

The Pathophysiological Significance of “Mitochondrial Ejection” from Cells

Abstract
:1. Introduction
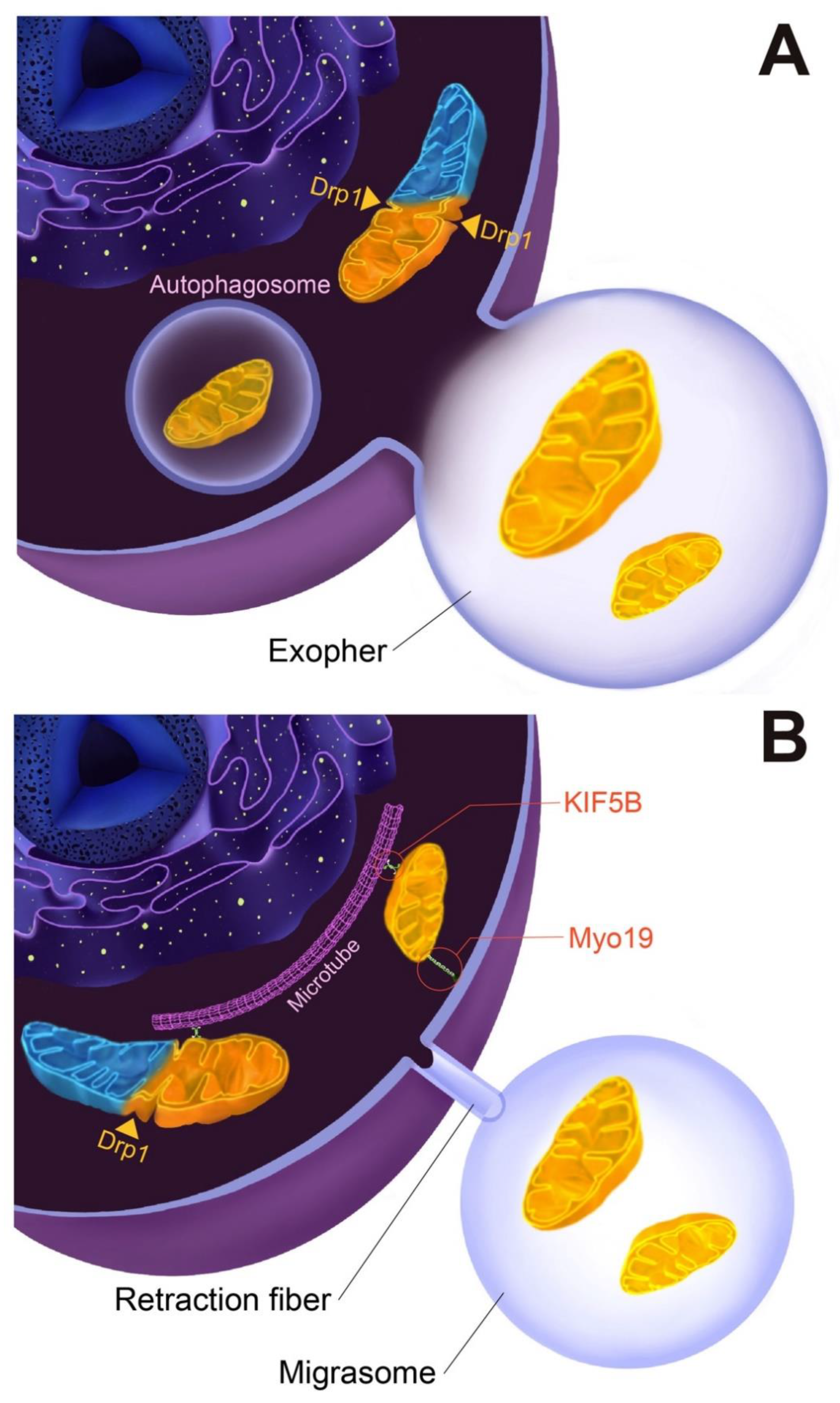
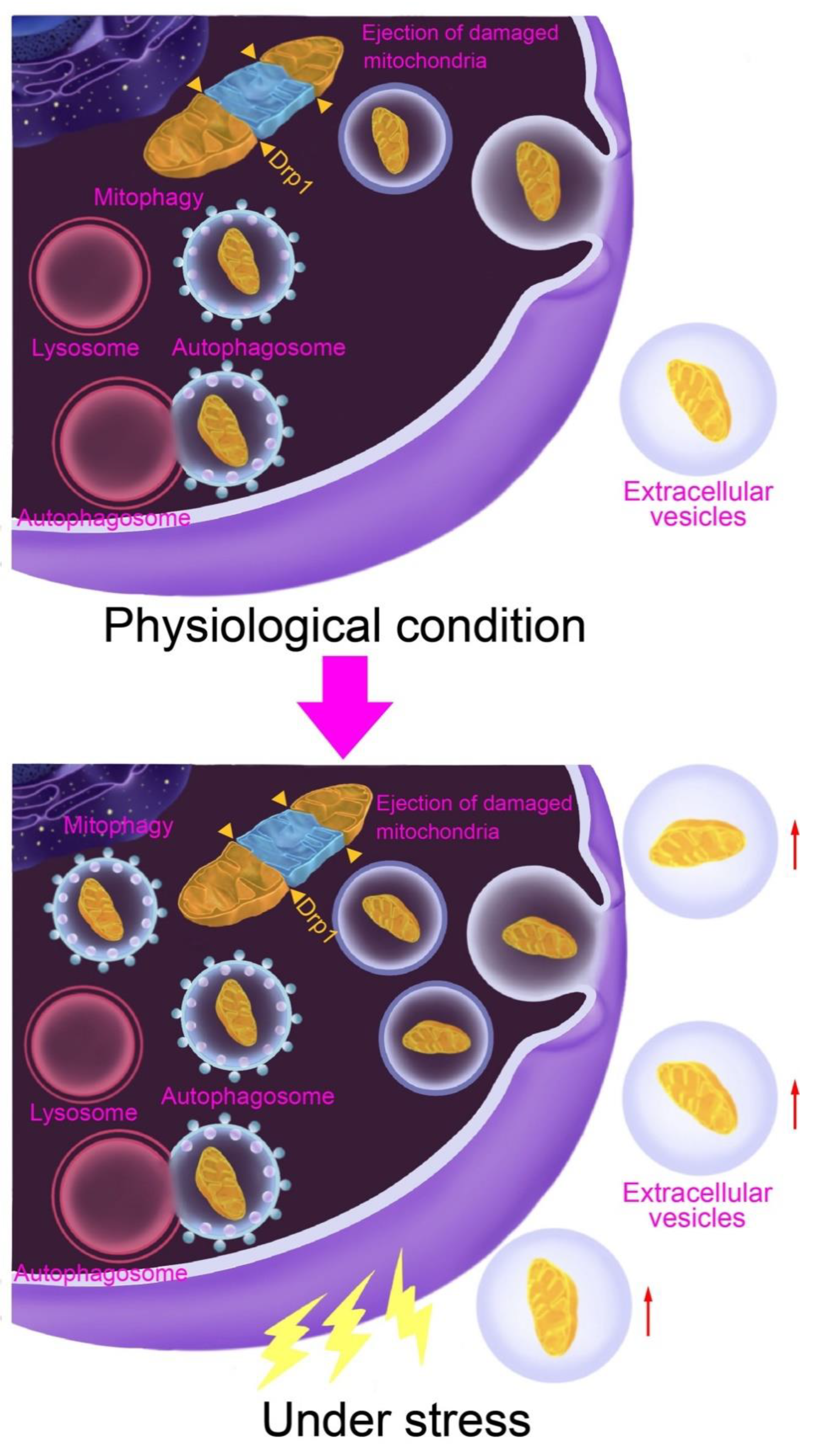
2. Disposal and Elimination of Damaged Mitochondria
3. Role of Released Mitochondria as a “Savior” of Target Cells
4. Released Mitochondria-Mediated Regulation of the Immune System
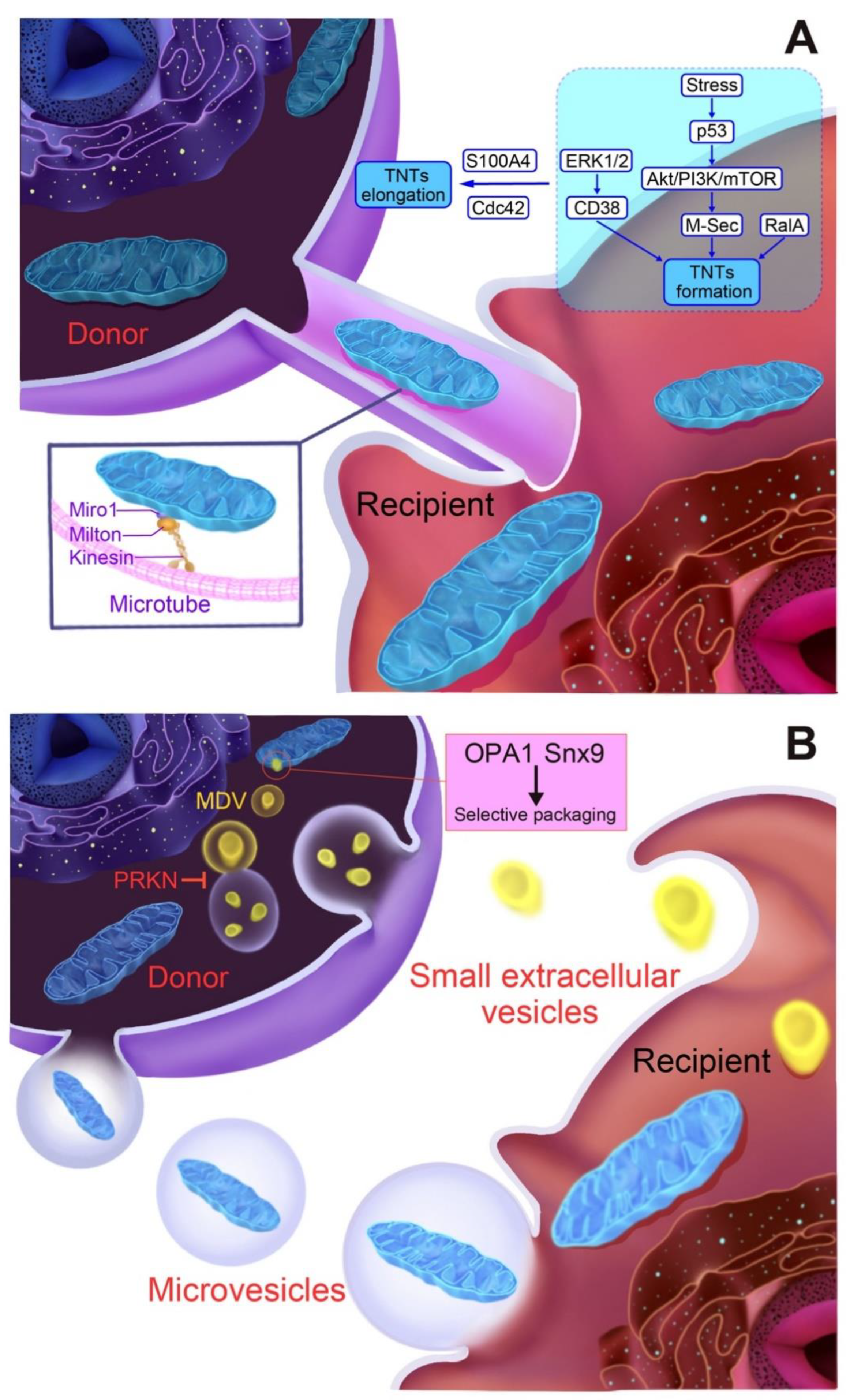
5. Mechanisms of Mitochondrial Release/Transfer from Cells
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Gabaldon, T.; Huynen, M.A. Shaping the mitochondrial proteome. Biochim. Biophys. Acta (BBA)-Bioenerg. 2004, 1659, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. CELL 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Yamazoe, M.; Sasano, T.; Ihara, K.; Takahashi, K.; Nakamura, W.; Takahashi, N.; Komuro, H.; Hamada, S.; Furukawa, T. Sparsely methylated mitochondrial cell free DNA released from cardiomyocytes contributes to systemic inflammatory response accompanied by atrial fibrillation. Sci. Rep. 2021, 11, 5837. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Williams, J.A.; Ding, W. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of mitochondrial biogenesis and function by the ubiquitin–proteasome system. Open Biol. 2017, 7, 170007. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2012, 18, 607–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Choong, C.; Okuno, T.; Ikenaka, K.; Baba, K.; Hayakawa, H.; Koike, M.; Yokota, M.; Doi, J.; Kakuda, K.; Takeuchi, T.; et al. Alternative mitochondrial quality control mediated by extracellular release. Autophagy 2021, 17, 2962–2974. [Google Scholar] [CrossRef]
- Rosina, M.; Ceci, V.; Turchi, R.; Chuan, L.; Borcherding, N.; Sciarretta, F.; Sánchez-Díaz, M.; Tortolici, F.; Karlinsey, K.; Chiurchiù, V.; et al. Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab. 2022, 34, 533–548.e12. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gao, Y.; Liu, J.; Huang, Y.; Yin, J.; Feng, Y.; Shi, L.; Meloni, B.P.; Zhang, C.; Zheng, M.; et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transduct. Target. Ther. 2021, 6, 65. [Google Scholar] [CrossRef]
