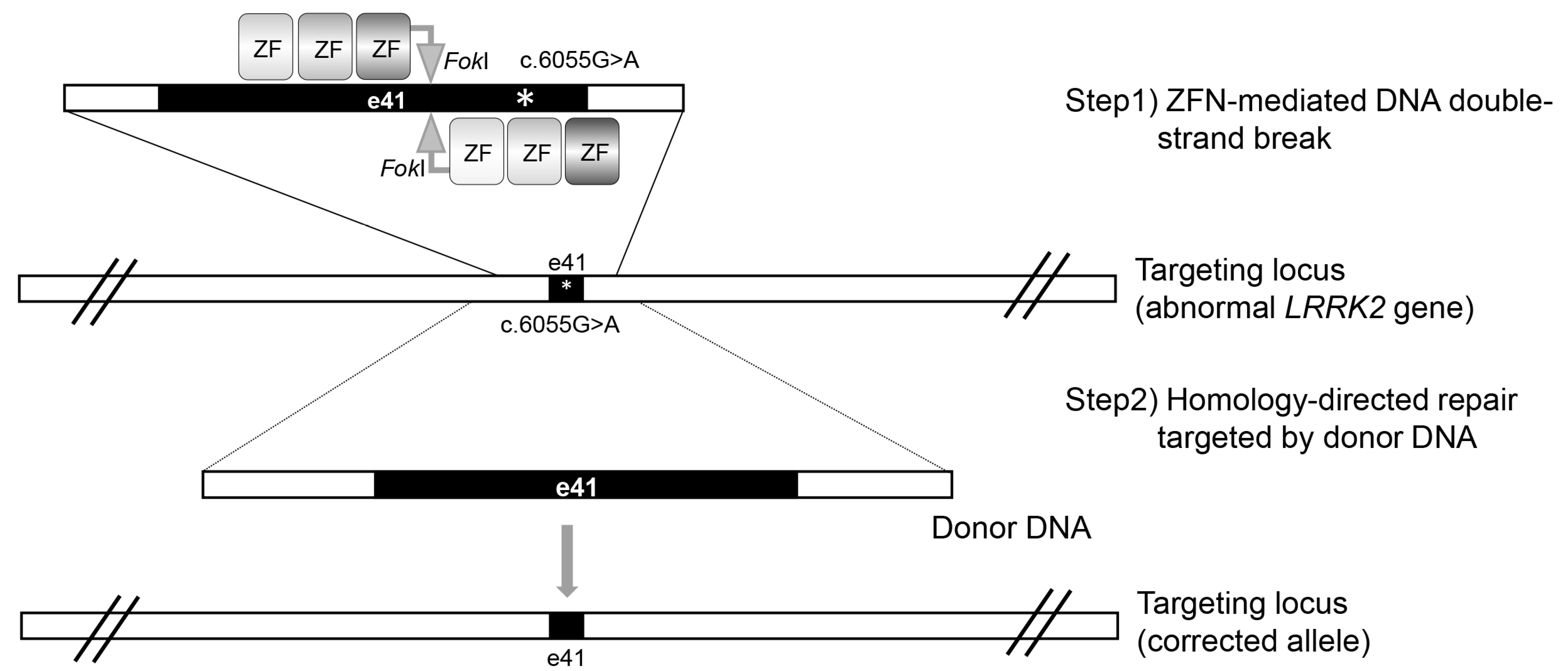
2. Zinc Finger Nuclease (ZFN)-Mediated LRRK2 Gene Targeting
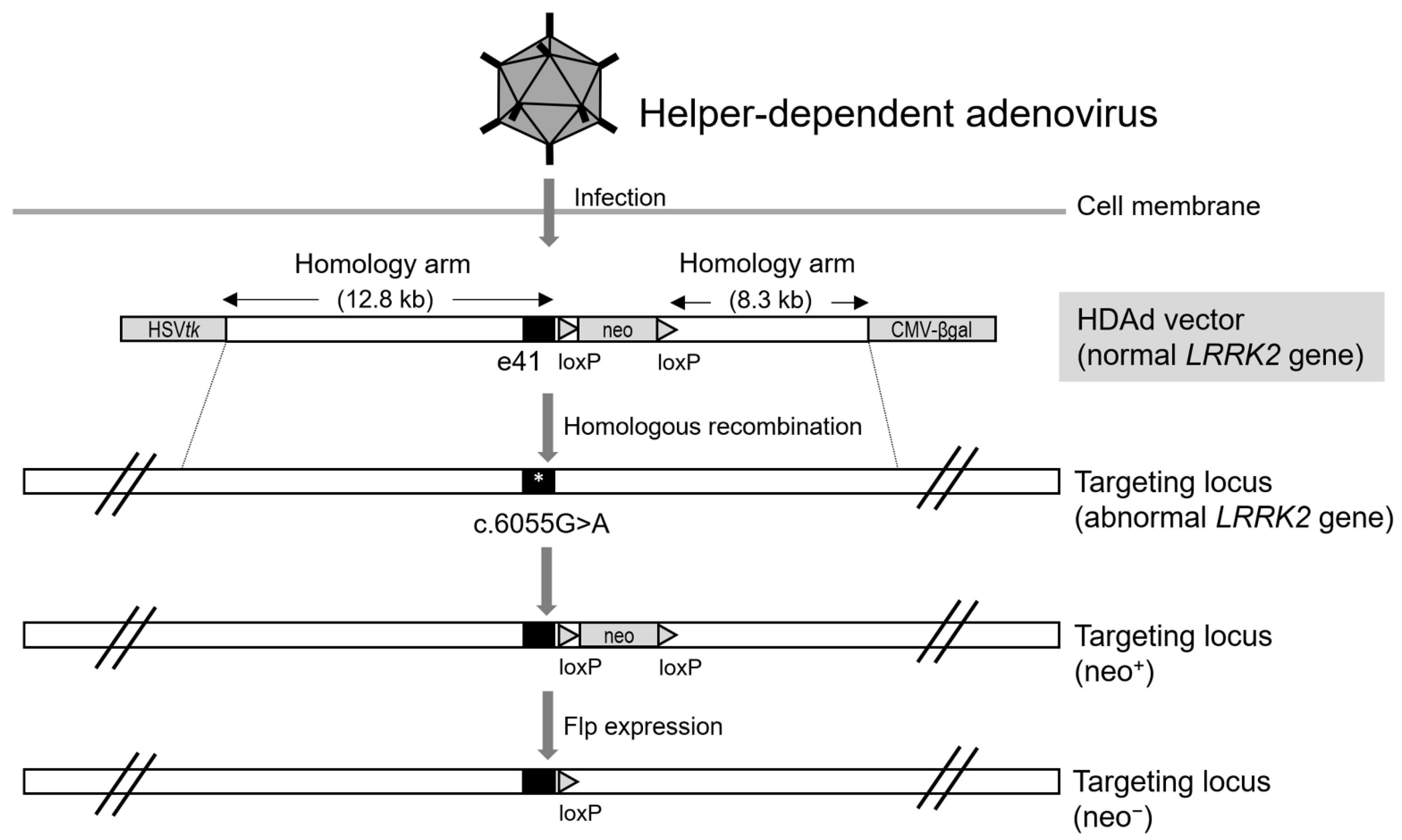
3. Helper-Dependent Adenoviral Vector (HDAdV)-Mediated LRRK2 Gene Targeting
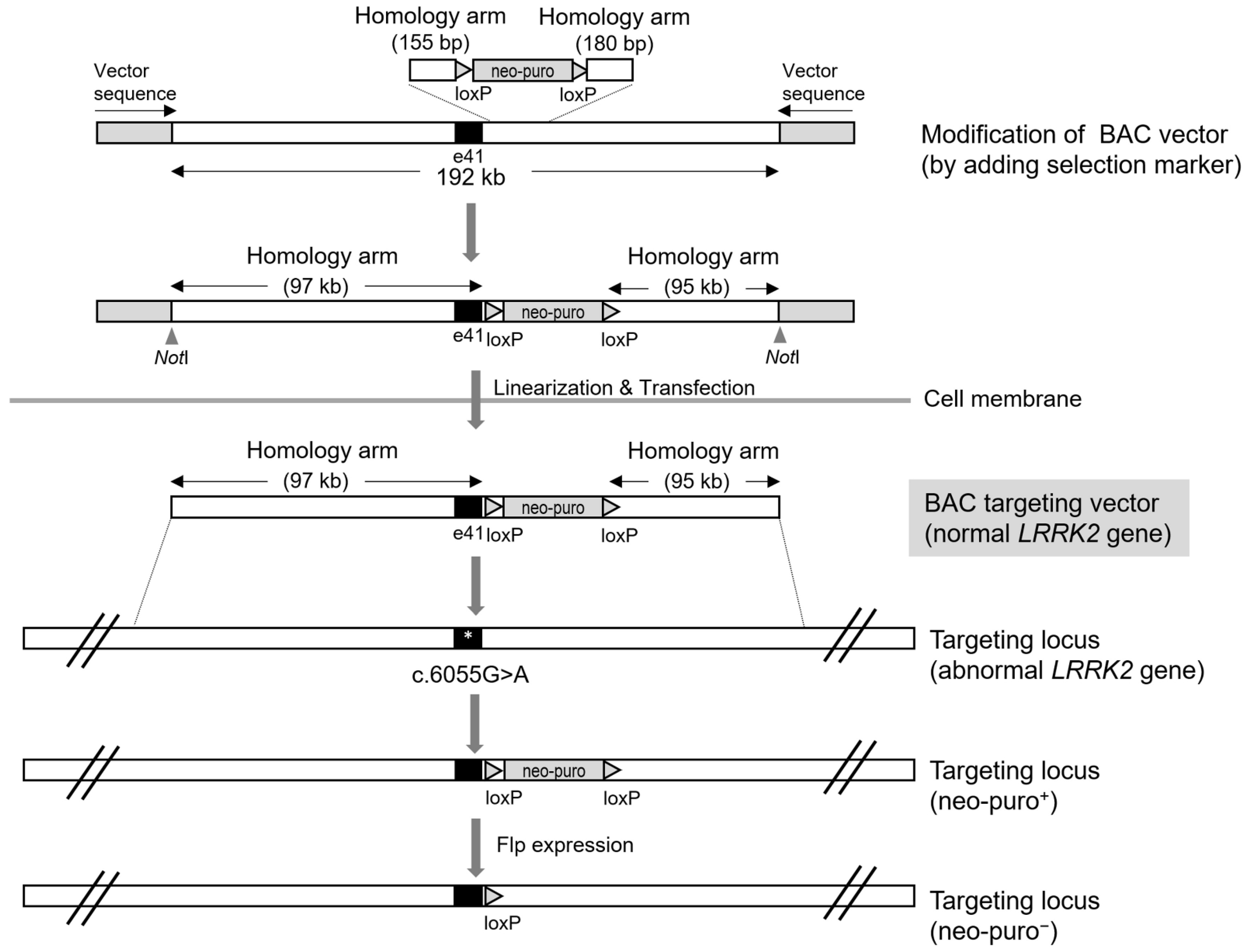
4. Bacterial Artificial Chromosome (BAC)-Mediated LRRK2 Gene Targeting
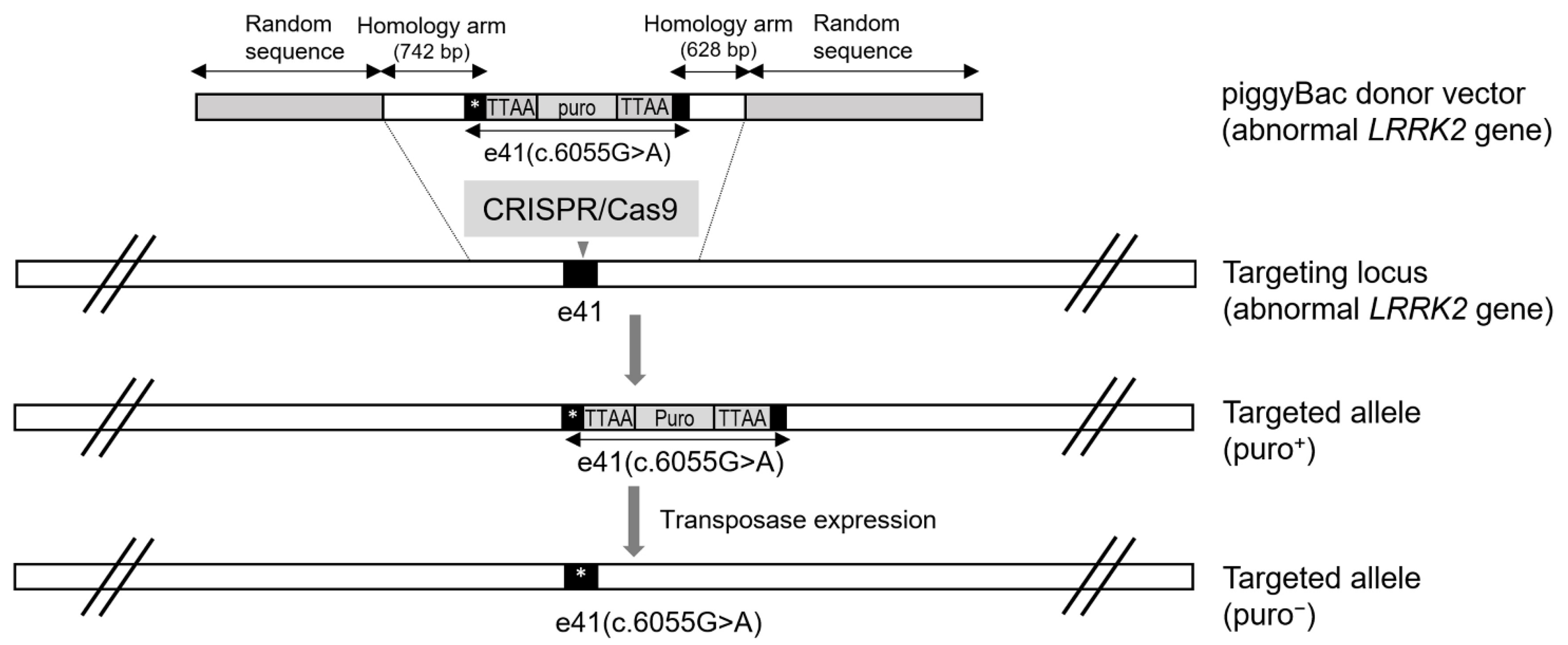
5. CRISPR/Cas9 and PiggyBac-Mediated LRRK2 Gene Targeting
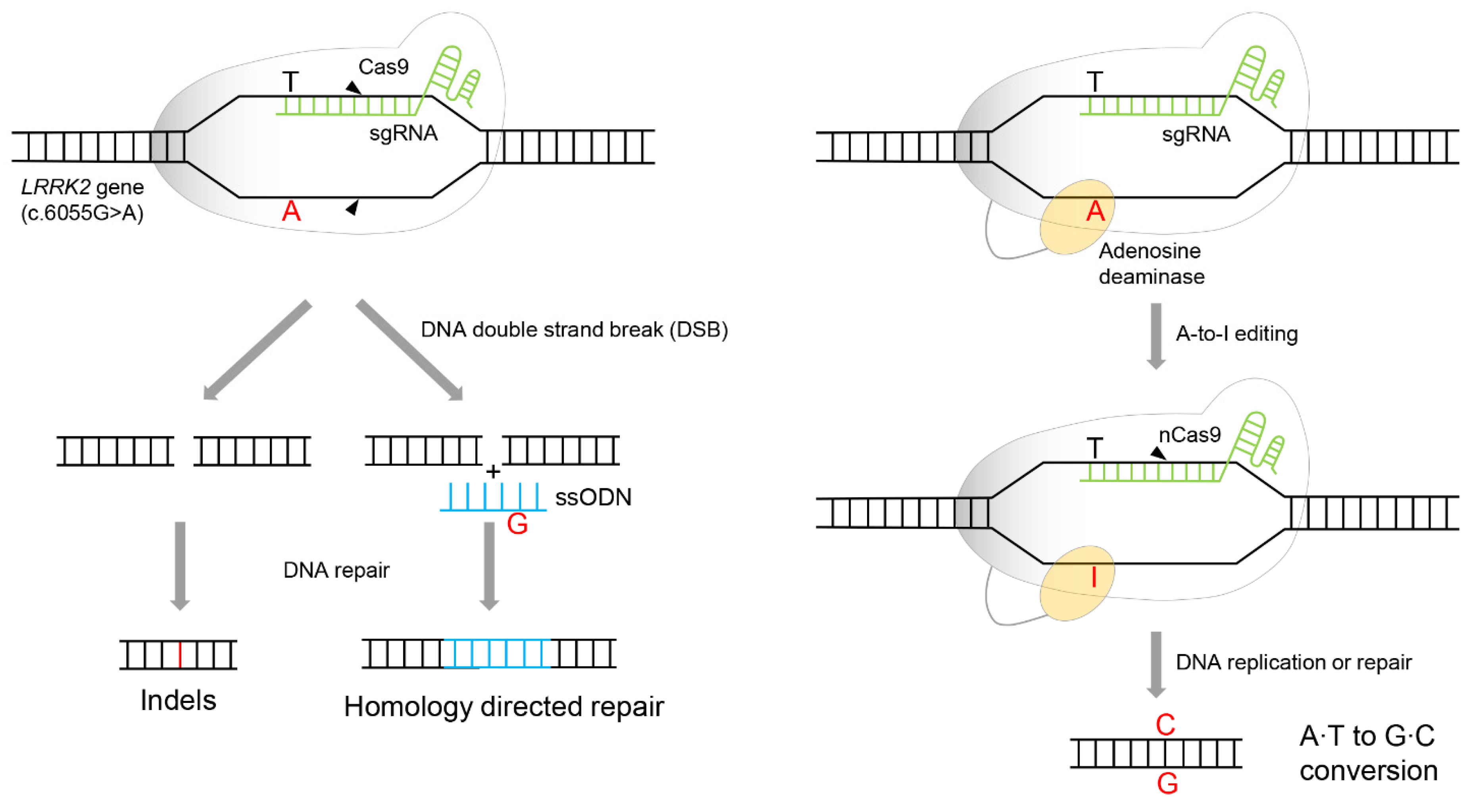
6. Adenine Base Editor (ABE)-Mediated LRRK2 Gene Targeting
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
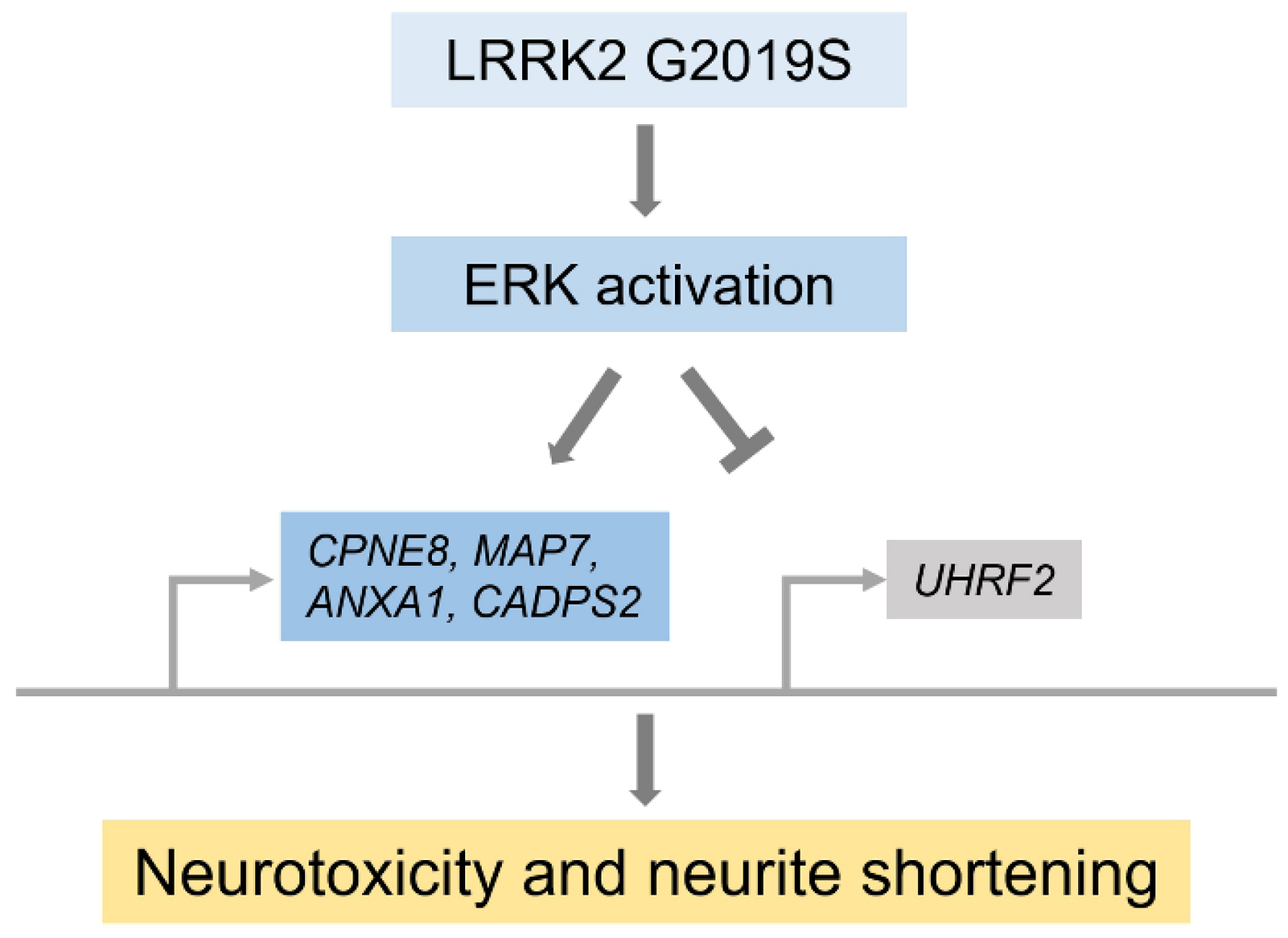
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic Correction of a LRRK2 Mutation in Human iPSCs Links Parkinsonian Neurodegeneration to ERK-Dependent Changes in Gene Expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Donaghy, P.C.; McKeith, I. The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis. Alzheimers Res. Ther. 2014, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obergasteiger, J.; Frapporti, G.; Lamonaca, G.; Pizzi, S.; Picard, A.; Lavdas, A.A.; Pischedda, F.; Piccoli, G.; Hilfiker, S.; Lobbestael, E.; et