
Elevated Extracellular HSP72 and Blunted Heat Shock Response in Severe COVID-19 Patients

 ,
,  ,
,  , , , , , and
, , , , , and 
Abstract
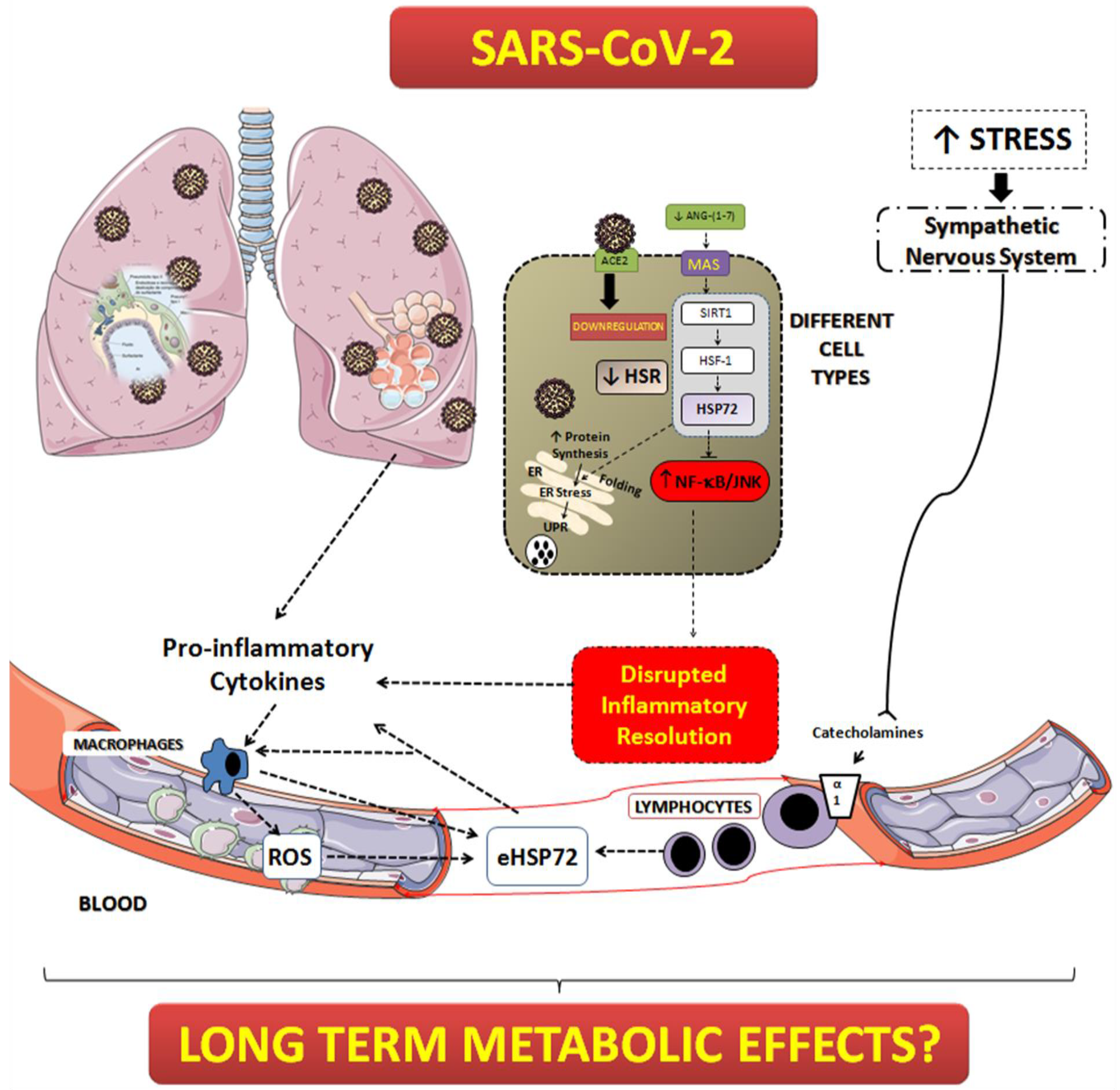
:1. Introduction
2. Methods and Materials
2.1. Study Design and Participants
2.2. Procedures and Biochemistry Measurements
2.3. Plasma Cytokine Quantification
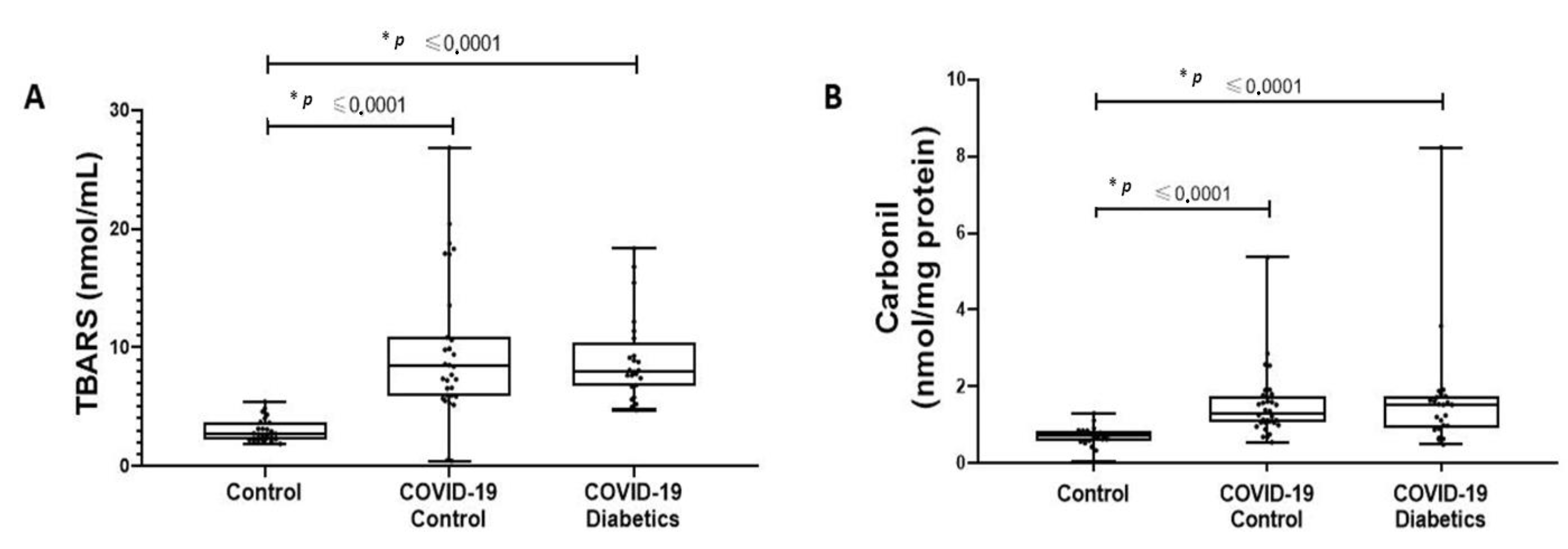
2.4. Oxidative Damage
2.5. Heat Shock Response Test
2.6. Extracellular HSP72 Quantification
2.7. Protein Quantification and Western Blotting for Intracellular HSP70 Immunocontent
2.8. Statistical Analysis and Sample Size
3. Results
3.1. Patient Characteristics and Cytokine Profile in Critically Ill Patients with Severe COVID-19 Pneumonia
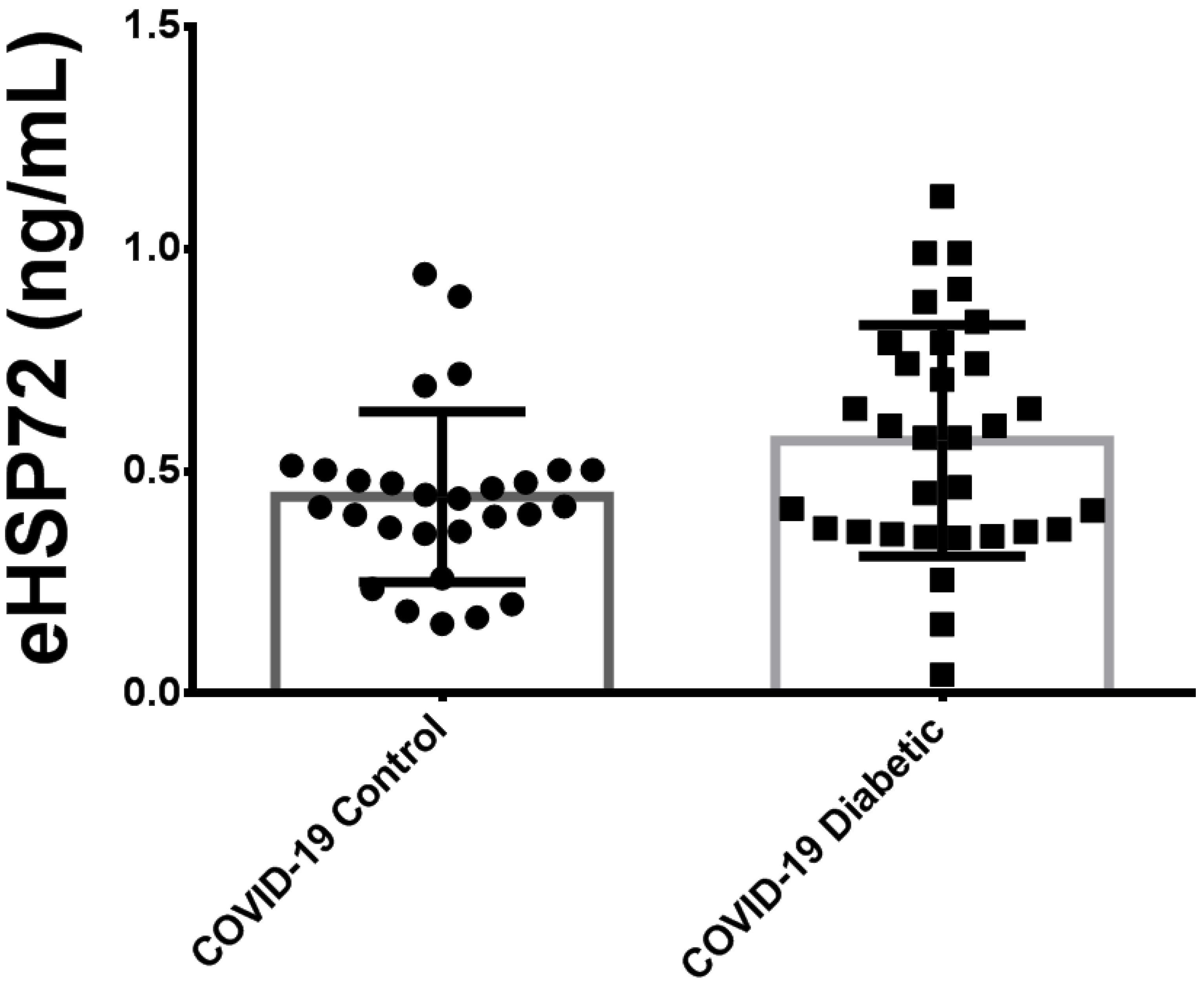
3.2. Plasma Extracellular HSP72 Concentration in Critically Ill Patients with COVID-19
3.3. Comparison of Plasma HSP72 among Different Groups: Noninfected Control Subjects, Noninfected Subjects with Diabetes and Critically Ill Patients with COVID-19 Pneumonia
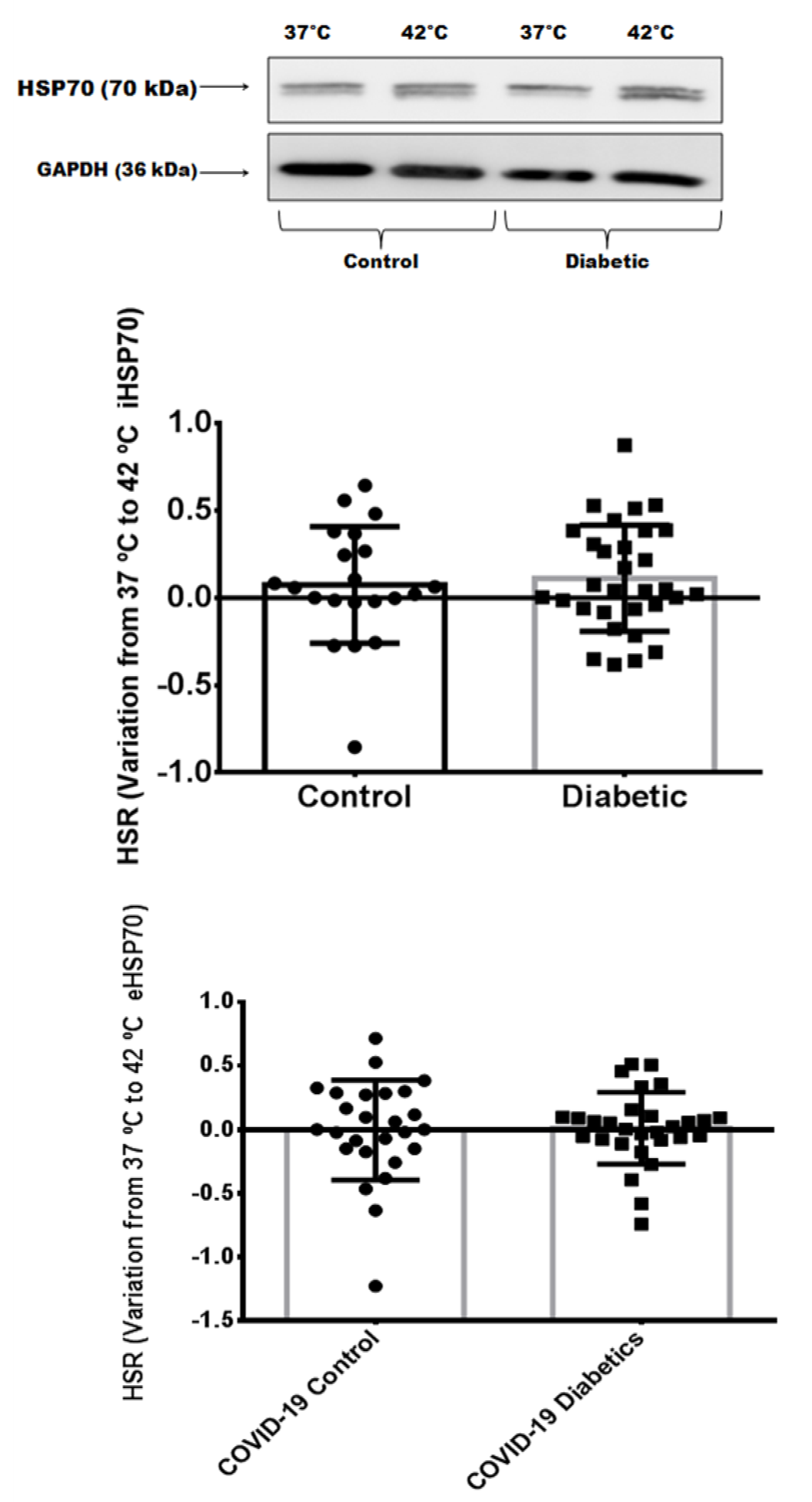
3.4. Heat Shock Response in Critically Ill Patients with Severe COVID-19 Pneumonia
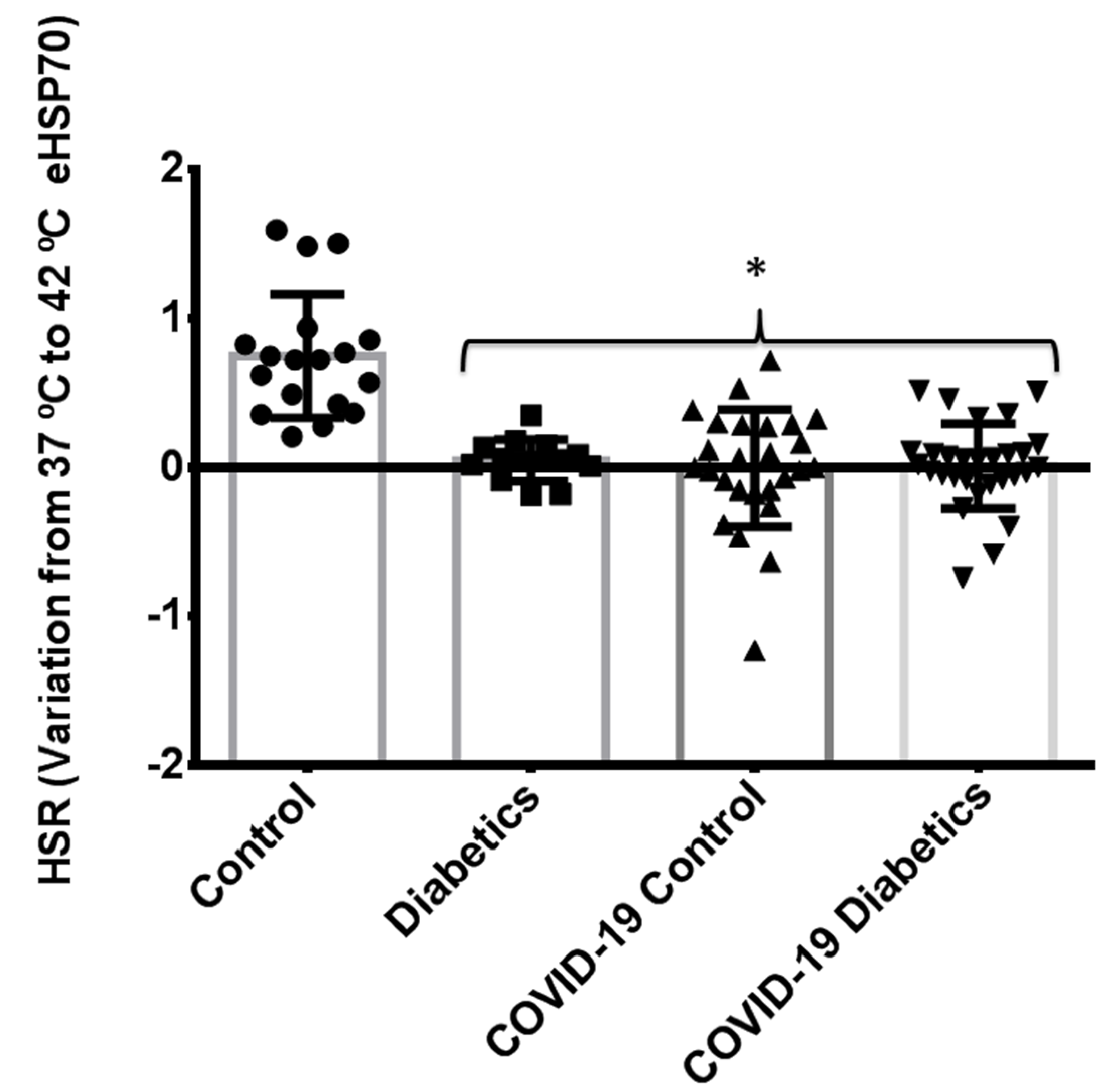
3.5. Comparison of Heat Shock Response between Different Groups: Noninfected Control Subjects, Noninfected Subjects with Controlled Diabetes, and Critically Ill Patients with Severe COVID-19 Pneumonia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef]
- John, A.E.; Joseph, C.; Jenkins, G.; Tatler, A.L. COVID-19 and pulmonary fibrosis: A potential role for lung epithelial cells and fibroblasts. Immunol. Rev. 2021, 302, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Li, X.; Shi, J.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Zhou, M.; Xu, S.; Wu, B.; et al. Gendered effects on inflammation reaction and outcome of COVID-19 patients in Wuhan. J. Med. Virol. 2020, 92, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The triumvirate of NF-kappaB, inflammation and cytokine storm in COVID-19. Int. Immunopharmacol. 2021, 101, 108255. [Google Scholar] [CrossRef]
