
Structural Insights into the Dimeric Form of Bacillus subtilis RNase Y Using NMR and AlphaFold
 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
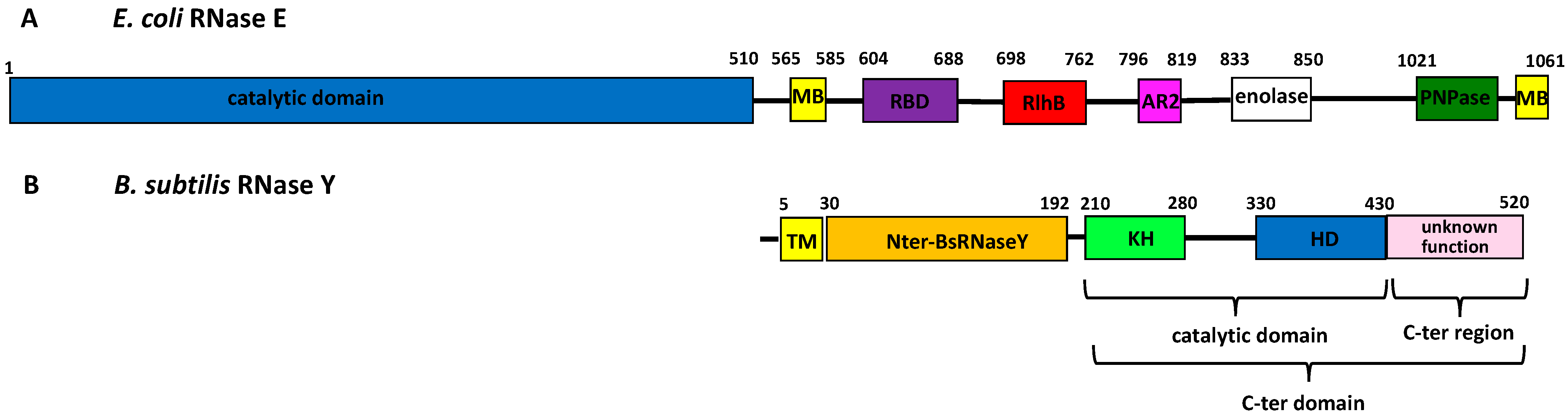
1. Introduction
2. Materials and Methods
2.1. Prediction of the Secondary Structure of Nter-BsRNaseY
2.2. Three-Dimensional Structure Prediction of Nter-BsRNaseY and Full-Length RNase Y Using AlphaFold
2.3. Production of Nter-BsRNaseY
2.4. Culture and Purification of 15N-Labeled and 13C-15N-2D-Labeled Nter-BsRNaseY
2.5. Circular Dichroism of Nter-BsRNaseY
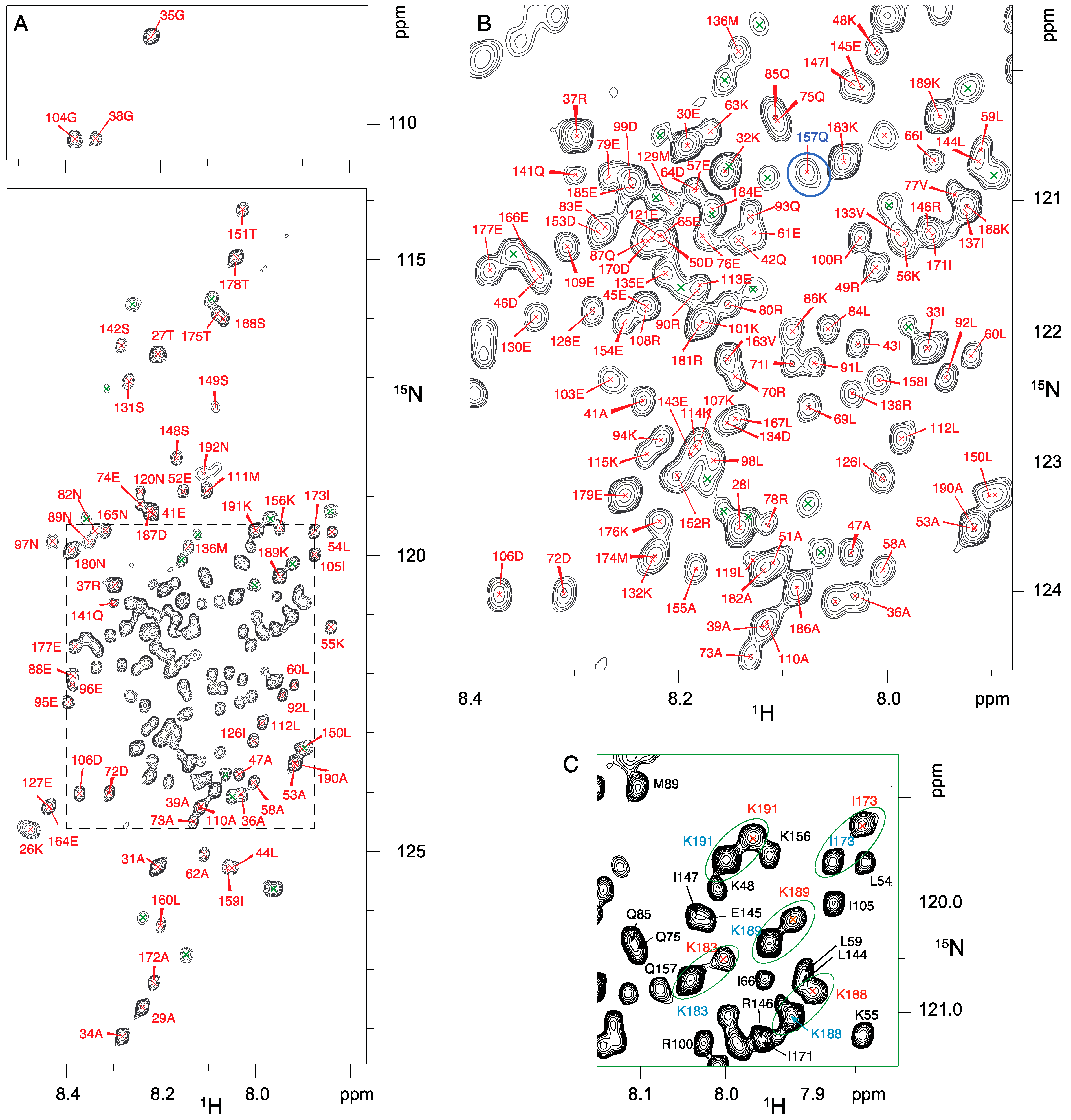
2.6. NMR Resonance Assignments for RNase Y Backbone
3. Results
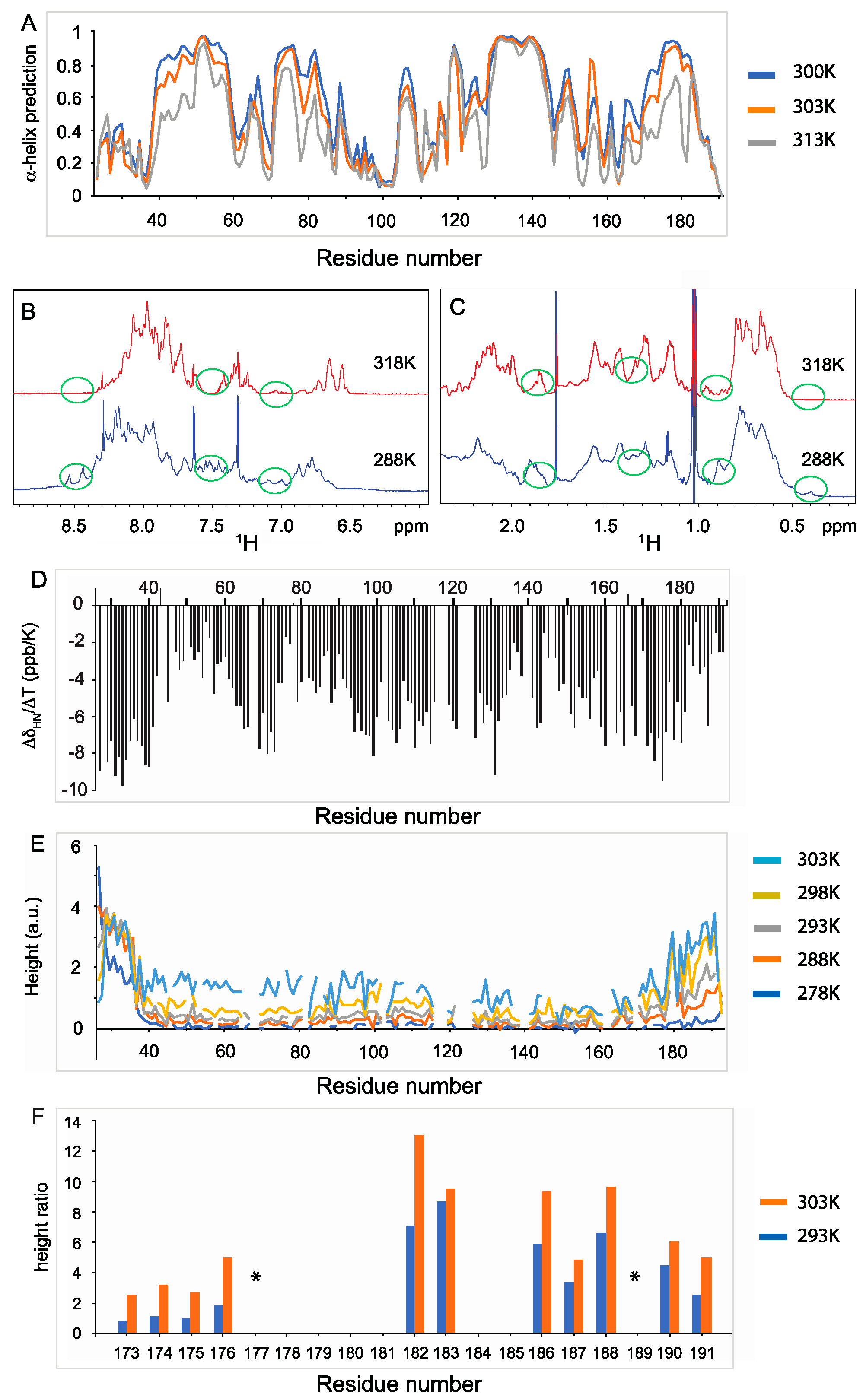
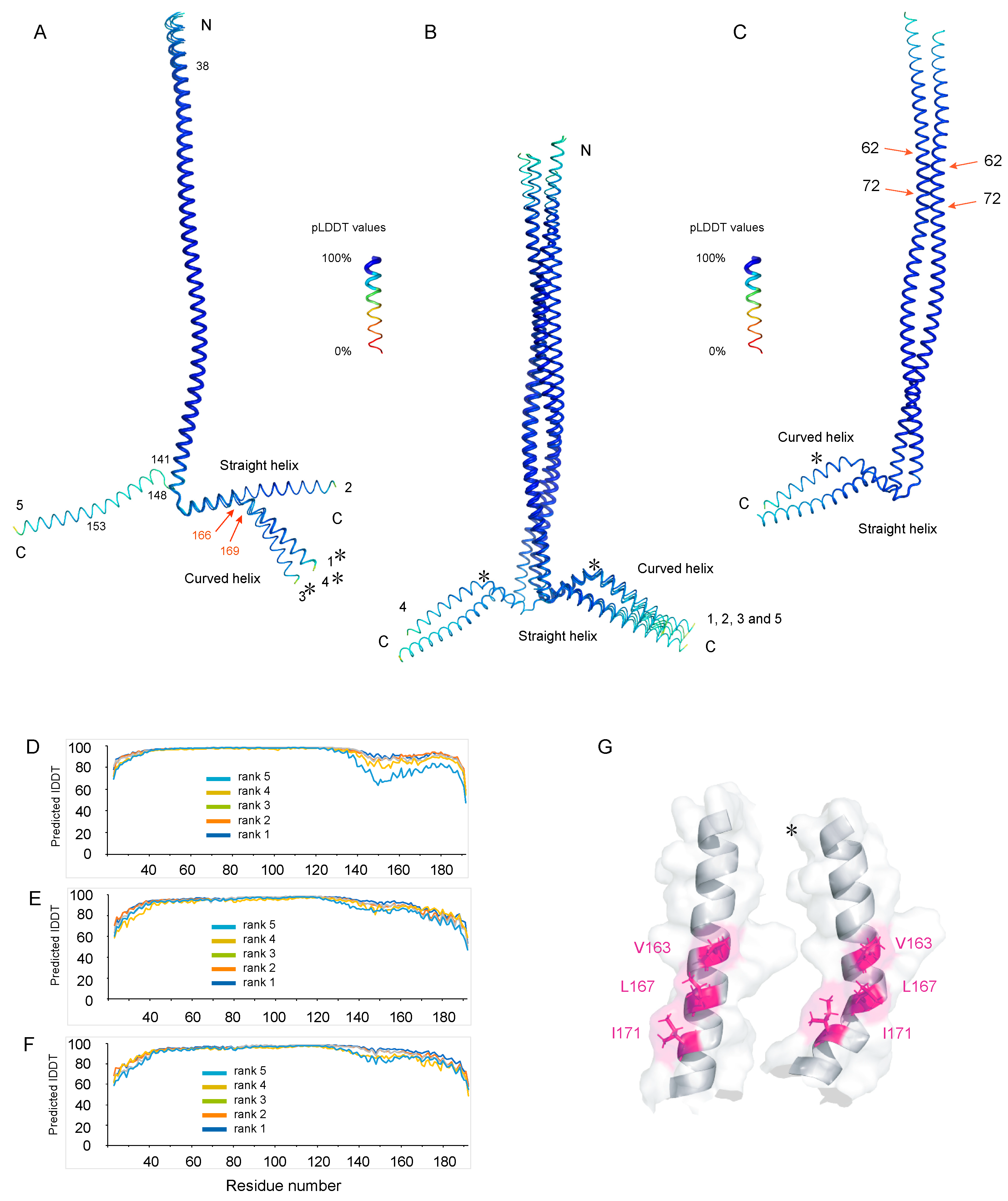
3.1. Several Regions of Nter-BsRNaseY Have a High Propensity to Form α-Helices
3.2. Two Main Conformations of the C-Terminal Extremity of Nter-BsRNaseY
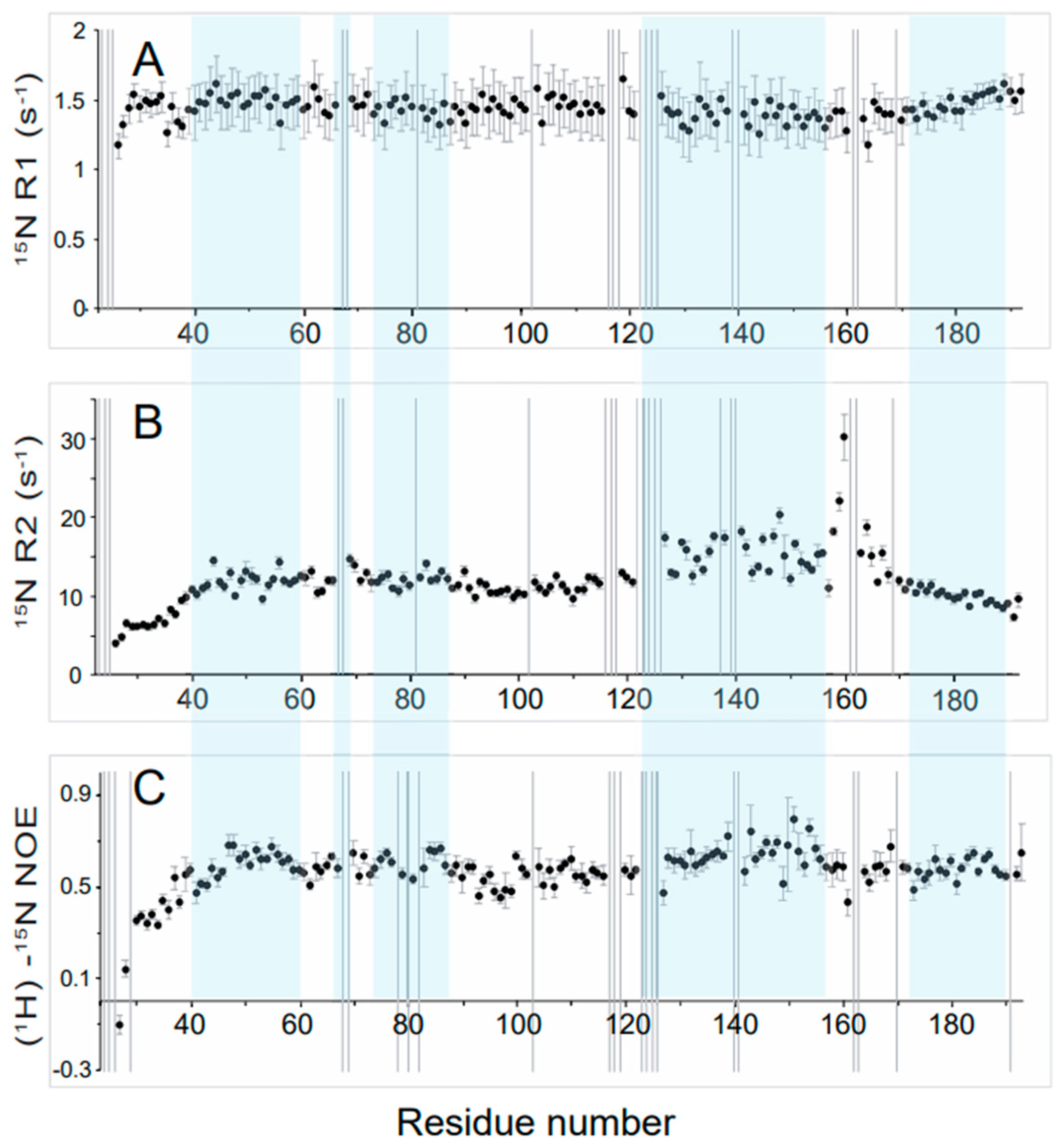
3.3. High Flexibility of the N- and C-Terminal Residues
3.4. AlphaFold Models of Nter-BsRNaseY
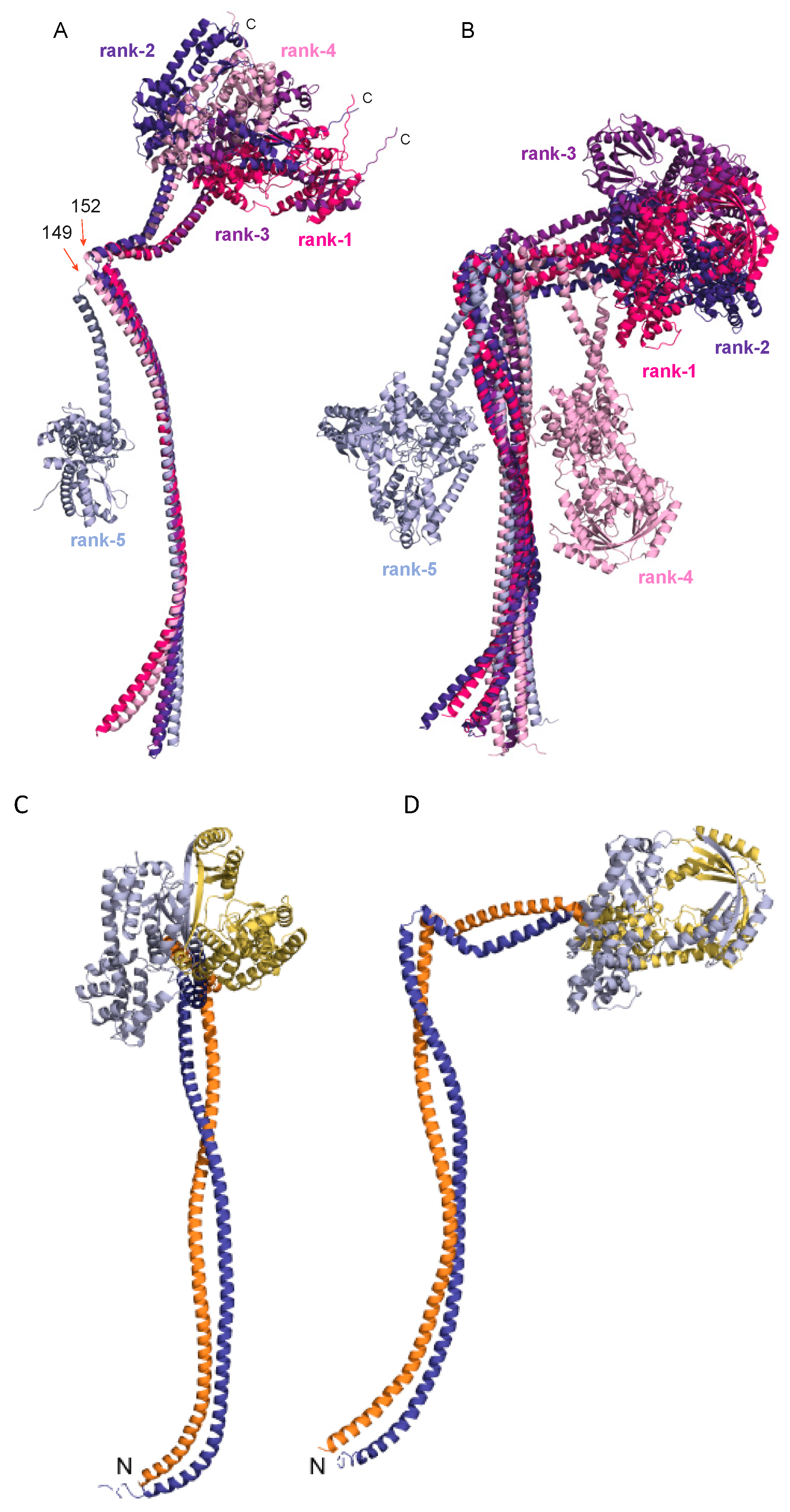
3.5. AlphaFold Model of the Full-Length B. subtilis RNase Y
4. Discussion
4.1. Interpretation of the NMR Data of Nter-BsRNaseY Using AlphaFold
4.2. AlphaFold Model of the Full-Length B. subtilis RNase Y
4.3. Interaction with Cellular Partners of the Degradosome Complex
