
Interfacial Peptides as Affinity Modulating Agents of Protein-Protein Interactions


Abstract
:1. Introduction
2. Main Structural and Biochemical Features of PPIs’ Interfaces
3. Current Approaches to the Design of Interfacial Peptides
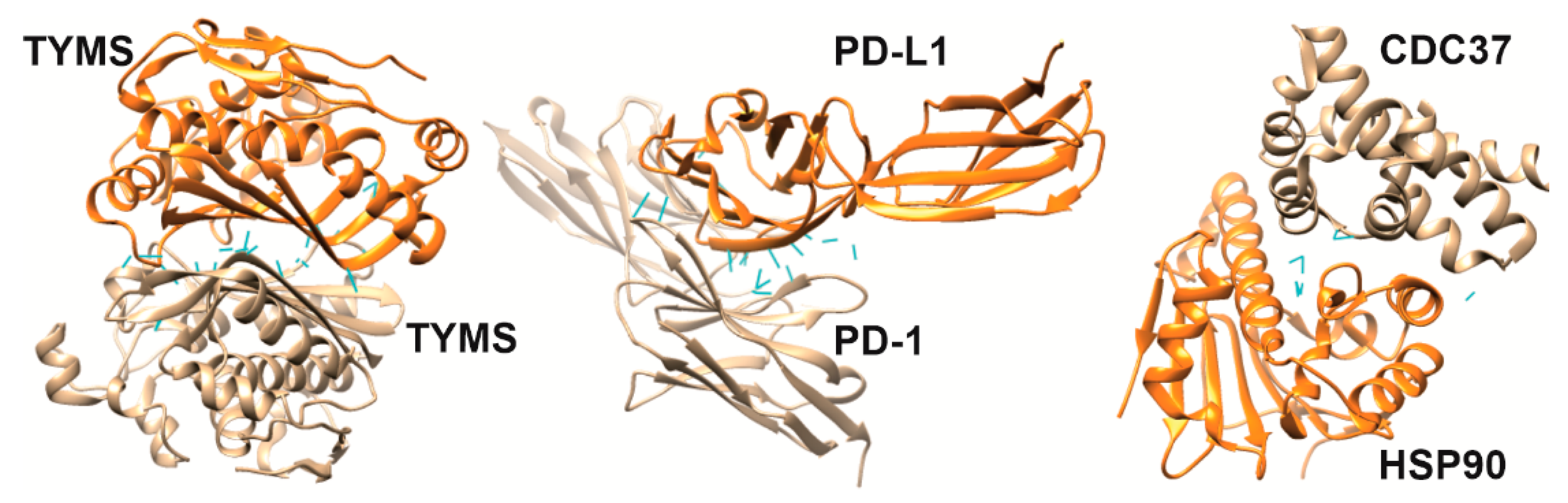
