

Structural Basis of Sequential and Concerted Cooperativity
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Human Hemoglobin
3.2. Aspartate Transcarbamylase
3.3. Phosphofructokinase
3.4. Glycogen Phosphorylase
3.5. The Chemotactic Asp Receptor from S. typhimurium and E. coli
3.6. Asp Semialdehyde Dehydrogenase
3.7. Bacterial D-Lactate Dehydrogenases
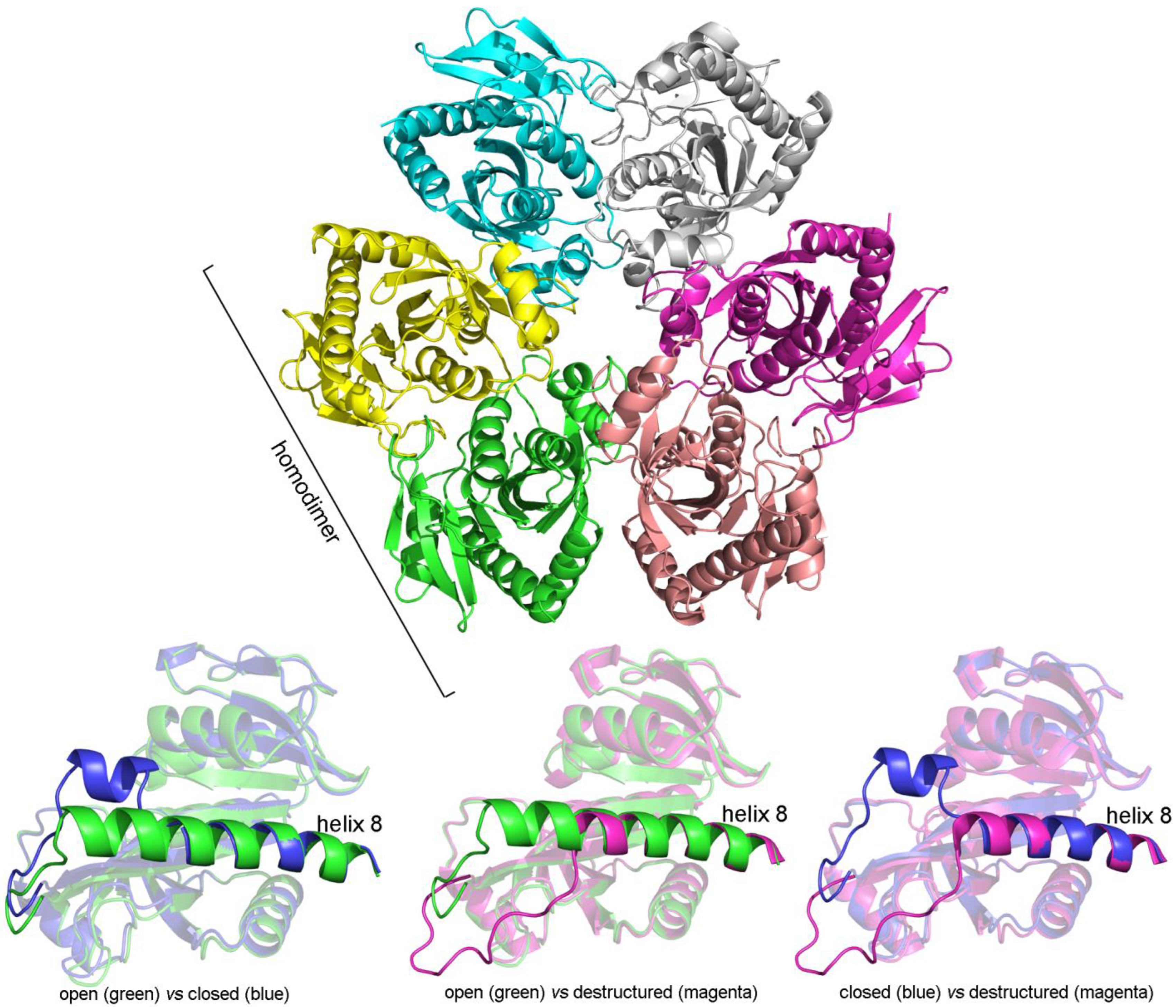
3.8. Bacterial Purine Nucleoside Phosphorylase
3.9. Other Enzymes Obeying a Sequential Reaction Mechanism
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wyman, J. The cybernetics of biological macromolecules. Biophys. Chem. 1981, 14, 135–146. [Google Scholar] [CrossRef]
- Monod, J.; Changeux, J.P.; Jacob, F. Allosteric proteins and cellular control systems. J. Mol. Biol. 1963, 6, 306–329. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.-P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Pauling, L. The Oxygen Equilibrium of Hemoglobin and Its Structural Interpretation. Proc. Natl. Acad. Sci. USA 1935, 21, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshland, D.E., Jr.; Némethy, G.; Filmer, D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 1966, 5, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F. Stereochemistry of cooperative effects in haemoglobin. Nature 1970, 228, 726–739. [Google Scholar] [CrossRef]
- Brunori, M.; Coletta, M.; Di Cera, E. A cooperative model for ligand binding to biological macromolecules as applied to oxygen carriers. Biophys. Chem. 1986, 23, 215–222. [Google Scholar] [CrossRef]
- Di Cera, E.; Robert, C.H.; Gill, S.J. Allosteric interpretation of the oxygen-binding reaction of human hemoglobin tetramers. Biochemistry 1987, 26, 4003–4008. [Google Scholar] [CrossRef]
- Ackers, G.K.; Doyle, M.L.; Myers, D.; Daugherty, M.A. Molecular Code for Cooperativity in Hemoglobin. Science 1992, 255, 54–63. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Storz, J.F. Hemoglobin; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Baldwin, J.; Chothia, C. Haemoglobin: The structural changes related to ligand binding and its allosteric mechanism. J. Mol. Biol. 1979, 129, 175–220. [Google Scholar] [CrossRef]
- Bellelli, A. Hemoglobin and cooperativity: Experiments and theories. Curr. Protein Pept. Sci. 2010, 11, 2–36. [Google Scholar] [CrossRef]
- Bucci, E.; Fronticelli, C. A new method for the preparation of the α and βsubunits of human hemoglobin. J. Biol. Chem. 1965, 240, PC551-2. [Google Scholar] [CrossRef]
- Gibson, Q.H.; Edelstein, S.J. Oxygen binding and subunit interaction of hemoglobin in relation to the two-state model. J. Biol. Chem. 1987, 262, 516–519. [Google Scholar] [CrossRef]