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laalami, S.; Putzer, H. mRNA degradation and maturation in prokaryotes: The global players. Biomol. Concepts 2011, 2, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Laalami, S.; Zig, L.; Putzer, H. Initiation of mRNA decay in bacteria. Cell Mol. Life Sci. 2014, 71, 1799–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, B.K.; Kushner, S.R. Regulation of mRNA Decay in Bacteria. Annu. Rev. Microbiol. 2016, 70, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Gorna, M.W.; Carpousis, A.J.; Luisi, B.F. From conformational chaos to robust regulation: The structure and function of the multi-enzyme RNA degradosome. Q. Rev. Biophys. 2012, 45, 105–145. [Google Scholar] [CrossRef] [PubMed]
- Mackie, G.A. RNase E: At the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013, 11, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Shahbabian, K.; Jamalli, A.; Zig, L.; Putzer, H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009, 28, 3523–3533. [Google Scholar] [CrossRef] [Green Version]
- Durand, S.; Tomasini, A.; Braun, F.; Condon, C.; Romby, P. sRNA and mRNA turnover in Gram-positive bacteria. FEMS Microbiol. Rev. 2015, 39, 316–330. [Google Scholar] [CrossRef] [Green Version]
- Kaito, C.; Kurokawa, K.; Matsumoto, Y.; Terao, Y.; Kawabata, S.; Hamada, S.; Sekimizu, K. Silkworm pathogenic bacteria infection model for identification of novel virulence genes. Mol. Microbiol. 2005, 56, 934–944. [Google Scholar] [CrossRef]
- Bonnin, R.A.; Bouloc, P. RNA Degradation in Staphylococcus aureus: Diversity of Ribonucleases and Their Impact. Int. J. Genom. 2015, 2015, 395753. [Google Scholar] [CrossRef] [Green Version]