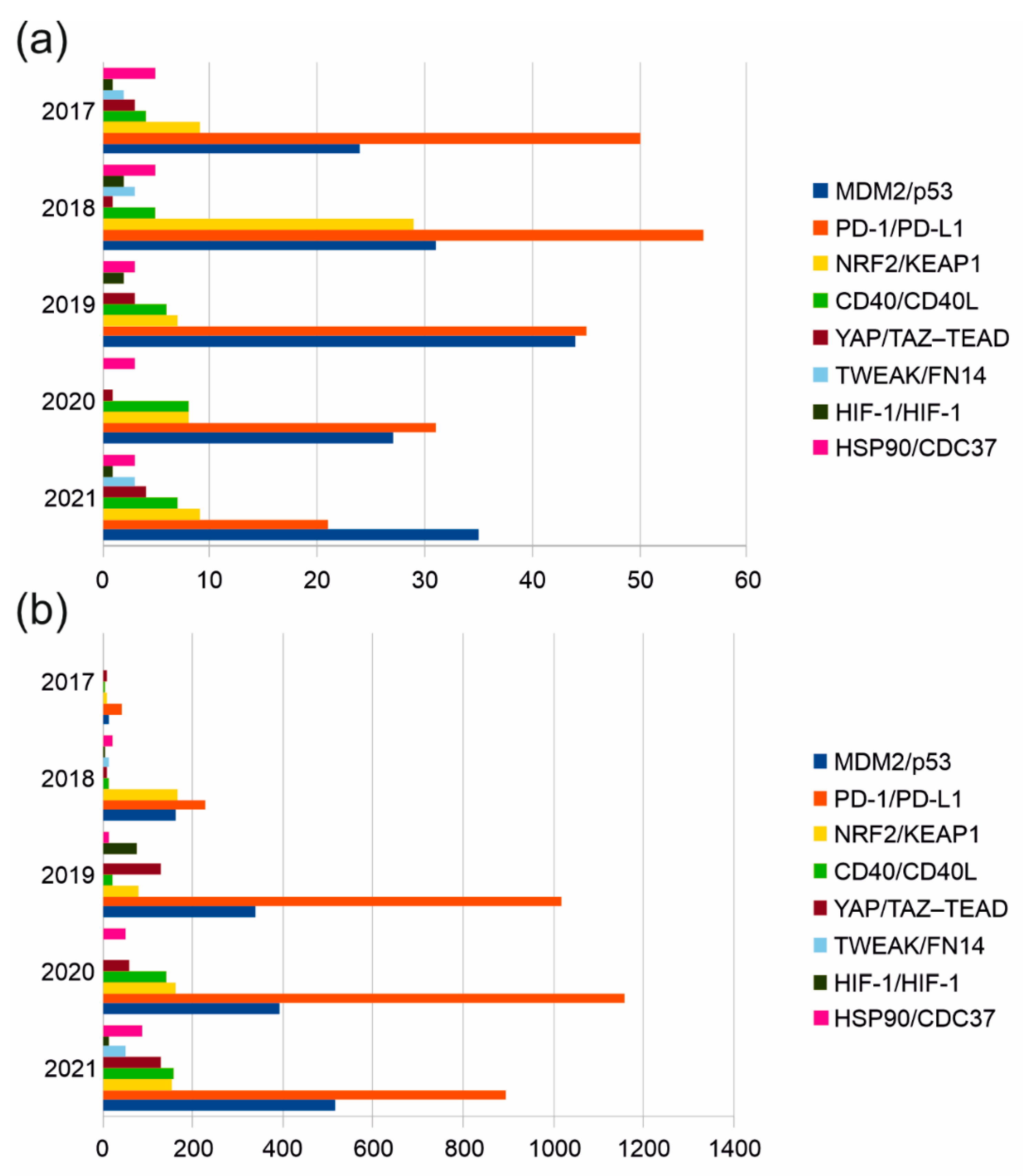
4. Targeting Clinically Significant PPIs with Interfacial Peptides
4.1. Targeting Homo-Oligomeric Protein-Protein Interactions with Interfacial Peptides
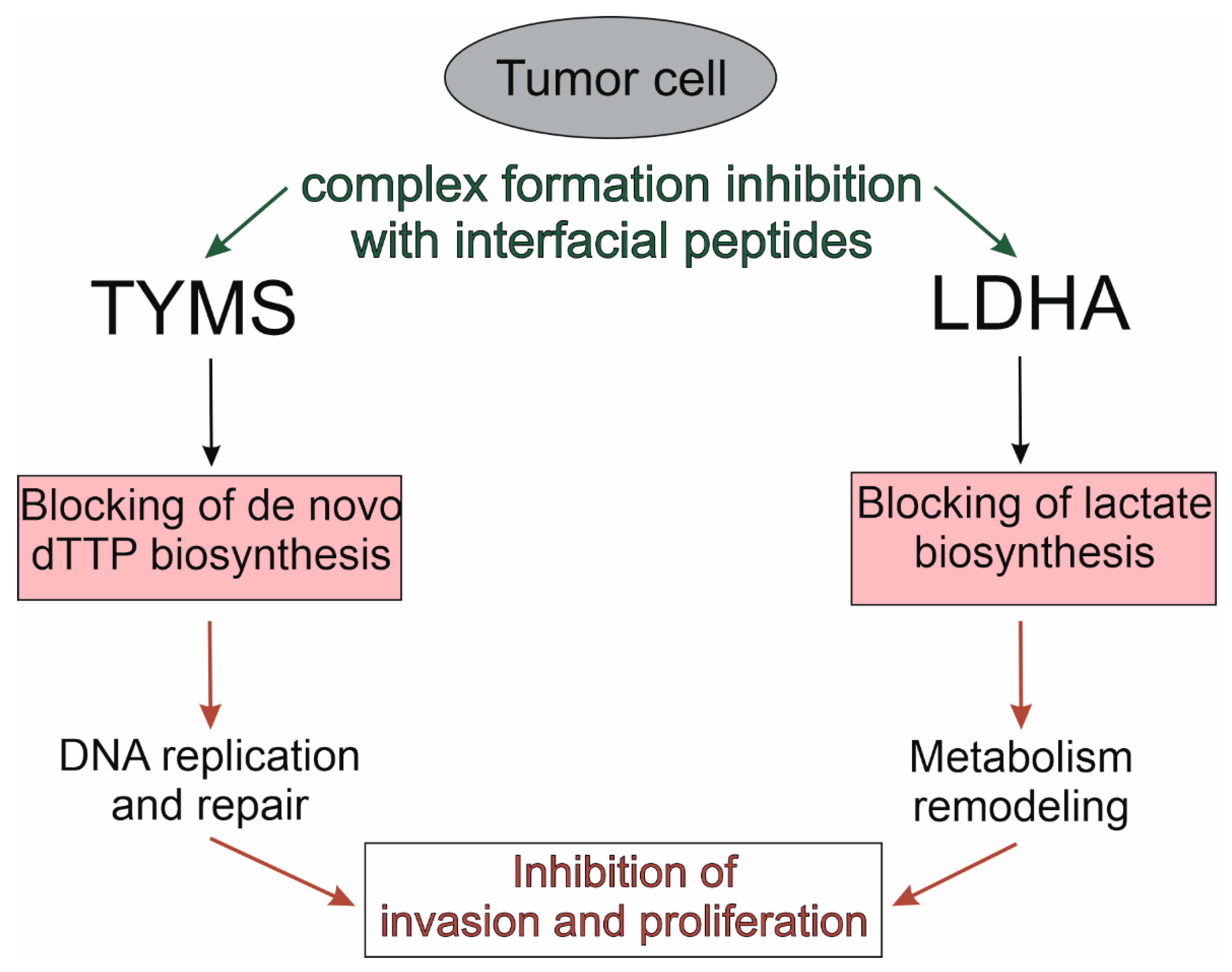
4.1.1. LDH5
4.1.2. TYMS
4.1.3. NFAT5
4.2. Targeting Hetero-Oligomeric Protein-Protein Interactions