- Kandasamy, M. NF-kappaB signalling as a pharmacological target in COVID-19: Potential roles for IKKbeta inhibitors. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 561–567. [Google Scholar] [CrossRef]
- Singh, I.S.; Hasday, J.D. Fever, hyperthermia and the heat shock response. Int. J. Hyperth. 2013, 29, 423–435. [Google Scholar] [CrossRef]
- de Thonel, A.; Le Mouel, A.; Mezger, V. Transcriptional regulation of small HSP-HSF1 and beyond. Int. J. Biochem. Cell Biol. 2012, 44, 1593–1612. [Google Scholar] [CrossRef]
- Tang, S.; Buriro, R.; Liu, Z.; Zhang, M.; Ali, I.; Adam, A.; Hartung, J.; Bao, E. Localization and expression of Hsp27 and alphaB-crystallin in rat primary myocardial cells during heat stress in vitro. PLoS ONE 2013, 8, e69066. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Heck, T.G.; Bittencourt, A.; Scomazzon, S.P.; Newsholme, P.; Curi, R.; de Bittencourt, P.I.H., Jr. The chaperone balance hypothesis: The importance of the extracellular to intracellular HSP70 ratio to inflammation-driven type 2 diabetes, the effect of exercise, and the implications for clinical management. Mediat. Inflamm. 2015, 2015, 249205. [Google Scholar] [CrossRef] [PubMed]
- Madden, L.A.; Sandstrom, M.E.; Lovell, R.J.; McNaughton, L. Inducible heat shock protein 70 and its role in preconditioning and exercise. Amino Acids 2008, 34, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Bock, P.M.; Takahashi, H.K.; De Bittencourt, P.I.H., Jr.; Newsholme, P. The regulatory roles of NADPH oxidase, intra- and extra-cellular HSP70 in pancreatic islet function, dysfunction and diabetes. Clin. Sci. 2015, 128, 789–803. [Google Scholar] [CrossRef]