- Jiao, H.; Jiang, D.; Hu, X.; Du, W.; Ji, L.; Yang, Y.; Li, X.; Sho, T.; Wang, X.; Li, Y.; et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell 2021, 184, 2896–2910.e13. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.O.; Kim, K.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Liu, F.; Hou, P.; Manaenko, A.; Xiao, Z.; Wang, F.; Xu, T.; Hu, Q. Neurons Release Injured Mitochondria as “Help-Me” Signaling After Ischemic Stroke. Front. Aging Neurosci. 2022, 14, 785761. [Google Scholar] [CrossRef]
- Falchi, A.M.; Sogos, V.; Saba, F.; Piras, M.; Congiu, T.; Piludu, M. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem. Cell Biol. 2012, 139, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolás-Ávila, J.A.; Pena-Couso, L.; Muñoz-Cánoves, P.; Hidalgo, A. Macrophages, Metabolism and Heterophagy in the Heart. Circ. Res. 2022, 130, 418–431. [Google Scholar] [CrossRef]
- Crewe, C.; Funcke, J.; Li, S.; Joffin, N.; Gliniak, C.M.; Ghaben, A.L.; An, Y.A.; Sadek, H.A.; Gordillo, R.; Akgul, Y.; et al. Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab. 2021, 33, 1853–1868.e11. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Koyanagi, M.; Brandes, R.P.; Haendeler, J.; Zeiher, A.M.; Dimmeler, S. Cell-to-Cell Connection of Endothelial Progenitor Cells with Cardiac Myocytes by Nanotubes: A Novel Mechanism for Cell Fate Changes? Circ. Res. 2005, 96, 1039–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial Transfer between Cells Can Rescue Aerobic Respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cselenyák, A.; Pankotai, E.; Horváth, E.M.; Kiss, L.; Lacza, Z. Mesenchymal stem cells rescue cardiomyoblasts from cell death in an in vitro ischemia model via direct cell-to-cell connections. BMC Cell Biol. 2010, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, E.Y.; Khryapenkova, T.G.; Vasileva, A.K.; Marey, M.V.; Galkina, S.I.; Isaev, N.K.; Sheval, E.V.; Polyakov, V.Y.; Sukhikh, G.T.; Zorov, D.B. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J. Cell. Mol. Med. 2008, 12, 1622–1631. [Google Scholar] [CrossRef]
- O’Brien, C.G.; Ozen, M.O.; Ikeda, G.; Vaskova, E.; Jung, J.H.; Bayardo, N.; Santoso, M.R.; Shi, L.; Wahlquist, C.; Jiang, Z.; et al. Mitochondria-Rich Extracellular Vesicles Rescue Patient-Specific Cardiomyocytes from Doxorubicin Injury: Insights Into the SENECA Trial. JACC CardioOncol. 2021, 3, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Fan, X.; Jiang, D.; Zhang, Y.; Li, X.; Xu, Z.; Fang, S.; Chiu, S.; Tse, H.; Lian, Q.; et al. Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Rep. 2018, 11, 1120–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.; Mak, J.C.W.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Khutornenko, A.A.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Improving the Post-Stroke Therapeutic Potency of Mesenchymal Multipotent Stromal Cells by Cocultivation With Cortical Neurons: The Role of Crosstalk Between Cells. Stem Cells Transl. Med. 2015, 4, 1011–1020. [Google Scholar] [CrossRef]