- Mozzarelli, A.; Rivetti, C.; Rossi, G.L.; Henry, E.R.; Eaton, W.A. Crystals of haemoglobin with the T quaternary structure bind oxygen noncooperatively with no Bohr effect. Nature 1991, 351, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Bonaccio, M.; Bettati, S.; Rivetti, C.; Viappiani, C.; Abbruzzetti, S.; Mozzarelli, A. High and low oxygen affinity conformations of T state hemoglobin. Protein Sci. 2008, 10, 2401–2407. [Google Scholar] [CrossRef] [Green Version]
- Bellelli, A.; Brunori, M. [5] Optical measurements of quaternary structural changes in hemoglobin. Methods Enzymol. 1994, 232, 56–71. [Google Scholar] [CrossRef]
- Brzozowski, A.; Derewenda, Z.; Dodson, E.; Dodson, G.; Grabowski, M.; Liddington, R.; Skarżyński, T.; Vallely, D. Bonding of molecular oxygen to T state human haemoglobin. Nature 1984, 307, 74–76. [Google Scholar] [CrossRef]
- Arnone, A.; Rogers, P.; Blough, N.V.; McGourty, J.L.; Hoffman, B.M. X-ray diffraction studies of a partially liganded hemoglobin, [α(FeII-CO)β(MnII)]2. J. Mol. Biol. 1986, 188, 693–706. [Google Scholar] [CrossRef]
- Luisi, B.; Shibayama, N. Structure of haemoglobin in the deoxy quaternary state with ligand bound at the α haems. J. Mol. Biol. 1989, 206, 723–736. [Google Scholar] [CrossRef]
- Luisi, B.; Liddington, B.; Fermi, G.; Shibayama, N. Structure of deoxy-quaternary haemoglobin with liganded beta subunits. J. Mol. Biol. 1990, 214, 7–14. [Google Scholar] [CrossRef]
- Gelin, B.R.; Karplus, M. Mechanism of tertiary structural change in hemoglobin. Proc. Natl. Acad. Sci. USA 1977, 74, 801–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelin, B.R.; Lee, A.W.-M.; Karplus, M. Hemoglobin tertiary structural change on ligand binding its role in the co-operative mechanism. J. Mol. Biol. 1983, 171, 489–559. [Google Scholar] [CrossRef]
- Paoli, M.; Liddington, R.; Tame, J.; Wilkinson, A.; Dodson, G. Crystal Structure of T State Haemoglobin with Oxygen Bound at All Four Haems. J. Mol. Biol. 1996, 256, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Howlett, G.J.; Blackburn, M.N.; Compton, J.G.; Schachman, H.K. Allosteric regulation of aspartate transcarbamoylase. Analysis of the structural and functional behavior in terms of a two-state model. Biochemistry 1977, 16, 5091–5099. [Google Scholar] [CrossRef]
- Fetler, L.; Kantrowitz, E.R.; Vachette, P. Direct observation in solution of a preexisting structural equilibrium for a mutant of the allosteric aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 2007, 104, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipscomb, W.N.; Kantrowitz, E.R. Structure and Mechanisms of Escherichia coli Aspartate Transcarbamoylase. Acc. Chem. Res. 2011, 45, 444–453. [Google Scholar] [CrossRef]
