
cGMP Signaling in the Neurovascular Unit—Implications for Retinal Ganglion Cell Survival in Glaucoma


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
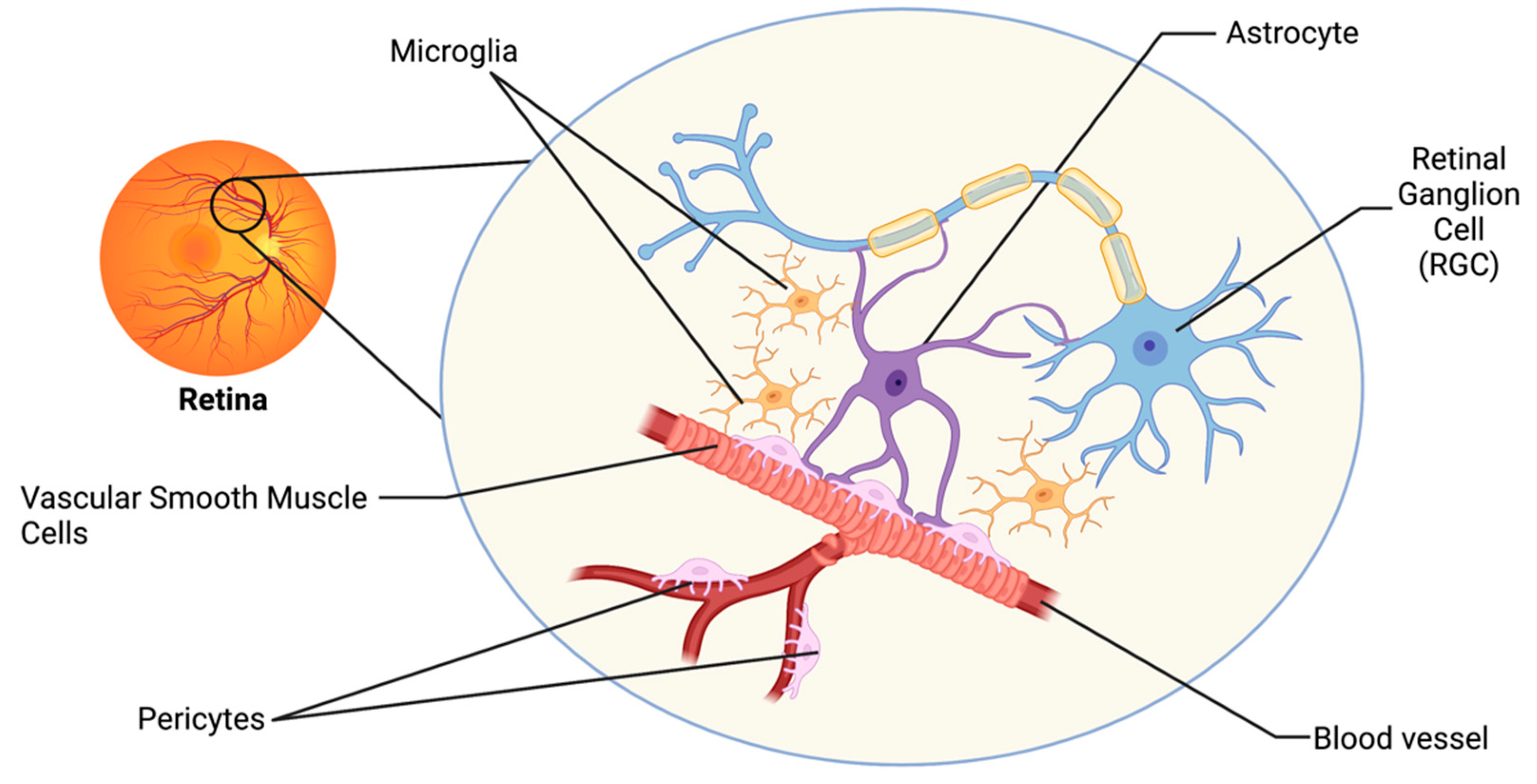
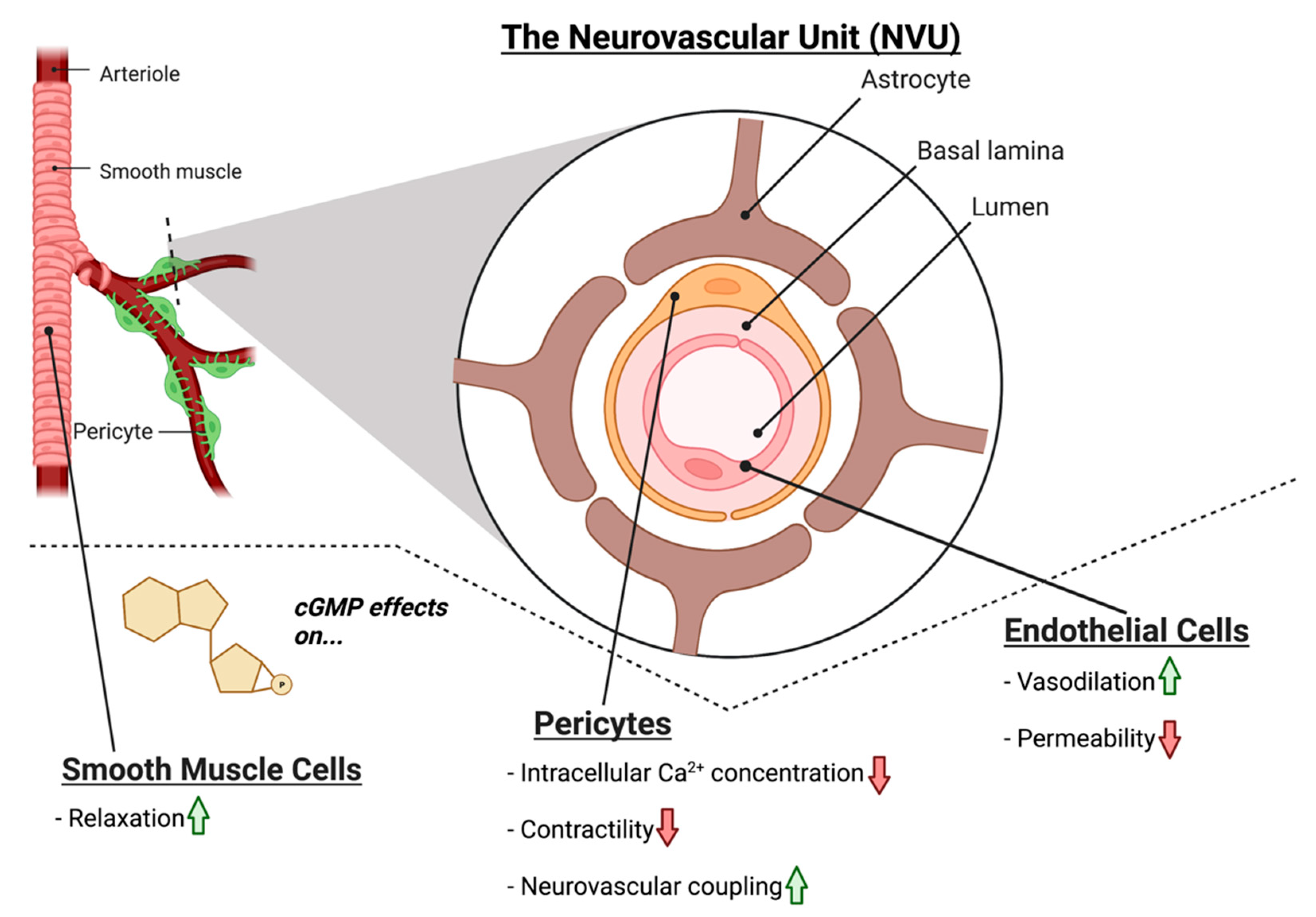
2. The Retinal Neurovascular Unit and Glaucoma
3. NO-cGMP Signaling in Glaucoma
4. NO-cGMP Signaling in Vascular Cells
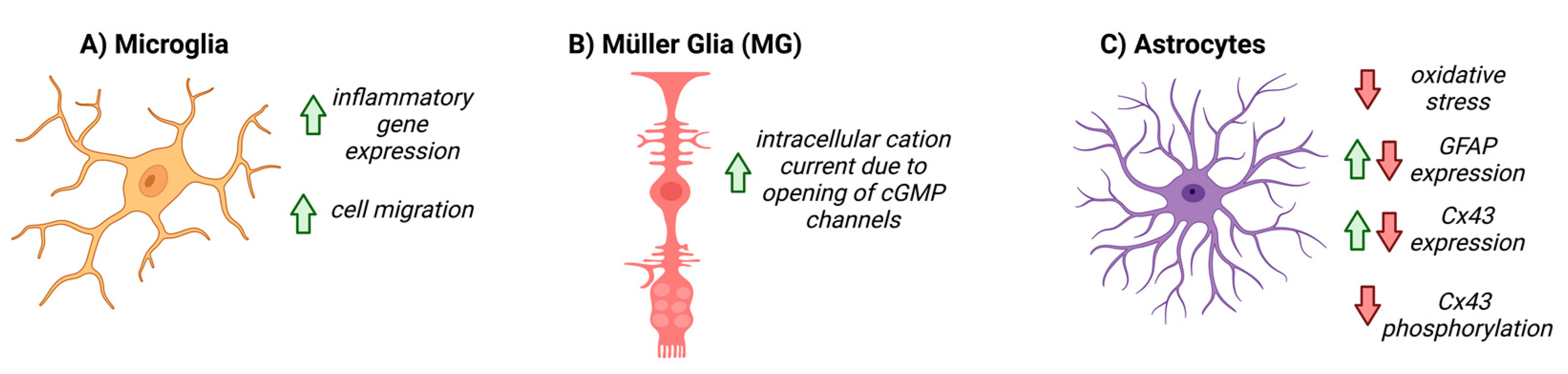
5. NO-cGMP Signaling in Glial Cells
6. NO-cGMP Signaling in Retinal Neurons
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Calkins, D.J. Adaptive responses to neurodegenerative stress in glaucoma. Prog. Retin. Eye Res. 2021, 84, 100953. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Calkins, D.J. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog. Retin. Eye Res. 2012, 31, 702–719. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Y.; Lai, Y.J.; Yen, Y.F.; Shen, Y.C.; Wang, C.Y.; Liang, C.Y.; Lin, K.H.; Fan, L.W. Association between normal tension glaucoma and the risk of Alzheimer’s disease: A nationwide population-based cohort study in Taiwan. BMJ Open 2018, 8, e022987. [Google Scholar] [CrossRef] [PubMed]
- Dascalu, A.M.; Stana, D.; Nicolae, V.A.; Cirstoveanu, C.; Vancea, G.; Serban, D.; Socea, B. Association between vascular comorbidity and glaucoma progression: A four-year observational study. Exp. Ther. Med. 2021, 21, 283. [Google Scholar] [CrossRef]
- Dienstbier, E.; Balik, J.; Kafka, H. A contribution to the theory of the vascular origin of glaucoma. Br. J. Ophthalmol. 1950, 34, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Wareham, L.K.; Calkins, D.J. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452. [Google Scholar] [CrossRef]
- Trivli, A.; Koliarakis, I.; Terzidou, C.; Goulielmos, G.N.; Siganos, C.S.; Spandidos, D.A.; Dalianis, G.; Detorakis, E.T. Normal-tension glaucoma: Pathogenesis and genetics. Exp. Ther. Med. 2019, 17, 563–574. [Google Scholar]
- Amerasinghe, N.; Aung, T.; Cheung, N.; Fong, C.W.; Wang, J.J.; Mitchell, P.; Saw, S.M.; Wong, T.Y. Evidence of retinal vascular narrowing in glaucomatous eyes in an Asian population. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5397–5402. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front. Cell Dev. Biol. 2021, 9, 2641. [Google Scholar] [CrossRef]
- Garhofer, G.; Zawinka, C.; Resch, H.; Huemer, K.H.; Schmetterer, L.; Dorner, G.T. Response of retinal vessel diameters to flicker stimulation in patients with early open angle glaucoma. J. Glaucoma 2004, 13, 340–344. [Google Scholar] [CrossRef]
- Garhofer, G.; Resch, H.; Weigert, G.; Lung, S.; Simader, C.; Schmetterer, L. Short-term increase of intraocular pressure does not alter the response of retinal and optic nerve head blood flow to flicker stimulation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1721–1725. [Google Scholar] [CrossRef] [Green Version]
- Alarcon-Martinez, L.; Shiga, Y.; Villafranca-Baughman, D.; Belforte, N.; Quintero, H.; Di Polo, A. Perciyte dysfunction and loss of inter-pericyte tunneling nanotubes promote neurovascular deficits in glaucoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2110329119. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Chapter Four—Nitric Oxide Signaling in Neurodegeneration and Cell Death. Adv. Pharmacol. 2018, 82, 57–83. [Google Scholar]