- Newsholme, P.; de Bittencourt, P.I., Jr. The fat cell senescence hypothesis: A mechanism responsible for abrogating the resolution of inflammation in chronic disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 295–305. [Google Scholar] [CrossRef]
- Krause, M.; Gerchman, F.; Friedman, R. Coronavirus infection (SARS-CoV-2) in obesity and diabetes comorbidities: Is heat shock response determinant for the disease complications? Diabetol. Metab. Syndr. 2020, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Bruxel, M.A.; Tavares, A.M.V.; Zavarize Neto, L.D.; de Souza Borges, V.; Schroeder, H.T.; Bock, P.M.; Rodrigues, M.I.L.; Bello-Klein, A.; de Bittencourt, P.I.H., Jr. Chronic whole-body heat treatment relieves atherosclerotic lesions, cardiovascular and metabolic abnormalities, and enhances survival time restoring the anti-inflammatory and anti-senescent heat shock response in mice. Biochimie 2019, 156, 33–46. [Google Scholar] [CrossRef]
- de Lemos Muller, C.H.; Rech, A.; Botton, C.E.; Schroeder, H.T.; Bock, P.M.; Farinha, J.B.; Lopez, P.; Scholer, C.M.; Grigolo, G.B.; Coelho, J.; et al. Heat-induced extracellular HSP72 release is blunted in elderly diabetic people compared with healthy middle-aged and older adults, but it is partially restored by resistance training. Exp. Gerontol. 2018, 111, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Di Naso, F.C.; Porto, R.R.; Fillmann, H.S.; Maggioni, L.; Padoin, A.V.; Ramos, R.J.; Mottin, C.C.; Bittencourt, A.; Marroni, N.A.; de Bittencourt, P.I., Jr. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity 2015, 23, 120–129. [Google Scholar] [CrossRef]
- Muller, C.H.D.L.; de Matos, J.R.; Grigolo, G.B.; Schroeder, H.T.; Rodrigues-Krause, J.; Krause, M. Exercise Training for the Elderly: Inflammaging and the Central Role for HSP70. J. Sci. Sport Exerc. 2019, 1, 10–25. [Google Scholar]