- Angelucci, F.; Bellelli, A.; Ardini, M.; Ippoliti, R.; Saccoccia, F.; Morea, V. One ring (or two) to hold them all—On the structure and function of protein nanotubes. FEBS J. 2015, 282, 2827–2845. [Google Scholar] [CrossRef]
- Beernink, P.T.; Endrizzi, J.A.; Alber, T.; Schachman, H.K. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 5388–5393. [Google Scholar] [CrossRef] [Green Version]
- Endrizzi, J.A.; Beernink, P.T.; Alber, T.; Schachman, H.K. Binding of bisubstrate analog promotes large structural changes in the unregulated catalytic trimer of aspartate transcarbamoylase: Implications for allosteric regulation. Proc. Natl. Acad. Sci. USA 2000, 97, 5077–5082. [Google Scholar] [CrossRef]
- Schirmer, T.; Evans, P.R. Structural basis of the allosteric behaviour of phosphofructokinase. Nature 1990, 343, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Hudson, P.J. Structure and control of phosphofructokinase from Bacillus stearothermophilus. Nature 1979, 279, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, G.; Teschner, W.; Deville-Bonne, D.; Garel, J.R. Urea-induced inactivation, dissociation, and unfolding of the allosteric phosphofructokinase from Escherichia coli. Biochemistry 1989, 28, 6836–6841. [Google Scholar] [CrossRef]
- Deville-Bonne, D.; Le Bras, V.; Teschner, W.; Garel, J.R. Ordered disruption of subunit interfaces during the stepwise reversible dissociation of Escherichia coli phosphofructokinase with KSCN. Biochemistry 1989, 28, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P. The Origins of Allostery: From Personal Memories to Material for the Future. J. Mol. Biol. 2013, 425, 1396–1406. [Google Scholar] [CrossRef] [Green Version]
- Edelstein, S.J.; Edsall, J.T. Linkage between ligand binding and the dimer-tetramer equilibrium in the Monod-Wyman-Changeux model of hemoglobin. Proc. Natl. Acad. Sci. USA 1986, 83, 3796–3800. [Google Scholar] [CrossRef] [Green Version]
- Barford, D.; Johnson, L.N. The allosteric transition of glycogen phosphorylase. Nature 1989, 340, 609–616. [Google Scholar] [CrossRef]
- Johnson, L.N.; Barford, D. Glycogen phosphorylase. The structural basis of the allosteric response and comparison with other allosteric proteins. J. Biol. Chem. 1990, 265, 2409–2412. [Google Scholar] [CrossRef]
- Leonidas, D.D.; Zographos, S.E.; Tsitsanou, K.E.; Skamnaki, V.T.; Stravodimos, G.; Kyriakis, E. Glycogen phosphorylase revisited: Extending the resolution of the R- and T-state structures of the free enzyme and in complex with allosteric activators. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2021, 77, 303–311. [Google Scholar] [CrossRef]
- Milburn, M.V.; Privé, G.G.; Milligan, D.L.; Scott, W.G.; Yeh, J.; Jancarik, J.; Koshland, D.E.; Kim, S.-H. Three-Dimensional Structures of the Ligand-Binding Domain of the Bacterial Aspartate Receptor with and Without a Ligand. Science 1991, 254, 1342–1347. [Google Scholar] [CrossRef]