- Knowles, R.G.; Palacios, M.; Palmer, R.M.; Moncada, S. Formation of nitric oxide from L-arginine in the central nervous system: A transduction mechanism for stimulation of the soluble guanylate cyclase. Proc. Natl. Acad. Sci. USA 1989, 86, 5159–5162. [Google Scholar] [CrossRef] [Green Version]
- Mergia, E.; Russwurm, M.; Zoidl, G.; Koesling, D. Major occurrence of the new alpha(2)beta(1) isoform of NO-sensitive guanylyl cyclase in brain. Cell Signal. 2003, 15, 189–195. [Google Scholar] [CrossRef]
- Sharina, I.G.; Jelen, F.; Bogatenkova, E.P.; Thomas, A.; Martin, E.; Murad, F. Alpha1 soluble guanylyl cyclase (sGC) splice forms as potential regulators of human sGC activity. J. Biol. Chem. 2008, 283, 15104–15113. [Google Scholar] [CrossRef] [Green Version]
- Zagotta, W.N.; Siegelbaum, S.A. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 1996, 19, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Sandner, P.; Schmidtko, A. cGMP: A unique 2nd messenger molecule—Recent developments in cGMP research and development. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Galassi, F.; Renieri, G.; Sodi, A.; Ucci, F.; Vannozzi, L.; Masini, E. Nitric oxide proxies and ocular perfusion pressure in primary open angle glaucoma. Br. J. Ophthalmol. 2004, 88, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Doganay, S.; Evereklioglu, C.; Turkoz, Y.; Er, H. Decreased nitric oxide production in primary open-angle glaucoma. Eur. J. Ophthalmol. 2002, 12, 44–48. [Google Scholar] [CrossRef]
- Buys, E.S.; Ko, Y.C.; Alt, C.; Hayton, S.R.; Jones, A.; Tainsh, L.T.; Ren, R.; Giani, A.; Clerte, M.; Abernathy, E.; et al. Soluble Guanylate Cyclase alpha1-Deficient Mice: A Novel Murine Model for Primary Open Angle Glaucoma. PLoS ONE 2013, 8, e60156. [Google Scholar] [CrossRef]
- Sappington, R.M.; Carlson, B.J.; Crish, S.D.; Calkins, D.J. The microbead occlusion model: A paradigm for induced ocular hypertension in rats and mice. Investig. Ophthalmol. Vis. Sci. 2010, 51, 207–216. [Google Scholar] [CrossRef]
- Wareham, L.K.; Dordea, A.C.; Schleifer, G.; Yao, V.; Batten, A.; Mertz, J.; Gregory-Ksander, M.; Pasquale, L.; Buys, E.S.; Sappington, R.M. Increased bioavailability of cyclic guanylate monophosphate prevents retinal ganglion cell degeneration. Neurobiol. Dis. 2019, 121, 65–75. [Google Scholar] [CrossRef]
- Holden, J.M.; Al Hussein Al Awamlh, S.; Croteau, L.-P.; Boal, A.M.; Rex, T.S.; Risner, M.L.; Calkins, D.J.; Wareham, L.K. Dysfunctional cGMP Signaling Leads to Age-Related Retinal Vascular Alterations and Astrocyte Remodeling in Mice. Int. J. Mol. Sci. 2022, 23, 3066. [Google Scholar] [CrossRef]
- Schneemann, A.; Leusink-Muis, A.; van den Berg, T.; Hoyng, P.F.; Kamphuis, W. Elevation of nitric oxide production in human trabecular meshwork by increased pressure. Graefes Arch. Clin. Exp. Ophthalmol. 2003, 241, 321–326. [Google Scholar] [CrossRef]