- Obana, N.; Nakamura, K.; Nomura, N. Role of RNase Y in Clostridium perfringens mRNA Decay and Processing. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef]
- Kang, S.O.; Caparon, M.G.; Cho, K.H. Virulence gene regulation by CvfA, a putative RNase: The CvfA-enolase complex in Streptococcus pyogenes links nutritional stress, growth-phase control, and virulence gene expression. Infect. Immun. 2010, 78, 2754–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnik-Habrink, M.; Newman, J.; Rothe, F.M.; Solovyova, A.S.; Rodrigues, C.; Herzberg, C.; Commichau, F.M.; Lewis, R.J.; Stulke, J. RNase Y in Bacillus subtilis: A Natively disordered protein that is the functional equivalent of RNase E from Escherichia coli. J. Bacteriol. 2011, 193, 5431–5441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, A.J.; Grossmann, J.G.; Redko, Y.U.; Ilag, L.L.; Moncrieffe, M.C.; Symmons, M.F.; Robinson, C.V.; McDowall, K.J.; Luisi, B.F. Quaternary structure and catalytic activity of the Escherichia coli ribonuclease E amino-terminal catalytic domain. Biochemistry 2003, 42, 13848–13855. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, A.J.; Marcaida, M.J.; Stead, J.A.; McDowall, K.J.; Scott, W.G.; Luisi, B.F. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 2005, 437, 1187–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koslover, D.J.; Callaghan, A.J.; Marcaida, M.J.; Garman, E.F.; Martick, M.; Scott, W.G.; Luisi, B.F. The crystal structure of the Escherichia coli RNase E apoprotein and a mechanism for RNA degradation. Structure 2008, 16, 1238–1244. [Google Scholar] [CrossRef]