- Koshland, D.E., Jr. The structural basis of negative cooperativity: Receptors and enzymes. Curr. Opin. Struct. Biol. 1996, 6, 757–761. [Google Scholar] [CrossRef]
- Yeh, J.I.; Biemann, H.-P.; Privé, G.G.; Pandit, J.; Koshland, D.E., Jr.; Kim, S.-H. High-resolution Structures of the Ligand Binding Domain of the Wild-type Bacterial Aspartate Receptor. J. Mol. Biol. 1996, 262, 186–201. [Google Scholar] [CrossRef]
- Milligan, D.L.; Koshland, D.E., Jr. Purification and characterization of the periplasmic domain of the aspartate chemoreceptor. J. Biol. Chem. 1993, 268, 19991–19997. [Google Scholar] [CrossRef]
- Biemann, H.P.; Koshland, D.E., Jr. Aspartate receptors of Escherichia coli and Salmonella typhimurium bind ligand with negative and half-of-the-sites cooperativity. Biochemistry 1994, 33, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.E.; Dhaliwal, B.; Lockyer, M.; Hawkins, A.R.; Stammers, D.K. High-resolution structures reveal details of domain closure and "half-of-sites-reactivity" in Escherichia coli aspartate beta-semialdehyde dehydrogenase. J. Mol. Biol. 2004, 341, 797–806. [Google Scholar] [CrossRef]
- Furukawa, N.; Miyanaga, A.; Nakajima, M.; Taguchi, H. Structural Basis of Sequential Allosteric Transitions in Tetrameric d-Lactate Dehydrogenases from Three Gram-Negative Bacteria. Biochemistry 2018, 57, 5388–5406. [Google Scholar] [CrossRef]
- Mao, C.; Cook, W.J.; Zhou, M.; Koszalka, G.W.; Krenitsky, T.A.; Ealick, S.E. The crystal structure of Escherichia coli purine nucleoside phosphorylase: A comparison with the human enzyme reveals a conserved topology. Structure 1997, 5, 1373–1383. [Google Scholar] [CrossRef] [Green Version]
- Koellner, G.; Luic, M.; Shugar, D.; Saenger, W.; Bzowska, A. Crystal Structure of the Ternary Complex of E. coli Purine Nucleoside Phosphorylase with Formycin B, a Structural Analogue of the Substrate Inosine, and Phosphate (Sulphate) at 2.1 A Resolution. J. Mol. Biol. 1998, 280, 153–166. [Google Scholar] [CrossRef]
- Koellner, G.; Bzowska, A.; Wielgus-Kutrowska, B.; Luić, M.; Steiner, T.; Saenger, W.; Stȩpiński, J. Open and closed conformation of the E. coli purine nucleoside phosphorylase active center and implications for the catalytic mechanism. J. Mol. Biol. 2002, 315, 351–371. [Google Scholar] [CrossRef]
- Stefancic, Z.; Narczyk, M.; Mikleusevic, G.; Karazic, S.; Bzowska, A.; Luic, M. Crystallographic snapshots of ligand binding to hexameric purine nucleotide phosphorylase and kinetic studies give insight into the mechanism of catalysis. Sci. Rep. 2018, 8, 15427. [Google Scholar] [CrossRef]