- Kotikoski, H.; Alajuuma, P.; Moilanen, E.; Salmenpera, P.; Oksala, O.; Laippala, P.; Vapaatalo, V. Comparison of nitric oxide donors in lowering intraocular pressure in rabbits: Role of cyclic GMP. J. Ocul. Pharmacol. Ther. 2002, 18, 11–23. [Google Scholar] [CrossRef]
- Kotikoski, H.; Vapaatalo, H.; Oksala, O. Nitric oxide and cyclic GMP enhance aqueous humor outflow facility in rabbits. Curr. Eye Res. 2003, 26, 119–123. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, L.R. Vascular and autonomic dysregulation in primary open-angle glaucoma. Curr. Opin. Ophthalmol. 2016, 27, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolluru, G.K.; Tamilarasan, K.P.; Rajkumar, A.S.; Geetha Priya, S.; Rajaram, M.; Saleem, N.K.; Majumder, S.; Ali, B.M.J.; Illavazagan, G.; Chatterjee, S. Nitric oxide/cGMP protects endothelial cells from hypoxia-mediated leakiness. Eur. J. Cell Biol. 2008, 87, 147–161. [Google Scholar] [CrossRef]
- Monica, F.Z.; Bian, K.; Murad, F. The Endothelium-Dependent Nitric Oxide-cGMP Pathway. Adv. Pharmacol. 2016, 77, 1–27. [Google Scholar] [PubMed]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp. Neurol. 2004, 190, 446–455. [Google Scholar] [CrossRef]
- Sugiyama, T.; Moriya, S.; Oku, H.; Azuma, I. Association of endothelin-1 with normal tension glaucoma: Clinical and fundamental studies. Surv. Ophthalmol. 1995, 39 (Suppl. S1), S49–S56. [Google Scholar] [CrossRef]
- Noske, W.; Hensen, J.; Wiederholt, M. Endothelin-like immunoreactivity in aqueous humor of patients with primary open-angle glaucoma and cataract. Graefes Arch. Clin. Exp. Ophthalmol. 1997, 235, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.M.; Kiu, K.Y.; Thambiraja, R.; Sulong, S.; Rasool, A.H.; Liza-Sharmini, A.T. Microvascular endothelial function and severity of primary open angle glaucoma. Eye 2016, 30, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resch, H.; Garhofer, G.; Fuchsjager-Mayrl, G.; Hommer, A.; Schmetterer, L. Endothelial dysfunction in glaucoma. Acta Ophthalmol. 2009, 87, 4–12. [Google Scholar] [CrossRef]
- Su, W.W.; Cheng, S.T.; Ho, W.J.; Tsay, P.K.; Wu, S.C.; Chang, S.H. Glaucoma is associated with peripheral vascular endothelial dysfunction. Ophthalmology 2008, 115, 1173–1178.e1. [Google Scholar] [CrossRef]
- Cellini, M.; Strobbe, E.; Gizzi, C.; Balducci, N.; Toschi, P.G.; Campos, E.C. Endothelin-1 plasma levels and vascular endothelial dysfunction in primary open angle glaucoma. Life Sci. 2012, 91, 699–702. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Pagano, C.; Baesso, I.; Kotsafti, O.; Doro, D.; de Kreutzenberg, S.V.; Avogaro, A.; Agostini, C.; Dorigo, M.T. Reduced endothelial progenitor cells and brachial artery flow-mediated dilation as evidence of endothelial dysfunction in ocular hypertension and primary open-angle glaucoma. Acta Ophthalmol. 2010, 88, 135–141. [Google Scholar] [CrossRef]