- Tseng, N.; Lambie, S.C.; Huynh, C.Q.; Sanford, B.; Patel, M.; Herson, P.S.; Ormond, D.R. Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1. J. Cereb. Blood Flow Metab. 2021, 41, 761–770. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 2018, 6, 139. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- English, K.; Shepherd, A.; Uzor, N.; Trinh, R.; Kavelaars, A.; Heijnen, C.J. Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol. Commun. 2020, 8, 36. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell. Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.; Burch, A.; Dave, K.M.; Sreeram, A.; Reynolds, M.J.; Dobbins, D.X.; Kamte, Y.S.; Zhao, W.; Sabatelle, C.; Joy, G.M.; et al. Microvesicles transfer mitochondria and increase mitochondrial function in brain endothelial cells. J. Control. Release 2021, 338, 505–526. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Shi, X.; Zhang, X.; Dang, S.; Ma, X.; Liu, F.; Xu, M.; Lv, Z.; Han, D.; Fang, X.; et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 2011, 92, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhang, J.; Xiao, H.; Wu, J.; He, K.; Lv, Z.; Li, Z.; Xu, M.; Zhang, Y. Mitochondria are transported along microtubules in membrane nanotubes to rescue distressed cardiomyocytes from apoptosis. Cell Death Dis. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, G.; Santoso, M.R.; Tada, Y.; Li, A.M.; Vaskova, E.; Jung, J.; O’Brien, C.; Egan, E.; Ye, J.; Yang, P.C. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll. Cardiol. 2021, 77, 1073–1088. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Kwon, S.H.; Jiang, K.; Ferguson, C.M.; Puranik, A.S.; Zhu, X.; Lerman, L.O. Renal scattered tubular-like cells confer protective effects in the stenotic murine kidney mediated by release of extracellular vesicles. Sci. Rep. 2018, 8, 1263. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Qin, A.; Liu, D.; Ruan, R.; Wang, Q.; Yuan, J.; Cheng, T.S.; Filipovska, A.; Papadimitriou, J.M.; Dai, K.; et al. Endoplasmic reticulum mediates mitochondrial transfer within the osteocyte dendritic network. Sci. Adv. 2019, 5, eaaw7215. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2022, 17, 98–106. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef]