- Anderson, A.C.; O’Neil, R.H.; DeLano, W.L.; Stroud, R.M. The Structural Mechanism for Half-the-Sites Reactivity in an Enzyme, Thymidylate Synthase, Involves a Relay of Changes between Subunits. Biochemistry 1999, 38, 13829–13836. [Google Scholar] [CrossRef] [PubMed]
- Cowan-Jacob, S.; Kaufmann, M.; Anselmo, A.N.; Stark, W.; Grutter, M.G. Structure of rabbit muscle glyceraldehyde–3-phosphate dehydrogenase. Acta Cryst. D 2003, 59, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.A.; Koshland, D.E., Jr. Positive and negative cooperativity in yeast glyceraldehyde 3-phosphate dehydrogenase. Biochemistry 1970, 9, 3337–3342. [Google Scholar] [CrossRef]
- Vivoli, M.; Pang, J.; Harmer, N.J. A half-site multimeric enzyme achieves its cooperativity without conformational changes. Sci. Rep. 2017, 7, 16529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellelli, A.; Brunori, M. Hemoglobin allostery: Variations on the theme. Biochim. Biophys. Acta 2011, 1807, 1262–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornish-Bowden, A. The physiological significance of negative cooperativity revised. J. Theor. Biol. 2013, 319, 144–147. [Google Scholar] [CrossRef]
- Sawicki, C.A.; Gibson, Q.H. Quaternary conformational changes in human hemoglobin studied by laser photolysis of carboxyhemoglobin. J. Biol. Chem. 1976, 251, 1533–1542. [Google Scholar] [CrossRef]
- Hofrichter, J.; Sommer, J.H.; Henry, E.R.; Eaton, W.A. Nanosecond absorption spectroscopy of hemoglobin: Elementary processes in kinetic cooperativity. Proc. Natl. Acad. Sci. USA 1983, 80, 2235–2239. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Olsen, K.W.; Nam, K.; Karplus, M. Unsuspected pathway of the allosteric transition in hemoglobin. Proc. Natl. Acad. Sci. USA 2011, 108, 5608–5613. [Google Scholar] [CrossRef] [Green Version]
- Altintel, B.; Acar, B.; Erman, B.; Haliloglu, T. Subsets of Slow Dynamic Modes Reveal Global Information Sources as Allosteric Sites. J. Mol. Biol. 2022, 434, 167644. [Google Scholar] [CrossRef]
- Daily, M.D.; Gray, J.J. Allosteric Communication Occurs via Networks of Tertiary and Quaternary Motions in Proteins. PLoS Comput. Biol. 2009, 5, e1000293. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
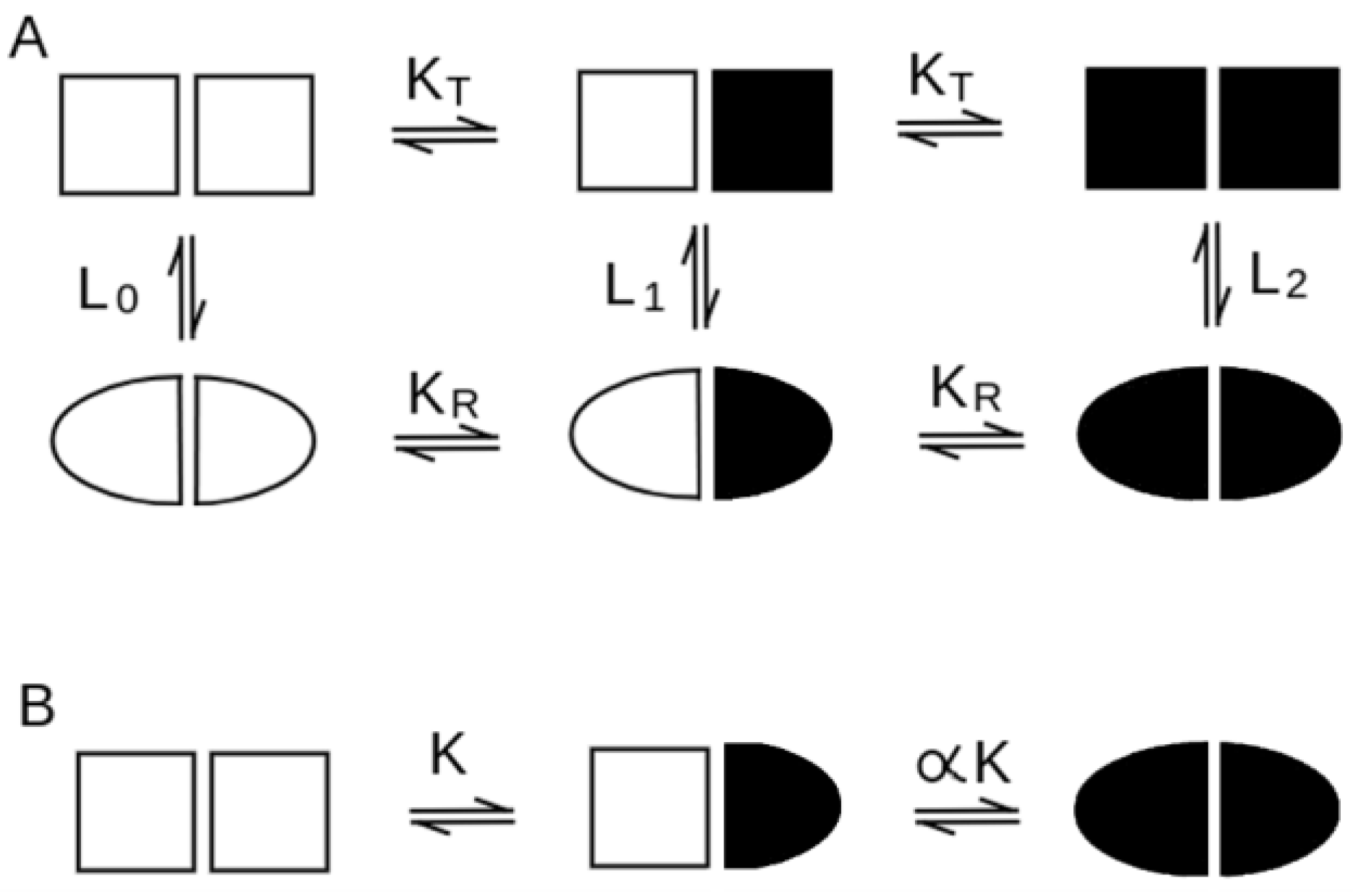
| Concerted Models | Sequential Models | |
|---|---|---|
| tertiary structure symmetry (all subunits share or do not share the same tertiary structure in the various ligation states) | necessary for unliganded, partially liganded and fully liganded states (asymmetry within T may be tolerated) | necessary asymmetry of ligation intermediates; asymmetry in the liganded state is tolerated |
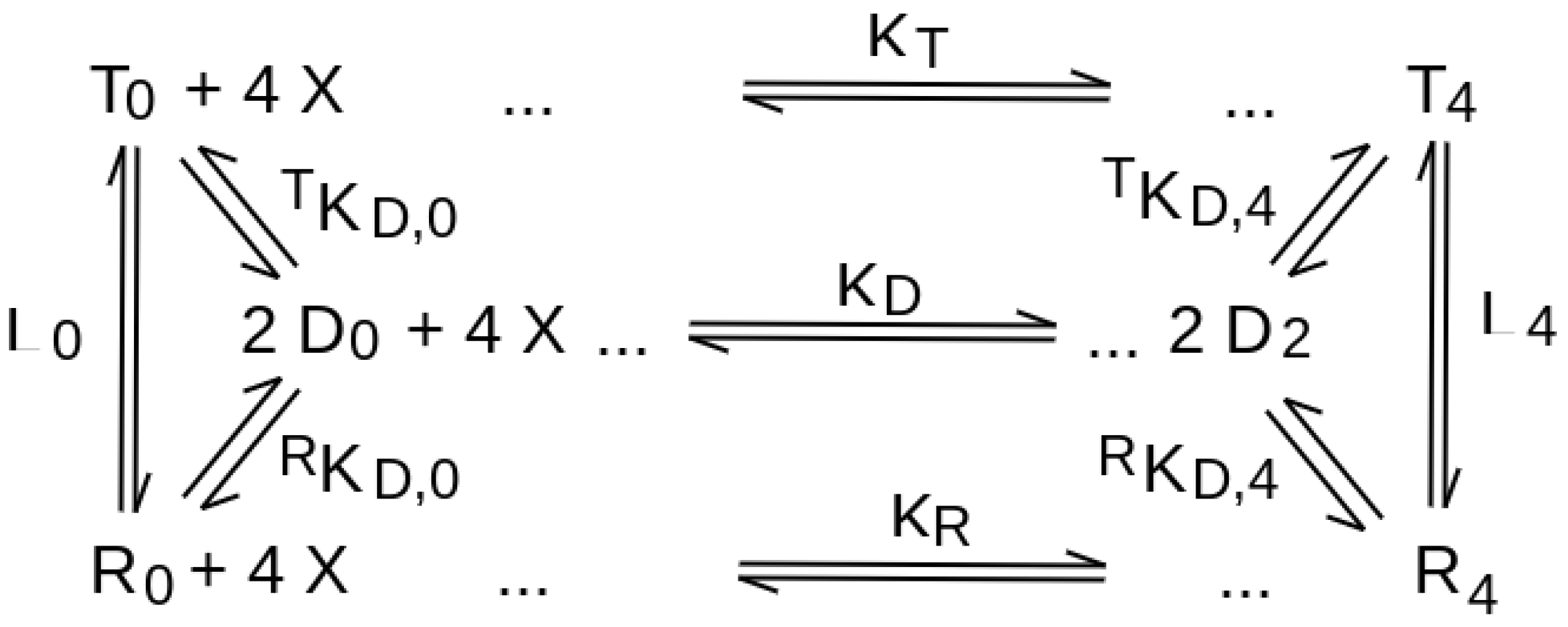