- Tong, L.; Hill, R.A.; Damisah, E.C.; Murray, K.N.; Yuan, P.; Bordey, A.; Grutzendler, J. Imaging and optogenetic modulation of vascular mural cells in the live brain. Nat. Protoc. 2021, 16, 472–496. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Henshall, T.L.; Keller, A.; He, L.; Johansson, B.R.; Wallgard, E.; Raschperger, E.; Mae, M.A.; Jin, S.; Bersholtz, C.; Lendahl, U. Notch3 is necessary for blood vessel integrity in the central nervous system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Kutcher, M.E.; Kolyada, A.Y.; Surks, H.K.; Herman, I.M. Pericyte Rho GTPase mediates both pericyte contractile phenotype and capillary endothelial growth state. Am. J. Pathol. 2007, 171, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Alarcon-Martinez, L.; Villafranca-Baughman, D.; Quintero, H.; Kacerovsky, J.B.; Dotigny, F.; Murai, K.K.; Prat, A.; Drapeau, P.; Di Polo, A. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 2020, 585, 91–95. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Alarcon-Martinez, L.; Yilmaz-Ozcan, S.; Yemisci, M.; Schallek, J.; Kılıç, K.; Villafranca-Baughman, D.; Can, A.; Di Polo, A.; Dalkara, T. Retinal ischemia induces α-SMA-mediated capillary pericyte contraction coincident with perivascular glycogen depletion. Acta Neuropathol. Commun. 2019, 7, 134. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.; Gardiner, T.; Hu, P.; Baxter, L.; Rosinova, E.; Chan-Ling, T. Altered pericyte-endothelial relations in the rat retina during aging: Implications for vessel stability. Neurobiol. Aging 2006, 27, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Orgül, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Reanrd, J.P.; Stefansson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Majumder, S.; Tamilarasan, K.P.; Kolluru, G.K.; Muley, A.; Nair, C.M.; Omanakuttan, A.; Murty, K.V.G.K.; Chatterjee, S. Activated pericyte attenuates endothelial functions: Nitric oxide-cGMP rescues activated pericyte-associated endothelial dysfunctions. Biochem. Cell Biol. 2007, 85, 709–720. [Google Scholar] [CrossRef]
- Zambach, S.A.; Cai, C.; Helms, H.C.C.; Hald, B.O.; Dong, Y.; Fordsmann, J.C.; Nielsen, R.M.; Hu, J.; Lonstrup, M.; Brodin, B.; et al. Precapillary sphincters and pericytes at first-order capillaries as key regulators for brain capillary perfusion. Proc. Natl. Acad. Sci. USA 2021, 118, e2023749118. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Kisler, K.; Nelson, A.R.; Rege, S.V.; Ramanathan, A.; Wang, Y.; Ahuja, A.; Lazic, D.; Tsai, P.S.; Zhao, Z.; Zhao, Y.; et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 2017, 20, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, K.; Kawamura, H.; Wu, D.M.; Puro, D.G. Nitric oxide/cGMP-induced inhibition of calcium and chloride currents in retinal pericytes. Microvasc. Res. 2001, 62, 196–203. [Google Scholar] [CrossRef]
- Newman, E.A. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140195. [Google Scholar] [CrossRef]
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The Translational Significance of the Neurovascular Unit. J. Biol. Chem. 2017, 292, 762–770. [Google Scholar] [CrossRef]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sanchez, M.C. A journey into the retina: Müller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef]