al. Kinase inhibition of G2019S-LRRK2 enhances autolysosome formation and function to reduce endogenous alpha-synuclein intracellular inclusions. Cell Death Discov. 2020, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Q.; Tan, L.; Yu, J.-T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideout, H.J.; Stefanis, L. The Neurobiology of LRRK2 and its Role in the Pathogenesis of Parkinson’s Disease. Neurochem. Res. 2014, 39, 576–592. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Nichols, W.C.; Pankratz, N.; Hernandez, D.; Paisan-Ruiz, C.; Jain, S.; Halter, C.A.; Michaels, V.E.; Reed, T.; Rudolph, A.; Shults, C.W.; et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005, 365, 410–412. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Rohe, C.F.; Ferreira, R.J.; Chien, H.F.; Vacca, L.; Stocchi, F.; Guedes, L.; Fabrizio, E.; Manfredi, M.; Vanacore, N.; et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 2005, 365, 412–415. [Google Scholar] [CrossRef]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. Common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Kachergus, J.; Mata, I.F.; Hulihan, M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Gibson, J.M.; Ross, O.A.; Lynch, T.; Wiley, J.; et al. Identification of a Novel LRRK2 Mutation Linked to Autosomal Dominant Parkinsonism: Evidence of a Common Founder across European Populations. Am. J. Hum. Genet. 2005, 76, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Dougall, P.; Gupta, P. Clinical Outcome of Flouoro-2, Deoxy-Glucose Positron Emmission Tomography/Computed Tomography [Fdg Pet/Ct] in Breast Cancer Patients-Study Based on Its Refferal Pattern. Value Health 2013, 16, A15–A16. [Google Scholar] [CrossRef] [Green Version]
- Ozelius, L.J.; Senthil, G.; Saunders-Pullman, R.; Ohmann, E.; Deligtisch, A.; Tagliati, M.; Hunt, A.L.; Klein, C.; Henick, B.; Hailpern, S.M.; et al. LRRK2 G2019S as a Cause of Parkinson’s Disease in Ashkenazi Jews. N. Engl. J. Med. 2006, 354, 424–425. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Qing, X.; Walter, J.; Jarazo, J.; Arias-Fuenzalida, J.; Hillje, A.L.; Schwamborn, J.C. CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and alpha-Synuclein modulation in dopaminergic neurons. Stem Cell Res. 2017, 24, 44–50. [Google Scholar] [CrossRef]