- Vanzo, N.F.; Li, Y.S.; Py, B.; Blum, E.; Higgins, C.F.; Raynal, L.C.; Krisch, H.M.; Carpousis, A.J. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 1998, 12, 2770–2781. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Malfavon, L.; Islas, L.D.; Luisi, B.F.; Garcia-Villegas, R.; Garcia-Mena, J. The assembly and distribution in vivo of the Escherichia coli RNA degradosome. Biochimie 2013, 95, 2034–2041. [Google Scholar] [CrossRef]
- Ait-Bara, S.; Carpousis, A.J.; Quentin, Y. RNase E in the gamma-Proteobacteria: Conservation of intrinsically disordered noncatalytic region and molecular evolution of microdomains. Mol. Genet. Genom. 2015, 290, 847–862. [Google Scholar] [CrossRef]
- Bruce, H.A.; Du, D.; Matak-Vinkovic, D.; Bandyra, K.J.; Broadhurst, R.W.; Martin, E.; Sobott, F.; Shkumatov, A.V.; Luisi, B.F. Analysis of the natively unstructured RNA/protein-recognition core in the Escherichia coli RNA degradosome and its interactions with regulatory RNA/Hfq complexes. Nucleic Acids Res. 2018, 46, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, A.J.; Aurikko, J.P.; Ilag, L.L.; Gunter Grossmann, J.; Chandran, V.; Kuhnel, K.; Poljak, L.; Carpousis, A.J.; Robinson, C.V.; Symmons, M.F.; et al. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J. Mol. Biol. 2004, 340, 965–979. [Google Scholar] [CrossRef]