- Pellerin, L.; Pellegri, G.; Bittar, P.G.; Charnay, Y.; Bouras, C.; Martin, J.L.; Stella, N.; Magistretti, P.J. Evidence Supporting the Existence of an Activity-Dependent Astrocyte-Neuron Lactate Shuttle. Dev. Neurosci. 1998, 20, 291–299. [Google Scholar] [CrossRef]
- Mason, S. Lactate Shuttles in Neuroenergetics—Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- Boal, A.M.; Risner, M.L.; Cooper, M.L.; Wareham, L.K.; Calkins, D.J. Astrocyte Networks as Therapeutic Targets in Glaucomatous Neurodegeneration. Cells 2021, 10, 1368. [Google Scholar] [CrossRef] [PubMed]
- Price, B.R.; Norris, C.M.; Sompol, P.; Wilcock, D.M. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 2018, 144, 644–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Glia of the human retina. Glia 2020, 68, 768–796. [Google Scholar] [CrossRef]
- Dixon, M.A.; Greferath, U.; Fletcher, E.L.; Jobling, A.I. The Contribution of Microglia to the Development and Maturation of the Visual System. Front. Cell Neurosci. 2021, 15, 659843. [Google Scholar] [CrossRef]
- Császár, E.; Lénárt, N.; Cserép, C.; Környei, Z.; Fekete, R.; Pósfai, B.; Balazsfi, D.; Hangya, B.; Schwarcz, A.D.; Szabadits, E.; et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J. Exp. Med. 2022, 219, e20211071. [Google Scholar] [CrossRef]
- Varela, H.J.; Hernandez, M.R. Astrocyte responses in human optic nerve head with primary open-angle glaucoma. J. Glaucoma 1997, 6, 303–313. [Google Scholar] [CrossRef]
- Hernandez, M.R.; Miao, H.; Lukas, T. Astrocytes in glaucomatous optic neuropathy. Prog. Brain Res. 2008, 173, 353–373. [Google Scholar]
- Akopian, A.; Kumar, S.; Ramakrishnan, H.; Roy, K.; Viswanathan, S.; Bloomfield, S.A. Targeting neuronal gap junctions in mouse retina offers neuroprotection in glaucoma. J. Clin. Investig. 2017, 127, 2647–2661. [Google Scholar] [CrossRef] [PubMed]
- Malone, P.; Miao, H.; Parker, A.; Juarez, S.; Hernandez, M.R. Pressure induces loss of gap junction communication and redistribution of connexin 43 in astrocytes. Glia 2007, 55, 1085–1098. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.L.; Crish, S.D.; Inman, D.M.; Horner, P.J.; Calkins, D.J. Early astrocyte redistribution in the optic nerve precedes axonopathy in the DBA/2J mouse model of glaucoma. Exp. Eye Res. 2016, 150, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.L.; Pasini, S.; Lambert, W.S.; D’Alessandro, K.B.; Yao, V.; Risner, M.L.; Calkins, D.J. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 18810–18821. [Google Scholar] [CrossRef]
- García-Bermúdez, M.Y.; Freude, K.K.; Mouhammad, Z.A.; Van Wijngaarden, P.; Martin, K.K.; Kolko, M. Glial cells in glaucoma: Friends, foes, and potential therapeutic targets. Front. Neurol. 2021, 12, 169. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Koizumi, S. Potential roles of astrocytes and Müller cells in the pathogenesis of glaucoma. J. Pharmacol. Sci. 2021, 145, 262–267. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, R.; Luo, X.; Wang, F.; Sun, X. The interaction between microglia and macroglia in glaucoma. Front. Neurosci. 2021, 15, 573. [Google Scholar]
- Pifarre, P.; Prado, J.; Giralt, M.; Molinero, A.; Hidalgo, J.; Garcia, A. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J. Neurochem. 2010, 112, 807–817. [Google Scholar] [CrossRef]