- Hough, K.P.; Trevor, J.L.; Strenkowski, J.G.; Wang, Y.; Chacko, B.K.; Tousif, S.; Chanda, D.; Steele, C.; Antony, V.B.; Dokland, T.; et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to T cells. Redox Biol. 2018, 18, 54–64. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [Green Version]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zhang, C.; Lv, N.; Liang, Z.; Ma, T.; Cheng, H.; Xia, Y.; Shi, L. AdMSC-derived exosomes alleviate acute lung injury via transferring mitochondrial component to improve homeostasis of alveolar macrophages. Theranostics 2022, 12, 2928–2947. [Google Scholar] [CrossRef]
- Luz-Crawford, P.; Hernandez, J.; Djouad, F.; Luque-Campos, N.; Caicedo, A.; Carrère-Kremer, S.; Brondello, J.; Vignais, M.; Pène, J.; Jorgensen, C. Mesenchymal stem cell repression of Th17 cells is triggered by mitochondrial transfer. Curr. Stem Cell Res. Ther. 2019, 10, 232. [Google Scholar] [CrossRef]
- Court, A.C.; Le-Gatt, A.; Luz-Crawford, P.; Parra, E.; Aliaga-Tobar, V.; Bátiz, L.F.; Contreras, R.A.; Ortúzar, M.I.; Kurte, M.; Elizondo-Vega, R.; et al. Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Reports 2020, 21, e48052. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Q.; Feng, X.; Zhang, Z.; Geng, L.; Xu, T.; Wang, D.; Sun, L. Umbilical Cord-Derived Mesenchymal Stem Cells Suppress Autophagy of T Cells in Patients with Systemic Lupus Erythematosus via Transfer of Mitochondria. Stem Cells Int. 2016, 2016, 4062789. [Google Scholar] [CrossRef]
- Rabas, N.; Palmer, S.; Mitchell, L.; Ismail, S.; Gohlke, A.; Riley, J.S.; Tait, S.W.G.; Gammage, P.; Soares, L.L.; Macpherson, I.R.; et al. PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J. Cell Biol. 2021, 220, 1. [Google Scholar] [CrossRef]
- Zhang, Z.; Sheng, H.; Liao, L.; Xu, C.; Zhang, A.; Yang, Y.; Zhao, L.; Duan, L.; Chen, H.; Zhang, B. Mesenchymal Stem Cell-Conditioned Medium Improves Mitochondrial Dysfunction and Suppresses Apoptosis in Okadaic Acid-Treated SH-SY5Y Cells by Extracellular Vesicle Mitochondrial Transfer. J. Alzheimer’s Dis. 2020, 78, 1161–1176. [Google Scholar] [CrossRef]