- Ortega, E.; Hinchado, M.D.; Martin-Cordero, L.; Asea, A. The effect of stress-inducible extracellular Hsp72 on human neutrophil chemotaxis: A role during acute intense exercise. Stress 2009, 12, 240–249. [Google Scholar] [CrossRef]
- Ortega, E.; Giraldo, E.; Hinchado, M.D.; Martinez, M.; Ibanez, S.; Cidoncha, A.; Collazos, M.E.; Garcia, J.J. Role of Hsp72 and norepinephrine in the moderate exercise-induced stimulation of neutrophils’ microbicide capacity. Eur. J. Appl. Physiol. 2006, 98, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Kalz, A.; Lim, C.L.; Pyne, D.; Saunders, P.; Mackinnon, L.; Peake, J.; Suzuki, K. Exercise-recruited NK cells display exercise-associated eHSP-70. Exerc. Immunol. Rev. 2007, 13, 100–111. [Google Scholar] [PubMed]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Fleshner, M. Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. J. Leukoc. Biol. 2006, 79, 425–434. [Google Scholar] [CrossRef]
- De Maio, A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: A form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones 2010, 16, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araujo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Krause, J.; Krause, M.; O’Hagan, C.; De Vito, G.; Boreham, C.; Murphy, C.; Newsholme, P.; Colleran, G. Divergence of intracellular and extracellular HSP72 in type 2 diabetes: Does fat matter? Cell Stress Chaperones 2012, 17, 293–302. [Google Scholar] [CrossRef]
- Trussardi Fayh, A.P.; de Carvalho Gomes, C.; Schroeder, H.T.; Henrique de Lemos Muller, C.; Maria de Araujo Moura Lemos, T.; Krause, M. Induction chemotherapy reduces extracellular heat shock protein 72 levels, inflammation, lipoperoxidation and changes insulin sensitivity in children and adolescents newly diagnosed with acute lymphoblastic leukemia. Oncotarget 2018, 9, 28784–28795. [Google Scholar] [CrossRef]