- Bonkale, W.L.; Winblad, B.; Ravid, R.; Cowburn, R.F. Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci. Lett 1995, 187, 5–8. [Google Scholar] [CrossRef]
- Baltrons, M.A.; Pifarré, P.; Ferrer, I.; Carot, J.M.; García, A. Reduced expression of NO-sensitive guanylyl cyclase in reactive astrocytes of Alzheimer disease, Creutzfeldt–Jakob disease, and multiple sclerosis brains. Neurobiol. Dis. 2004, 17, 462–472. [Google Scholar] [CrossRef]
- Kusaka, S.; Dabin, I.; Barnstable, C.J.; Puro, D.G. cGMP-mediated effects on the physiology of bovine and human retinal Müller (glial) cells. J. Physiol. 1996, 497 Pt 3, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Paris, D.; Town, T.; Parker, T.A.; Tan, J.; Humphrey, J.; Crawford, F.; Mullan, M. Inhibition of Alzheimer’s β-amyloid induced vasoactivity and proinflammatory response in microglia by a cGMP-dependent mechanism. Exp. Neurol. 1999, 157, 211–221. [Google Scholar] [CrossRef]
- Paris, D.; Town, T.; Mullan, M. Novel strategies for opposing murine microglial activation. Neurosci. Lett. 2000, 278, 5–8. [Google Scholar] [CrossRef]
- Choi, S.H.; Choi, D.H.; Song, K.S.; Shin, K.H.; Chun, B.G. Zaprinast, an inhibitor of cGMP-selective phosphodiesterases, enhances the secretion of TNF-α and IL-1β and the expression of iNOS and MHC class II molecules in rat microglial cells. J. Neurosci. Res. 2002, 67, 411–421. [Google Scholar] [CrossRef]
- Pilz, R.B.; Broderick, K.E. Role of cyclic GMP in gene regulation. Front. Biosci. 2005, 10, 1239–1268. [Google Scholar] [CrossRef] [Green Version]
- Vollmar, A.M. The role of atrial natriuretic peptide in the immune system. Peptides 2005, 26, 1086–1094. [Google Scholar] [CrossRef]
- Moriyama, N.; Taniguchi, M.; Miyano, K.; Miyoshi, M.; Watanabe, T. ANP inhibits LPS-induced stimulation of rat microglial cells by suppressing NF-kappaB and AP-1 activations. Biochem. Biophys Res. Commun. 2006, 350, 322–328. [Google Scholar]
- Roy, A.; Fung, Y.K.; Liu, X.; Pahan, K. Up-regulation of microglial CD11b expression by nitric oxide. J. Biol. Chem. 2006, 281, 14971–14980. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Haugabook, S.J.; Sahley, C.L.; Muller, K.J. Methylene blue blocks cGMP production and disrupts directed migration of microglia to nerve lesions in the leech CNS. J. Neurobiol. 2003, 57, 183–192. [Google Scholar] [CrossRef]
- Scheiblich, H.; Roloff, F.; Singh, V.; Stangel, M.; Stern, M.; Bicker, G. Nitric oxide/cyclic GMP signaling regulates motility of a microglial cell line and primary microglia in vitro. Brain Res. 2014, 1564, 9–21. [Google Scholar] [CrossRef]
- Borán, M.S.; García, A. The cyclic GMP-protein kinase G pathway regulates cytoskeleton dynamics and motility in astrocytes. J. Neurochem. 2007, 102, 216–230. [Google Scholar] [CrossRef]
- Brahmachari, S.; Fung, Y.K.; Pahan, K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 2006, 26, 4930–4939. [Google Scholar] [CrossRef] [Green Version]
- Sticozzi, C.; Belmonte, G.; Frosini, M.; Pessina, F. Nitric Oxide/Cyclic GMP-Dependent Calcium Signalling Mediates IL-6- and TNF-α-Induced Expression of Glial Fibrillary Acid Protein. J. Mol. Neurosci. 2021, 71, 854–866. [Google Scholar]