- Marcaida, M.J.; DePristo, M.A.; Chandran, V.; Carpousis, A.J.; Luisi, B.F. The RNA degradosome: Life in the fast lane of adaptive molecular evolution. Trends Biochem. Sci. 2006, 31, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Khemici, V.; Poljak, L.; Luisi, B.F.; Carpousis, A.J. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 2008, 70, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, S.; McKay, A.R.; Robinson, C.V.; Luisi, B.F. Molecular recognition between Escherichia coli enolase and ribonuclease E. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 1036–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, V.; Luisi, B.F. Recognition of enolase in the Escherichia coli RNA degradosome. J. Mol. Biol. 2006, 358, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, S.; Vaidialingam, B.; Callaghan, A.J.; Luisi, B.F. Crystal structure of Escherichia coli polynucleotide phosphorylase core bound to RNase E, RNA and manganese: Implications for catalytic mechanism and RNA degradosome assembly. J. Mol. Biol. 2009, 389, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V. KH domain: One motif, two folds. Nucleic Acids Res. 2001, 29, 638–643. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Laalami, S.; Cavaiuolo, M.; Roque, S.; Chagneau, C.; Putzer, H. Escherichia coli RNase E can efficiently replace RNase Y in Bacillus subtilis. Nucleic Acids Res. 2021, 49, 4643–4654. [Google Scholar] [CrossRef]
- Lehnik-Habrink, M.; Schaffer, M.; Mader, U.; Diethmaier, C.; Herzberg, C.; Stulke, J. RNA processing in Bacillus subtilis: Identification of targets of the essential RNase Y. Mol. Microbiol. 2011, 81, 1459–1473. [Google Scholar] [CrossRef]