- de Souza, D.C.; Matos, V.A.F.; Dos Santos, V.O.A.; Medeiros, I.F.; Marinho, C.S.R.; Nascimento, P.R.P.; Dorneles, G.P.; Peres, A.; Muller, C.H.; Krause, M.; et al. Effects of High-Intensity Interval and Moderate-Intensity Continuous Exercise on Inflammatory, Leptin, IgA, and Lipid Peroxidation Responses in Obese Males. Front. Physiol. 2018, 9, 567. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- da Silva Rossato, J.; Krause, M.; Fernandes, A.J.; Fernandes, J.R.; Seibt, I.L.; Rech, A.; de Bittencourt, P.I.H., Jr. Role of alpha- and beta-adrenoreceptors in rat monocyte/macrophage function at rest and acute exercise. J. Physiol. Biochem. 2014, 70, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Galbinski, S.; Kowalewski, L.S.; Grigolo, G.B.; da Silva, L.R.; Jimenez, M.F.; Krause, M.; Frantz, N.; Bos-Mikich, A. Comparison between two cryopreservation techniques of human ovarian cortex: Morphological aspects and the heat shock response (HSR). Cell Stress Chaperones 2022, 27, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Keane, K.; Rodrigues-Krause, J.; Crognale, D.; Egan, B.; De Vito, G.; Murphy, C.; Newsholme, P. Elevated levels of extracellular heat-shock protein 72 (eHSP72) are positively correlated with insulin resistance in vivo and cause pancreatic beta-cell dysfunction and death in vitro. Clin. Sci. 2013, 126, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis. Eur. Respir. J. 2020, 55, 2000547. [Google Scholar] [CrossRef]
- Oglesbee, M.J.; Herdman, A.V.; Passmore, G.G.; Hoffman, W.H. Diabetic ketoacidosis increases extracellular levels of the major inducible 70-kDa heat shock protein. Clin. Biochem. 2005, 38, 900–904. [Google Scholar] [CrossRef]