| quaternary conformation equilibrium in the absence of the ligand | necessary, for all ligation intermediates, described by the allosteric constant L (see Equation (2)). | absent (see Equation (1)) |
| cooperativity in the absence of quaternary structure change | impossible | possible |
| positive homotropic cooperativity | possible | possible |
| negative homotropic cooperativity | usually impossible (some exceptions may be considered) | possible |
| heterotropic regulation | almost always present | possible but not common |
| quaternary enhancement/constraint | usually quaternary constraint (but enhancement possible) | both possible |
| structural differences between the fully liganded and fully unliganded states | present (the main difference occurs between the R and T state, irrespective of ligation) | present |
| Protein | Reaction Mechanism | Available Information |
|---|---|---|
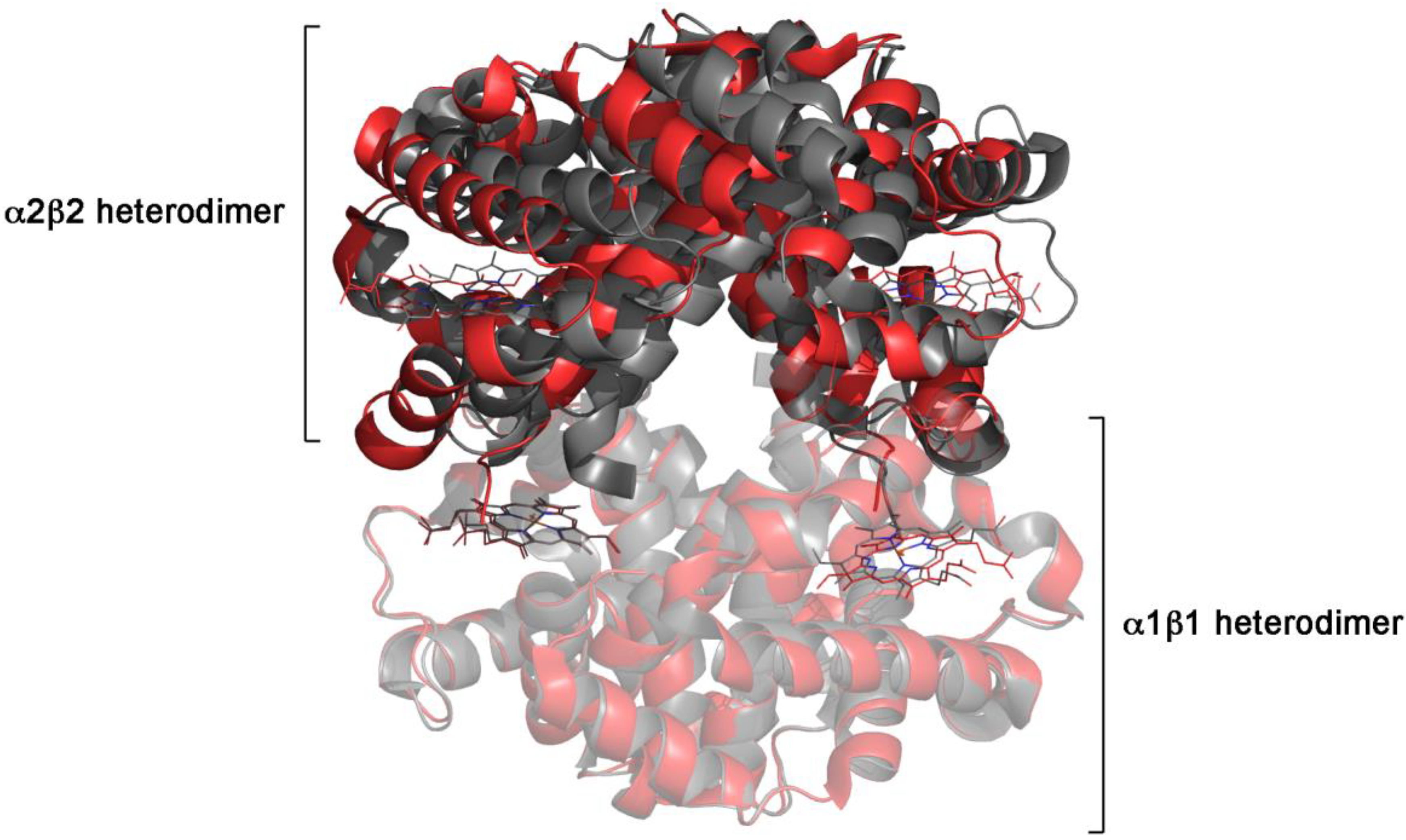
| vertebrate hemoglobins | concerted | -positive homotropic cooperativity -ligand-independent allosteric equilibrium -symmetry of ligation intermediates -absence of cooperativity under conditions that prevent the quaternary structure change -quaternary constraint |
| Asp transcarbamylase | concerted | -positive homotropic cooperativity -ligand-independent allosteric equilibrium -quaternary constraint |
| Phosphofructokinase | likely concerted | -positive homotropic cooperativity -quaternary enhancement -T state is only induced by heterotropic effector (s) -Ligand binds only to the R state |
| Glycogen phosphorylase B | likely concerted | -positive homotropic cooperativity |
| Bacterial Asp receptor | sequential | -negative homotropic cooperativity -asymmetry of ligation intermediates |
| dTMP synthase | sequential | -negative homotropic cooperativity -asymmetry of ligation intermediates |
| Asp semialdehyde dehydrogenase | sequential | -negative homotropic cooperativity -asymmetry of ligation intermediates |
| Bacterial D-Lactate dehydrogenases | sequential | -negative or positive homotropic cooperativity -strong asymmetry in either the liganded or unliganded state |
| Bacterial Purine nucleoside phosphorylase | sequential | -negative homotropic cooperativity -strong asymmetry in the liganded state |
| Ligation State RMSD | |
|---|---|
| Human Hemoglobin (Concerted, MWC-like) | |
| R Hb(CO)4, PDB 2DN3, vs. T Hb, PDB 2DN2 | tertiary, average: 0.67 Å overall (tertiary + quaternary): 2.43 Å |
| α1 subunit in T Hb, PDB 2DN2, vs. α2 subunit in THb, PDB 2DN2 | tertiary: 0.23 Å |
| β1 subunit in T Hb, PDB 2DN2, vs. β2 subunit in T Hb, PDB 2DN2 | tertiary: 0.3 Å |
| R Hb(CO)4, PDB 2DN3, vs. T αFeCO2 βCo2, PDB 1COH | tertiary, average: 0.67 Å |
| R Hb(O2)4, PDB 2DN1, vs. THb(O2)4, PDB 1GZX | overall (tertiary + quaternary): 2.57 Å |