- Chang, K.-H.; Huang, C.-Y.; Ou-Yang, C.-H.; Ho, C.-H.; Lin, H.-Y.; Hsu, C.-L.; Chen, Y.-T.; Chou, Y.-C.; Chen, Y.-J.; Chen, Y.; et al. In vitro genome editing rescues parkinsonism phenotypes in induced pluripotent stem cells-derived dopaminergic neurons carrying LRRK2 p.G2019S mutation. Stem Cell Res. Ther. 2021, 12, 508. [Google Scholar] [CrossRef]
- Song, H.; Chung, S.-K.; Xu, Y. Modeling Disease in Human ESCs Using an Efficient BAC-Based Homologous Recombination System. Cell Stem Cell 2010, 6, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-Y.; Park, J.-H.; Jeong, S.; Kim, B.-Y.; Kang, Y.-K.; Xu, Y.; Chung, S.-K. K120R mutation inactivates p53 by creating an aberrant splice site leading to nonsense-mediated mRNA decay. Oncogene 2019, 38, 1597–1610. [Google Scholar] [CrossRef]
- Chung, S.-K.; Zhu, S.; Xu, Y.; Fu, X. Functional analysis of the acetylation of human p53 in DNA damage responses. Protein Cell 2014, 5, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-N.; Chung, S.