- Nakhjavani, M.; Morteza, A.; Khajeali, L.; Esteghamati, A.; Khalilzadeh, O.; Asgarani, F.; Outeiro, T.F. Increased serum HSP70 levels are associated with the duration of diabetes. Cell Stress Chaperones 2010, 15, 959–964. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef]
- Njemini, R.; Demanet, C.; Mets, T. Inflammatory status as an important determinant of heat shock protein 70 serum concentrations during aging. Biogerontology 2004, 5, 31–38. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Lee, Y.H.; Giraud, J.; Davis, R.J.; White, M.F. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J. Biol. Chem. 2003, 278, 2896–2902. [Google Scholar] [CrossRef]
- Barrett, C.E.; Koyama, A.K.; Alvarez, P.; Chow, W.; Lundeen, E.A.; Perrine, C.G.; Pavkov, M.E.; Rolka, D.B.; Wiltz, J.L.; Bull-Otterson, L.; et al. Risk for Newly Diagnosed Diabetes >30 Days After SARS-CoV-2 Infection Among Persons Aged < 18 Year—United States, 1 March 2020–28 June 2021. MMWR. Morb. Mortal. Wkly. Rep. 2022, 71, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat. Metab. 2021, 3, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Narayan, K.M.V.; Staimez, L.R. Rising diabetes diagnosis in long COVID. Lancet Diabetes Endocrinol. 2022, 10, 298–299. [Google Scholar] [CrossRef]
- Karpe, P.A.; Tikoo, K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1-7) signaling. Diabetes 2013, 63, 1124–1139. [Google Scholar] [CrossRef]
- Krause, M.; Ludwig, M.S.; Heck, T.G.; Takahashi, H.K. Heat shock proteins and heat therapy for type 2 diabetes: Pros and cons. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 374–380. [Google Scholar] [CrossRef]
- Liao, Y.; Yuan, Q.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 2006, 349, 264–275. [Google Scholar] [CrossRef]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef]