| T Hb, PDB 2DN2, vs. T Hb(O2)4, PDB 1GZX | tertiary, α subunits: 0.28 Å tertiary, β subunits: 0.41 Å overall (tertiary + quaternary): 0.51 Å |
| E. coli Asp transcarbamylase (concerted, MWC-like) | |
| Unliganded T ATC, PDB 6AT1 vs. PALA-liganded R ATC, PDB 8ATC | tertiary (catalytic subunits): 1.85 Å tertiary (regulatory subunits): 1.70 Å overall (tertiary + quaternary): 6.20 Å |
| Unliganded T ATC, PDB 6AT1 vs. unliganded T ATC, PDB 6AT1 | tertiary (catalytic subunits): 0.56 Å tertiary (regulatory subunits): 1.34 Å |
| PALA-liganded R ATC, PDB 8ATC vs. PALA-liganded R ATC, PDB 8ATC | tertiary (catalytic subunits): 0.32 Å tertiary (regulatory subunits): 1.43 Å |
| Geobacillus stearothermophylus phosphofructokinase (concerted, MWC-like ?) | |
| PGC-inhibited T PFK, PDB 6PFK, vs. unliganded R PFK, PDB 3PFK | tertiary, average: 0.94 Å overall (tertiary + quaternary): 1.55 Å |
| PGC-inhibited T PFK, PDB 6PFK, vs. PGC-inhibited T PFK, PDB 6PFK | tertiary, average: 0.28 Å |
| Rabbit (O. cuniculus) muscle glycogen phosphorylase b (concerted, MWC-like ?) | |
| R GPb, PDB 7GPB vs. T GPb, PDB 8GPB | tertiary, average: 1.33 Å overall (tertiary + quaternary): 2.61 Å |
| R GPb, PDB 7GPB vs. R GPb, PDB 7GPB | tertiary, average: 0.55 Å |
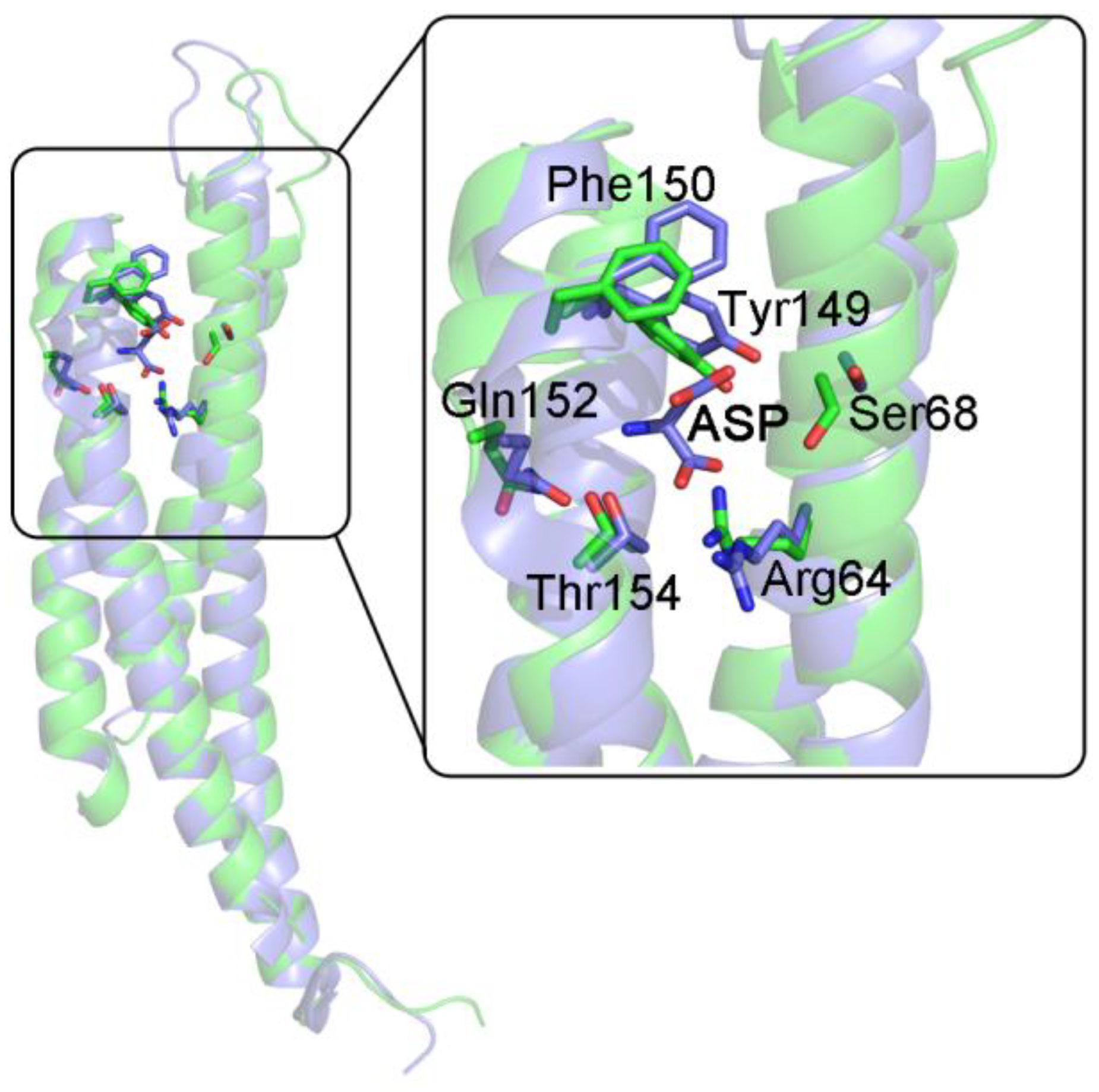
| S. typhimurium Asp receptor (sequential, KNF-like) | |
| unliganded, PDB 1LIH vs. liganded, PDB 2LIG | tertiary 1LIH vs. 2LIG A: 2.58 Å tertiary 1LIH vs. 2LIG B: 2.42 Å overall (tertiary + quaternary): 2.85 Å |
| liganded, PDB 2LIG A (high affinity), vs. liganded, PDB 2LIG B (low affinity) | tertiary: 2.44 Å |
| E. coli Asp semialdehyde dehydrogenase (sequential, KNF-like) | |
| unliganded, PDB 1GL3, vs. half-liganded, PDB 1T4B | tertiary, liganded subunit of 1T4B: 1.3 Å tertiary, unliganded subunit of 1T4B: 0.94 Å overall (tertiary + quaternary): 1.49 Å |
| unliganded, PDB 1GL3, vs. unliganded, PDB 1GL3 | tertiary: 0.27 Å |
| half-liganded, PDB 1T4B, vs. half-liganded, PDB 1T4B | tertiary: 0.62 Å |
| P. aeruginosa D-lactate dehydrogenase (sequential, KNF-like) | |
| unliganded PDB 6ABJ, vs. liganded PDB 5Z20 | tertiary (average): 2.35 Å overall (tertiary + quaternary): 3.30 Å |
| unliganded PDB 6ABJ, vs. unliganded PDB 6ABJ | tertiary (average): 0.31 Å |
| liganded PDB 5Z20, vs. liganded PDB 5Z20 | tertiary (average): 0.50 Å |
| F. nucleatum D-lactate dehydrogenase (sequential, KNF-like) | |
| unliganded PDB 6ABI, vs. liganded PDB 5Z21 | tertiary (average): 4.7 Å overall (tertiary + quaternary): 6.44 Å |
| unliganded PDB 6ABI, vs. unliganded PDB 6ABI | tertiary (average): 1.9 Å |
| liganded PDB 5Z21, vs. liganded PDB 5Z21 | tertiary (average): 0.25 Å |
| E. coli purine nucleoside phosphorylase (sequential, KNF-like) | |