- Karimova, G.; Pidoux, J.; Ullmann, A.; Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756. [Google Scholar] [CrossRef]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef]
- Hardouin, P.; Velours, C.; Bou-Nader, C.; Assrir, N.; Laalami, S.; Putzer, H.; Durand, D.; Golinelli-Pimpaneau, B. Dissociation of the Dimer of the Intrinsically Disordered Domain of RNase Y upon Antibody Binding. Biophys. J. 2018, 115, 2102–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commichau, F.M.; Rothe, F.M.; Herzberg, C.; Wagner, E.; Hellwig, D.; Lehnik-Habrink, M.; Hammer, E.; Volker, U.; Stulke, J. Novel activities of glycolytic enzymes in Bacillus subtilis: Interactions with essential proteins involved in mRNA processing. Mol. Cell Proteom. 2009, 8, 1350–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnik-Habrink, M.; Lewis, R.J.; Mader, U.; Stulke, J. RNA degradation in Bacillus subtilis: An interplay of essential endo- and exoribonucleases. Mol. Microbiol. 2012, 84, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Lehnik-Habrink, M.; Pfortner, H.; Rempeters, L.; Pietack, N.; Herzberg, C.; Stulke, J. The RNA degradosome in Bacillus subtilis: Identification of CshA as the major RNA helicase in the multiprotein complex. Mol. Microbiol. 2010, 77, 958–971. [Google Scholar] [CrossRef]
- Newman, J.A.; Hewitt, L.; Rodrigues, C.; Solovyova, A.S.; Harwood, C.R.; Lewis, R.J. Dissection of the network of interactions that links RNA processing with glycolysis in the Bacillus subtilis degradosome. J. Mol. Biol. 2012, 416, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Salvo, E.; Alabi, S.; Liu, B.; Schlessinger, A.; Bechhofer, D.H. Interaction of Bacillus subtilis Polynucleotide Phosphorylase and RNase Y: Structural Mapping and effect on mRNA turnover. J. Biol. Chem. 2016, 291, 6655–6663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.H. The Structure and Function of the Gram-Positive Bacterial RNA Degradosome. Front. Microbiol. 2017, 8, 154. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Jeffery, C.J. Moonlighting Proteins in the Fuzzy Logic of Cellular Metabolism. Molecules 2020, 25, 3440. [Google Scholar] [CrossRef]
- Antikainen, J.; Kuparinen, V.; Lahteenmaki, K.; Korhonen, T.K. Enolases from Gram-positive bacterial pathogens and commensal lactobacilli share functional similarity in virulence-associated traits. FEMS Immunol. Med. Microbiol. 2007, 51, 526–534. [Google Scholar] [CrossRef]
- Agarwal, S.; Kulshreshtha, P.; Bambah Mukku, D.; Bhatnagar, R. alphaa-Enolase binds to human plasminogen on the surface of Bacillus anthracis. Biochim Biophys. Acta 2008, 1784, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Esgleas, M.; Li, Y.; Hancock, M.A.; Harel, J.; Dubreuil, J.D.; Gottschalk, M. Isolation and characterization of alpha-enolase, a novel fibronectin-binding protein from Streptococcus suis. Microbiology 2008, 154, 2668–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Grigoryan, G.; Keating, A.E. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 2008, 18, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: https://pymol.org/2/ (accessed on 22 September 2022).