- Langouche, L.; Vander Perre, S.; Wouters, P. Insulin signaling in critical illness: Intensive versus conventional insulin therapy. In Proceedings of the 25th International Symposium on Intensive Care and Emergency Medicine, Brussels, Belgium, 21–25 March 2005; p. 384. [Google Scholar]
- Literati-Nagy, B.; Kulcsar, E.; Literati-Nagy, Z.; Buday, B.; Peterfai, E.; Horvath, T.; Tory, K.; Kolonics, A.; Fleming, A.; Mandl, J.; et al. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: A proof of concept randomized double-blind clinical trial. Horm. Metab. Res. 2009, 41, 374–380. [Google Scholar] [CrossRef]
- Literati-Nagy, B.; Peterfai, E.; Kulcsar, E.; Literati-Nagy, Z.; Buday, B.; Tory, K.; Mandl, J.; Sumegi, B.; Fleming, A.; Roth, J.; et al. Beneficial effect of the insulin sensitizer (HSP inducer) BGP-15 on olanzapine-induced metabolic disorders. Brain Res. Bull. 2010, 83, 340–344. [Google Scholar] [CrossRef]
- Smuder, A.J.; Morton, A.B.; Hall, S.E.; Wiggs, M.P.; Ahn, B.; Wawrzyniak, N.R.; Sollanek, K.J.; Min, K.; Kwon, O.S.; Nelson, W.B.; et al. Effects of exercise preconditioning and HSP72 on diaphragm muscle function during mechanical ventilation. J. Cachexia Sarcopenia Muscle 2019, 10, 767–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristic | Critically Ill Infected COVID-19: Control (n = 27) | Critically Ill Infected COVID-19: Diabetics (n = 32) | p Value |
|---|---|---|---|
| Sex (M/F) | (13/14) | (18/14) | |
| Age (years) | 58.2 ± 13.5 | 63.5 ± 11.2 | p = 0.522 |
| Body Mass (kg) | 90.8 ± 18.42 | 89.3 ± 20 | p = 0.762 |
| BMI (kg/m2) | 33 ± 6.7 | 33.6 ± 7.9 | p = 0.631 |
| Glycemia (mg/dL) | 171 ± 48 | 235 ± 79 # | p = 0.006 |
| HbA1C (%) | 5.9 ± 0.5 | 8.9 ± 2.2 # | p < 0.0001 |
| TNF-α (pg/mL) | 26.6 [9.36–32.39] | 17.7 [12.77–27.41] | p = 0.532 |
| IL-10 (pg/mL) | 2.86 [1.42–4.57] | 2.0 [1.42–6.87] | p = 0.682 |