- van der Vlist, M.; Raoof, R.; Willemen, H.L.D.M.; Prado, J.; Versteeg, S.; Martin Gil, C.; Vos, M.; Lokhorst, R.E.; Pasterkamp, R.J.; Kojima, T.; et al. Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. Neuron 2022, 110, 613–626.e9. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; van den Bosch, A.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021, 19, e3001166. [Google Scholar] [CrossRef]
- Levoux, J.; Prola, A.; Lafuste, P.; Gervais, M.; Chevallier, N.; Koumaiha, Z.; Kefi, K.; Braud, L.; Schmitt, A.; Yacia, A.; et al. Platelets facilitate the wound-healing capability of mesenchymal stem cells by mitochondrial transfer and metabolic reprogramming. Cell Metab. 2021, 33, 688–690. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.; Chai, Y.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Takatsu, H.; Kawano, S.; Watarai, H.; Kimura, S.; Yeaman, C.; Ohno, H.; Ohmae, M.; Kitamura, H.; Hase, K.; Hazelett, C.C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef]
- Wang, Y.; Ni, J.; Gao, T.; Gao, C.; Guo, L.; Yin, X. Activation of astrocytic sigma-1 receptor exerts antidepressant-like effect via facilitating CD38-driven mitochondria transfer. Glia 2020, 68, 2415–2426. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.; Su, X.; Wang, L.; Zhang, Y. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef] [Green Version]
- MacAskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 Is a Calcium Sensor for Glutamate Receptor-Dependent Localization of Mitochondria at Synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Sugiura, A.; McLelland, G.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.L.; Eng, S.P.; Hafez, P.; Abdul Karim, N.; Law, J.X.; Ng, M.H. Mesenchymal Stromal Cell Mitochondrial Transfer as a Cell Rescue Strategy in Regenerative Medicine: A Review of Evidence in Preclinical Models. Stem Cells Transl. Med. 2022, 11, 814. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Donor | Acceptor | Stimulation | Components | Outcome | Routes | Ref. |
|---|---|---|---|---|---|---|
| Adipocytes | Cardiomyocytes | Mitochondrial stress | Damaged mitochondria | Burst of ROS; protect the heart through hormesis | EVs | [24] |
| hMSCs | A549 cell line (NSCLC-derived) | Mitochondria and mtDNA | Rescued aerobic respiration | [27] | ||
| MSCs | Cardiomyoblasts | Ischemia | Mitochondria | Reduced cell death | [28] | |
| BMMSCs | H9c2 cell line | Ischemia/reperfusion | Mitochondria | Reduced apoptosis | TNTs | [29] |
| MSCs | Cardiomyocytes | Mitochondria | [30] | |||