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Trainor, K.; Palumbo, J.A.; MacKenzie, D.W.S.; Meiering, E.M. Temperature dependence of NMR chemical shifts: Tracking and statistical analysis. Protein Sci. 2020, 29, 306–314. [Google Scholar] [CrossRef]
- Das, A.; Mukhopadhyay, C. Urea-mediated protein denaturation: A consensus view. J. Phys. Chem. B 2009, 113, 12816–12824. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Cierpicki, T.; Otlewski, J. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 2001, 21, 249–261. [Google Scholar] [CrossRef]
- Ishima, R. Recent developments in (15)N NMR relaxation studies that probe protein backbone dynamics. Top Curr. Chem. 2012, 326, 99–122. [Google Scholar] [CrossRef]
- Mason, J.M.; Arndt, K.M. Coiled coil domains: Stability, specificity, and biological implications. Chembiochem 2004, 5, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Basavalingappa, V.; Jacoby, G.; Shimon, L.J.W.; Beck, R.; Gazit, E. Functional Coiled-Coil-like Assembly by Knob-into-Hole Packing of Single Heptad Repeat. ACS Nano 2019, 13, 12630–12637. [Google Scholar] [CrossRef]
- Chao, J.A.; Prasad, G.S.; White, S.A.; Stout, C.D.; Williamson, J.R. Inherent protein structural flexibility at the RNA-binding interface of L30e. J. Mol. Biol. 2003, 326, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Zsolyomi, F.; Guharoy, M.; Tompa, P. Functional Advantages of Conserved Intrinsic Disorder in RNA-Binding Proteins. PLoS ONE 2015, 10, e0139731. [Google Scholar] [CrossRef] [PubMed]
- Baudin, A.; Moreno-Romero, A.K.; Xu, X.; Selig, E.E.; Penalva, L.O.F.; Libich, D.S. Structural Characterization of the RNA-Binding Protein SERBP1 Reveals Intrinsic Disorder and Atypical RNA Binding Modes. Front. Mol. Biosci. 2021, 8, 744707. [Google Scholar] [CrossRef]
- Weichsel, A.; Kievenaar, J.A.; Curry, R.; Croft, J.T.; Montfort, W.R. Instability in a coiled-coil signaling helix is conserved for signal transduction in soluble guanylyl cyclase. Protein Sci. 2019, 28, 1830–1839. [Google Scholar] [CrossRef]
- Horst, B.G.; Yokom, A.L.; Rosenberg, D.J.; Morris, K.L.; Hammel, M.; Hurley, J.H.; Marletta, M.A. Allosteric activation of the nitric oxide receptor soluble guanylate cyclase mapped by cryo-electron microscopy. eLife 2019, 8, e50634. [Google Scholar] [CrossRef]
- Holm, L. Using Dali for Protein Structure Comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [CrossRef]
- Kantsadi, A.L.; Hatzopoulos, G.N.; Gönczy, P.; Vakonakis, I. Structures of SAS-6 coiled coil hold implications for the polarity of the centriolar cartwheel. Structure 2022, 30, 671–684.e5. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Büttner, K.; Wenig, K.; Hopfner, K.P. Structural framework for the mechanism of archaeal exosomes in RNA processing. Mol. Cell 2005, 20, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeytuni, N.; Uebe, R.; Maes, M.; Davidov, G.; Baram, M.; Raschdorf, O.; Nadav-Tsubery, M.; Kolusheva, S.; Bitton, R.; Goobes, G.; et al. Cation diffusion facilitators transport initiation and regulation is mediated by cation induced conformational changes of the cytoplasmic domain. PLoS ONE 2014, 9, e92141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Chai, J.; Fu, D. Structural basis for autoregulation of the zinc transporter YiiP. Nat. Struct. Mol. Biol. 2009, 16, 1063–1067. [Google Scholar] [CrossRef]
- Schraidt, O.; Marlovits, T.C. Three-dimensional model of Salmonella’s needle complex at subnanometer resolution. Science 2011, 331, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Fong, Y.H.; Deme, J.C.; Furlong, E.J.; Kuhlen, L.; Lea, S.M. Symmetry mismatch in the MS-ring of the bacterial flagellar rotor explains the structural coordination of secretion and rotation. Nat. Microbiol. 2020, 5, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Carpousis, A.J. The RNA degradosome of Escherichia coli: An mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007, 61, 71–87. [Google Scholar] [CrossRef]
- Truebestein, L.; Leonard, T.A. Coiled-coils: The long and short of it. Bioessays 2016, 38, 903–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrakis, A.; Sixma, T.K. AI revolutions in biology: The joys and perils of AlphaFold. EMBO Rep. 2021, 22, e54046. [Google Scholar] [CrossRef] [PubMed]
- Van Breugel, M.; Rosa, E.; Silva, I.; Andreeva, A. Structural validation and assessment of AlphaFold2 predictions for centrosomal and centriolar proteins and their complexes. Commun. Biol. 2022, 5, 312. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021, 10.04.463034. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morellet, N.; Hardouin, P.; Assrir, N.; van Heijenoort, C.; Golinelli-Pimpaneau, B. Structural Insights into the Dimeric Form of Bacillus subtilis RNase Y Using NMR and AlphaFold. Biomolecules 2022, 12, 1798. https://doi.org/10.3390/biom12121798
Morellet N, Hardouin P, Assrir N, van Heijenoort C, Golinelli-Pimpaneau B. Structural Insights into the Dimeric Form of Bacillus subtilis RNase Y Using NMR and AlphaFold. Biomolecules. 2022; 12(12):1798. https://doi.org/10.3390/biom12121798
Chicago/Turabian StyleMorellet, Nelly, Pierre Hardouin, Nadine Assrir, Carine van Heijenoort, and Béatrice Golinelli-Pimpaneau. 2022. "Structural Insights into the Dimeric Form of Bacillus subtilis RNase Y Using NMR and AlphaFold" Biomolecules 12, no. 12: 1798. https://doi.org/10.3390/biom12121798