| TNF-α/IL-10 | 6.85 [3.35–13.28] | 5.06 [2.38–10.43] | p = 0.405 |
| Patient Characteristic | Noninfected Control (n = 19) | Noninfected Diabetics (n = 22) | Critically Ill Infected COVID-19: Control (n = 27) | Critically Ill Infected COVID-19: Diabetics (n = 32) |
|---|---|---|---|---|
| Age (years) | 54.5 ± 8.3 | 68.9 ± 7.8 | 58.2 ± 13.5 | 63.5 ± 11.2 |
| Body Mass (kg) | 68.1 ± 9.3 | 79.12 ± 10.8 | 90.8 ± 18.42 * | 89.3 ± 20 * |
| Height (m) | 1.63 ± 0.08 | 1.66 ± 0.8 | 1.66 ± 0.1 | 1.67 ± 0.1 |
| BMI (kg/m2) | 25.6 ± 2.5 | 28.7 ± 3.1 | 33 ± 6.7 * | 33.6 ± 7.9 * |
| Glycaemia (mg/dL) | 102.5 ± 12.4 | 133.1 ± 21.4 | 171.1 ± 48.38 *‡ | 235.7 ± 79.4 *‡# |
| HbA1C (%) | - | 6.75 ± 0.6 | 5.94 ± 0.51 | 8.9 ± 2.2 ‡# |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borges Russo, M.K.; Kowalewski, L.S.; da Natividade, G.R.; de Lemos Muller, C.H.; Schroeder, H.T.; Bock, P.M.; Ayres, L.R.; Cardoso, B.U.; Zanotto, C.; Schein, J.T.; et al. Elevated Extracellular HSP72 and Blunted Heat Shock Response in Severe COVID-19 Patients. Biomolecules 2022, 12, 1374. https://doi.org/10.3390/biom12101374
Borges Russo MK, Kowalewski LS, da Natividade GR, de Lemos Muller CH, Schroeder HT, Bock PM, Ayres LR, Cardoso BU, Zanotto C, Schein JT, et al. Elevated Extracellular HSP72 and Blunted Heat Shock Response in Severe COVID-19 Patients. Biomolecules. 2022; 12(10):1374. https://doi.org/10.3390/biom12101374
Chicago/Turabian StyleBorges Russo, Mariana Kras, Lucas Stahlhöfer Kowalewski, Gabriella Richter da Natividade, Carlos Henrique de Lemos Muller, Helena Trevisan Schroeder, Patrícia Martins Bock, Layane Ramos Ayres, Bernardo Urbano Cardoso, Caroline Zanotto, Julia Tsao Schein, and et al. 2022. "Elevated Extracellular HSP72 and Blunted Heat Shock Response in Severe COVID-19 Patients" Biomolecules 12, no. 10: 1374. https://doi.org/10.3390/biom12101374