| MSCs | ICMs | Doxorubicin administration | Mitochondria | Improved ICM contractility and mitochondrial biogenesis | EVs | [31] |
| BMSCs | Alveolar epithelia | Acute lung injury (ALI) caused by lipopolysaccharide (LPS) | Mitochondria | Reduced acute lung injury, increased alveolar ATP concentrations | Gap junctions and MVs | [32] |
| iPSC-MSCs | Epithelial cells | Mitochondria | Improved mitochondrial function, reduced inflammation | TNTs | [33] | |
| iPSC-MSCs | Epithelial cells | Cigarette smoke | Mitochondria | Alleviates alveolar fibrosis | TNTs | [34] |
| MMSCs | Neuronal cells | Mitochondria | Alleviated stroke-induced damage | [35] | ||
| MSCs | Mouse neurons | Hydrogen peroxide exposure | Mitochondria | Increased neuronal survival and improved metabolism | [36] | |
| MMSCs | Neurons | Oxygen-glucose deprivation | Mitochondria | Restored Cell Proliferation and respiration | TNTs | [37] |
| MSCs | Neural stem cells | Co-culture with cisplatin damaged NSCs | Mitochondria | Normalized mitochondrial membrane potential, prevented cell death | TNTs | [38] |
| Astrocytes | Neurons | Ischemia | Functional mitochondria | Increased neuronal viability and ATP levels | MVs | [39] |
| Astrocytes | Neurons | Cisplatin treatment | Mitochondria | Increased neuronal survival, restored neuronal mitochondrial membrane potential, and normalized neuronal calcium dynamics | [40] | |
| Astrocytes or neuronal cells | Astrocytes | Mitochondria | Elevated mitochondrial membrane potential | [41] | ||
| hCMEC/D3 (human brain EC cell line) | Brain ECs and neurons | Ischemia | Polarized mitochondria | Increased ATP levels | EVs | [42] |
| Cardiomyocytes | Cardiofibroblasts | Mitochondria | TNTs | [43] | ||
| Cardiac myofibroblasts | Cardiomyocytes | Hypoxia/reoxygenation | Mitochondria | Reduced apoptosis | TNTs | [44] |
| Human-iPS cell-derived cardiomyocytes (iCMs) | Cardiomyocytes | Mitochondria | Restoration of bioenergetics and mitochondrial biogenesis | EVs | [45] | |
| Renal scattered tubular cells | Tubular epithelial cells | Mitochondria | Improved mitochondrial function (in vitro), perfusion and oxygenation(in vivo) | EVs | [46] | |
| B16ρ0 mouse melanoma cells | Mitochondria | Rescued mitochondrial function | [50] | |||
| BMSCs | Multiple myeloma cells | Mitochondria | Increased ATP levels and proliferation | TNTs | [49] | |