-K.; Xu, Z.; Xu, Y. Oct4 Maintains the Pluripotency of Human Embryonic Stem Cells by Inactivating p53 Through Sirt1-Mediated Deacetylation. Stem Cells 2014, 32, 157–165. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Chung, S.-K. Generation of gene-corrected iPSC line, KIOMi002-A, from Parkinson’s disease patient iPSC with LRRK2 G2019S mutation using BAC-based homologous recombination. Stem Cell Res. 2019, 41, 101649. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The Familial Parkinsonism Gene LRRK2 Regulates Neurite Process Morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badiola, N.; de Oliveira, R.M.; Herrera, F.; Guardia-Laguarta, C.; Gonçalves, S.; Pera, M.; Suárez-Calvet, M.; Clarimón, J.; Outeiro, T.F.; Lleó, A. Tau Enhances α-Synuclein Aggregation and Toxicity in Cellular Models of Synucleinopathy. PLoS ONE 2011, 6, e26609. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J., 3rd; Steer, E.; Gusdon, A.M.; Kiselyov, K.; Chu, C.T. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 2013, 182, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo-Betancor, O.; Galosi, L.; Bonfili, L.; Eleuteri, A.M.; Cecarini, V.; Verin, R.; Dini, F.; Attili, A.; Berardi, S.; Biagini, L.; et al. Homozygous CADPS2 Mutations Cause Neurodegenerative Disease with Lewy Body-like Pathology in Parrots. Mov. Disord. 2022. [Google Scholar] [CrossRef]
- Tymanskyj, S.R.; Yang, B.H.; Verhey, K.J.; Ma, L. MAP7 regulates axon morphogenesis by recruiting kinesin-1 to microtubules and modulating organelle transport. eLife 2018, 7, e36374. [Google Scholar] [CrossRef]
- Darvish, H.; Azcona, L.J.; Taghavi, S.; Firouzabadi, S.G.; Tafakhori, A.; Alehabib, E.; Mohajerani, F.; Zardadi, S.; Paisán-Ruiz, C. ANXA1 with Anti-Inflammatory Properties Might Contribute to Parkinsonism. Ann. Neurol. 2021, 90, 319–323. [Google Scholar] [CrossRef]
- Wu, T.-F.; Zhang, W.; Su, Z.-P.; Chen, S.-S.; Chen, G.-L.; Wei, Y.-X.; Sun, T.; Xie, X.-S.; Li, B.; Zhou, Y.-X.; et al. UHRF2 mRNA expression is low in malignant glioma but silencing inhibits the growth of U251 glioma cells in vitro. Asian Pac. J. Cancer Prev. 2012, 13, 5137–5142. [Google Scholar] [CrossRef]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schule, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.K.; Song, B.; Bae, S. Current