| unliganded, PDB 1ECP, vs. fully liganded, PDB 1A69 | tertiary (average) 0.88 Å tertiary (open vs. open) 0.31 Å tertiary (open vs. closed) 1.0–1.32 Å overall (tertiary + quaternary) 0.84 Å |
| unliganded, PDB 1ECP, vs. partially liganded, PDB 4TTA | tertiary (average) 1.3 Å tertiary (open vs. open) 0.47 Å tertiary (open vs. closed) 1.37 Å tertiary (open vs. destructured) 2.75 Å overall (tertiary + quaternary) 1.50 Å |
| fully liganded, PDB 1A69, vs. partially liganded, PDB 4TTA | tertiary (average) 1.44 Å tertiary (open vs. open) 0.45 Å tertiary (open vs. closed) 1.5 Å tertiary (closed vs. destructured) 3.5 Å tertiary (open vs. destructured) 2.75 Å overall (tertiary + quaternary) 1.71 Å |
| unliganded, PDB 1ECP, vs. unliganded, PDB 1ECP | tertiary (average) 0.32 Å |
| fully liganded, PDB 1A69, vs. fully liganded, PDB 1A69 | tertiary (average) 1.35 Å tertiary (A-closed vs. B-open) 1.32 Å tertiary (A-closed vs. C-open) 1.76 Å tertiary (B-open vs. C-open) 0.98 Å |
| partially liganded, PDB 4TTA, vs. partially liganded, PDB 4TTA | tertiary (A-closed vs. F-closed) 0.17 Å tertiary (B-open vs. D-open) 0.45 Å tertiary (A/F-closed vs. B/D-open) 1.39 Å tertiary (A/F-closed vs. E-destruct.) 3.52 Å tertiary (B/D-open vs. E-destruct.) 2.82 Å |
| Protein | Reaction Scheme | R1 | R2 | R3 |
|---|---|---|---|---|
| human HbA (2DN2/2DN3) | concerted | 0.28 | 0.11 | 0 |
| E. coli ATC (6AT1/8ATC) | concerted | 0.28 | 0.09 * | 0.05 * |
| G. stearothermophyilus PFK (6PFK/3PFK) | likely concerted | 0.60 | 0.18 | 0 |
| rabbit glycogen phosphorylase b (7GPB/8GPB) | likely concerted | 0.51 | 0 | 0.21 |
| S. typhimurium Asp receptor (1VLT/1VLS) | sequential | 0.88 | 0 | 0.85 |
| E. coli Asp semialdehyde dehydrogenase (1T4B/1GL3) | sequential | 0.75 | 0.18 | 0.42 |
| Pseudomonas aeruginosa D-lactate dehydrogenase (6ABJ/5Z20) | sequential | 0.71 | 0.09 | 0.15 |
| Fusobacterium nucleatum D-lactate dehydrogenase (6ABI/5Z21) | sequential | 0.73 | 0.29 | 0.04 |
| E. coli purine nucleotide phosphorylase (1ECP/1A69) | sequential | 1.05 | 0.38 | 1.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morea, V.; Angelucci, F.; Tame, J.R.H.; Di Cera, E.; Bellelli, A. Structural Basis of Sequential and Concerted Cooperativity. Biomolecules 2022, 12, 1651. https://doi.org/10.3390/biom12111651
Morea V, Angelucci F, Tame JRH, Di Cera E, Bellelli A. Structural Basis of Sequential and Concerted Cooperativity. Biomolecules. 2022; 12(11):1651. https://doi.org/10.3390/biom12111651
Chicago/Turabian StyleMorea, Veronica, Francesco Angelucci, Jeremy R. H. Tame, Enrico Di Cera, and Andrea Bellelli. 2022. "Structural Basis of Sequential and Concerted Cooperativity" Biomolecules 12, no. 11: 1651. https://doi.org/10.3390/biom12111651