- Ampey, B.C.; Ampey, A.C.; Lopez, G.E.; Bird, I.M.; Magness, R.R. Cyclic Nucleotides Differentially Regulate Cx43 Gap Junction Function in Uterine Artery Endothelial Cells From Pregnant Ewes. Hypertension 2017, 70, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Hiramatsu, N.; Zhu, Y.; Morioka, T.; Takeda, M.; Oite, T.; Kitamura, M. Nitric oxide-mediated regulation of connexin43 expression and gap junctional intercellular communication in mesangial cells. J. Am. Soc. Nephrol. 2005, 16, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crassous, P.A.; Shu, P.; Huang, C.; Gordan, R.; Brouckaert, P.; Lampe, P.D.; Xie, L.H.; Beuve, A. Newly Identified NO-Sensor Guanylyl Cyclase/Connexin 43 Association Is Involved in Cardiac Electrical Function. J. Am. Heart Assoc. 2017, 6, e006397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, C.N.; Martin, D.N.; Shaver, P.; Madamanchi, C.; Muller-Borer, B.J.; Tulis, D.A. Control of vascular smooth muscle cell growth by connexin 43. Front. Physiol. 2012, 3, 220. [Google Scholar] [CrossRef] [Green Version]
- Podda, M.V.; Leone, L.; Piacentini, R.; Cocco, S.; Mezzogori, D.; D’Ascenzo, M.; Grassi, C. Expression of olfactory-type cyclic nucleotide-gated channels in rat cortical astrocytes. Glia 2012, 60, 1391–1405. [Google Scholar] [CrossRef]
- Zarate, S.M.; Huntington, T.E.; Bagher, P.; Srinivasan, R. Aging reduces calreticulin expression and alters spontaneous calcium signals in astrocytic endfeet of the mouse dorsolateral striatum. bioRxiv 2021. [Google Scholar] [CrossRef]
- Woart, N.M. Enhanced cGMP-Dependent Signaling in Astrocytes: Novel Therapeutic Target in Alzheimer’s Disease; Philadelphia College of Osteopathic Medicine: Philadelphia, PA, USA, 2015. [Google Scholar]
- Jehle, A.; Garaschuk, O. The Interplay between cGMP and Calcium Signaling in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 7048. [Google Scholar] [CrossRef]
- Burette, A.; Zabel, U.; Weinberg, R.J.; Schmidt, H.H.; Valtschanoff, J.G. Synaptic localization of nitric oxide synthase and soluble guanylyl cyclase in the hippocampus. J. Neurosci. 2002, 22, 8961–8970. [Google Scholar] [CrossRef]
- Wareham, L.K.; Buys, E.S.; Sappington, R.M. The nitric oxide-guanylate cyclase pathway and glaucoma. Nitric Oxide 2018, 77, 75–87. [Google Scholar] [CrossRef]
- Fiscus, R.R.; Johlfs, M.G. Protein kinase G (PKG): Involvement in Promoting Neural Cell Survival, Proliferation, Synaptogenesis, and Synaptic Plasticity and the Use of New Ultrasensitive Capillary-Electrophoresis-Based Methodologies for Measuring PKG Expression and Molecular Actions. In Protein Kinase Technologies; Mukai, H., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 319–347. [Google Scholar]
- Nagai, A.; Nakamura, M.; Mukuno, H.; Negi, A.; Seigel, G.M. Nipradilol rescues retinal neurons from apoptosis by nitric oxide donation through a cGMP/PKG pathway in culture. Investig. Ophthalmol. Vis. Sci. 2004, 45, 914. [Google Scholar]