Status and Challenges of DNA Base Editing Tools. Mol. Ther. 2020, 28, 1938–1952. [Google Scholar] [CrossRef]
- Liang, P.; Xie, X.; Zhi, S.; Sun, H.; Zhang, X.; Chen, Y.; Chen, Y.; Xiong, Y.; Ma, W.; Liu, D.; et al. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat. Commun. 2019, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Munawar, N.; Khan, Z.; Qusmani, A.T.; Khan, S.H.; Jamil, A.; Ashraf, S.; Ghouri, M.Z.; Aslam, S.; Mubarik, M.S.; et al. An Outlook on Global Regulatory Landscape for Genome-Edited Crops. Int. J. Mol. Sci. 2021, 22, 11753. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5429. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Benamer, H.T.; de Silva, R. LRRK2 G2019S in the North African population: A review. Eur. Neurol. 2010, 63, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Van De Vrugt, H.J.; Harmsen, T.; Riepsaame, J.; Alexantya, G.; Van Mil, S.E.; De Vries, Y.; Bin Ali, R.; Huijbers, I.J.; Dorsman, J.C.; Wolthuis, R.M.F.; et al. Effective CRISPR/Cas9-mediated correction of a Fanconi anemia defect by error-prone end joining or templated repair. Sci. Rep. 2019, 9, 768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavani, G.; Fabiano, A.; Laurent, M.; Amor, F.; Cantelli, E.; Chalumeau, A.; Maule, G.; Tachtsidi, A.; Concordet, J.P.; Cereseto, A.; et al. Correction of beta-thalassemia by CRISPR/Cas9 editing of the alpha-globin locus in human hematopoietic stem cells. Blood Adv. 2021, 5, 1137–1153. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise Correction of the Dystrophin Gene in Duchenne Muscular Dystrophy Patient Induced Pluripotent Stem Cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, S.-K.; Lee, S.-Y. Advances in Gene Therapy Techniques to Treat LRRK2 Gene Mutation. Biomolecules 2022, 12, 1814. https://doi.org/10.3390/biom12121814
Chung S-K, Lee S-Y. Advances in Gene Therapy Techniques to Treat LRRK2 Gene Mutation. Biomolecules. 2022; 12(12):1814. https://doi.org/10.3390/biom12121814
Chicago/Turabian StyleChung, Sun-Ku, and Seo-Young Lee. 2022. "Advances in Gene Therapy Techniques to Treat LRRK2 Gene Mutation" Biomolecules 12, no. 12: 1814. https://doi.org/10.3390/biom12121814