| MSCs | Jurkat cells | Chemotherapeutic drugs | Mitochondria | Chemoresistance | TNTs | [51] |
| MSCs | Primary bone marrow cells of ALL patients | Chemotherapeutic drugs | Mitochondria | Reduced apoptosis | TNTs | [52] |
| NKT cells (DN32.D3, mouse T cells) | Cancer cells (MDA-MB-231, 4T1, TALL-104) | Mitochondria | Enhanced cancer cell activity and caused death of immune cells | TNTs | [53] | |
| MDA-MB-231 breast cancer cells | MDA-MB-231 breast cancer cells | Activation of mGluR3 by extracellular glutamate | mtDNA | Promote endosomal trafficking and invasiveness | MVs | [63] |
| Human umbilical cord derived MSC-CM | SH-SY5Y cells | OA(okadaic acid) treatment | Mitochondria | Alleviated oxidative stress, suppressed apoptosis, improved mitochondrial function | MVs | [64] |
| MSCs | Macrophage | Acute respiratory distress syndrome (ARDS) | Mitochondria | Promote an anti-inflammatory and highly phagocytic macrophage phenotype | MVs | [58] |
| Macrophages | Sensory neurons | Inflammation | Mitochondria | Resolution of inflammatory pain | EVs | [65] |
| Airway myeloid-derived regulatorycells | T cells | Polarized mitochondria and mtDNA | EVs | [56] | ||
| Neural stem cell | mtDNA-deficient L929 Rho0 cells (mononuclear phagocytes) | Functional mitochondria | Rescued mitochondrial function, increased cell survival, reduced the expression of pro-inflammatory markers | EVs | [66] | |
| Platelets | MSC | Mitochondria | Improved the regenerative capacity | EVs | [67] | |
| MSCs | Epithelial cell | Stress induction by rotenone or TNF-α | Mitochondria | Reduced ROS production | TNTs | [68] |
| MSCs | cardiomyocytes | Anthracycline or Dox | Mitochondria | Rescued damage | TNTs | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Maejima, Y.; Wei, L.; Nakagama, S.; Shiheido-Watanabe, Y.; Sasano, T. The Pathophysiological Significance of “Mitochondrial Ejection” from Cells. Biomolecules 2022, 12, 1770. https://doi.org/10.3390/biom12121770
Fan Q, Maejima Y, Wei L, Nakagama S, Shiheido-Watanabe Y, Sasano T. The Pathophysiological Significance of “Mitochondrial Ejection” from Cells. Biomolecules. 2022; 12(12):1770. https://doi.org/10.3390/biom12121770
Chicago/Turabian StyleFan, Qintao, Yasuhiro Maejima, Lai Wei, Shun Nakagama, Yuka Shiheido-Watanabe, and Tetsuo Sasano. 2022. "The Pathophysiological Significance of “Mitochondrial Ejection” from Cells" Biomolecules 12, no. 12: 1770. https://doi.org/10.3390/biom12121770