- Mastrodimou, N.; Kiagiadaki, F.; Thermos, K. The Role of Nitric Oxide and cGMP in Somatostatin’s Protection against Retinal Ischemia. Investig. Ophthalmol. Vis. Sci. 2008, 49, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Mejía-García, T.A.; Portugal, C.C.; Encarnação, T.G.; Prado, M.A.M.; Paes-de-Carvalho, R. Nitric oxide regulates AKT phosphorylation and nuclear translocation in cultured retinal cells. Cell. Signal. 2013, 25, 2424–2439. [Google Scholar] [CrossRef]
- Estévez, A.G.; Spear, N.; Thompson, J.A.; Cornwell, T.L.; Radi, R.; Barbeito, L.; Beckman, J.S. Nitric Oxide-Dependent Production of cGMP Supports the Survival of Rat Embryonic Motor Neurons Cultured with Brain-Derived Neurotrophic Factor. J. Neurosci. 1998, 18, 3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolone, A.; Haq, W.; Fachinger, A.; Rentsch, A.; Herberg, F.W.; Schwede, F.; Paquet-Durand, F. Retinal degeneration: Multilevel protection of photoreceptor and ganglion cell viability and function with the novel PKG inhibitor CN238. bioRxiv 2021. [Google Scholar] [CrossRef]
- Roy, A.; Groten, J.; Marigo, V.; Tomar, T.; Hilhorst, R. Identification of Novel Substrates for cGMP Dependent Protein Kinase (PKG) through Kinase Activity Profiling to Understand Its Putative Role in Inherited Retinal Degeneration. Int. J. Mol. Sci. 2021, 22, 1180. [Google Scholar] [CrossRef]
- Kawa, F.; Sterling, P. cGMP modulates spike responses of retinal ganglion cells via a cGMP-gated current. Vis. Neurosci. 2002, 19, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Leinders-Zufall, T.; Kocsis, J.D.; Shepherd, G.M.; Zufall, F.; Barnstable, C.J. Retinal ganglion cells express a cGMP-gated cation conductance activatable by nitric oxide donors. Neuron 1994, 12, 155–165. [Google Scholar] [CrossRef]
- Grimes, M.T.; Powell, M.; Gutierrez, S.M.; Darby-King, A.; Harley, C.W.; McLean, J.H. Epac activation initiates associative odor preference memories in the rat pup. Learn. Mem. 2015, 22, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Nagai-Kusuhara, A.; Nakamura, M.; Mukuno, H.; Kanamori, A.; Negi, A.; Seigel, G.M. cAMP-responsive element binding protein mediates a cGMP/protein kinase G-dependent anti-apoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp. Eye Res. 2007, 84, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirooka, K.; Kourennyi, D.E.; Barnes, S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J. Neurophysiol. 2000, 83, 198–206. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haider, A.A.; Rex, T.S.; Wareham, L.K. cGMP Signaling in the Neurovascular Unit—Implications for Retinal Ganglion Cell Survival in Glaucoma. Biomolecules 2022, 12, 1671. https://doi.org/10.3390/biom12111671
Haider AA, Rex TS, Wareham LK. cGMP Signaling in the Neurovascular Unit—Implications for Retinal Ganglion Cell Survival in Glaucoma. Biomolecules. 2022; 12(11):1671. https://doi.org/10.3390/biom12111671
Chicago/Turabian StyleHaider, Ameer A., Tonia S. Rex, and Lauren K. Wareham. 2022. "cGMP Signaling in the Neurovascular Unit—Implications for Retinal Ganglion Cell Survival in Glaucoma" Biomolecules 12, no. 11: 1671. https://doi.org/10.3390/biom12111671