4.2.1. MDM2/p53
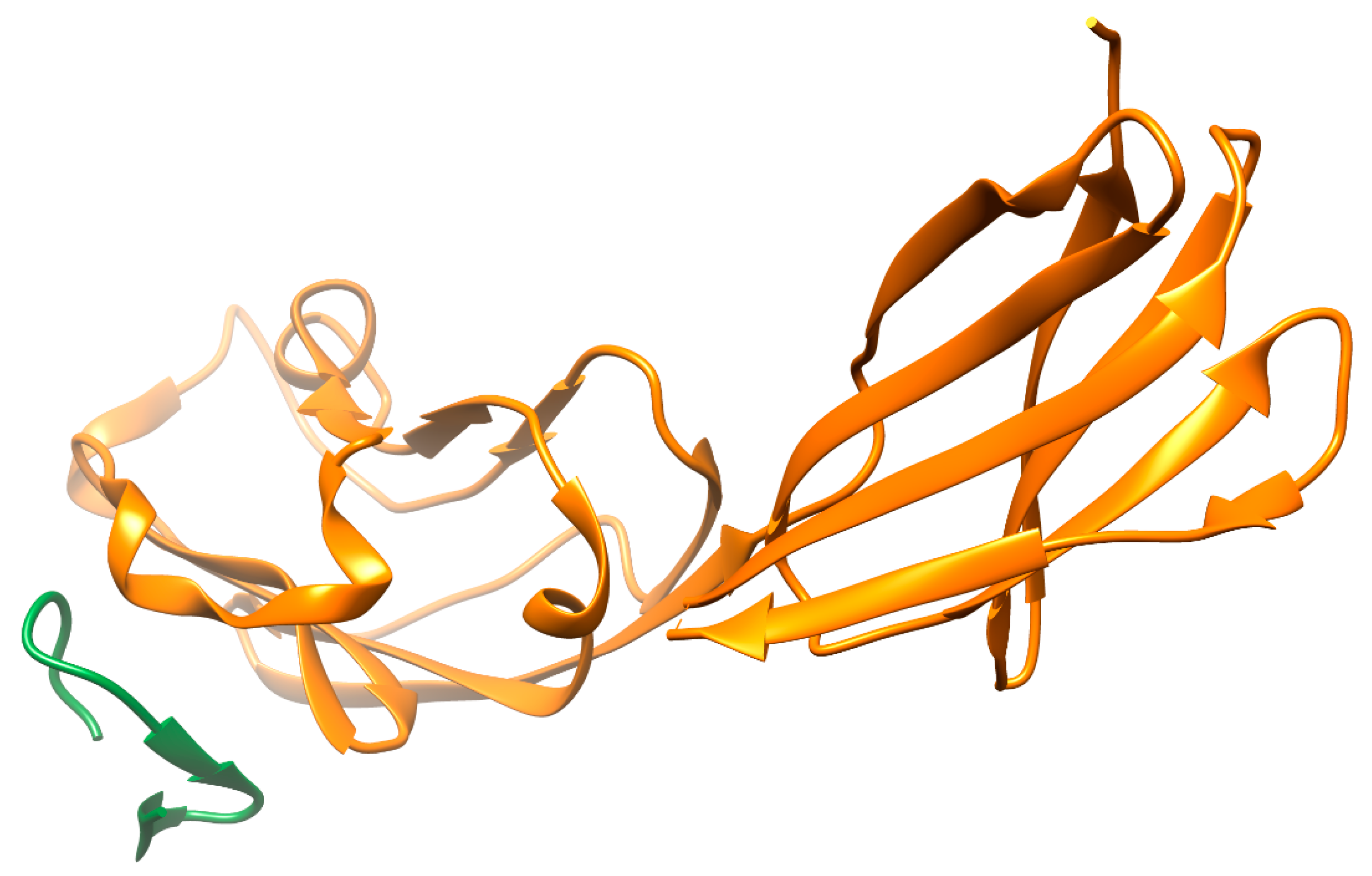
4.2.2. PD-1/PD-L1
4.2.3. HIF-1
4.2.4. NRF2/KEAP1
4.2.5. RbAp48/MTA1
4.2.6. Interaction of SRPK1 with Its Protein Substrates
4.2.7. HSP90/CDC37
4.2.8. BIRC5/CRM1
4.2.9. BIRC5/XIAP
4.2.10. YAP/TAZ–TEAD
4.2.11. TWEAK/FN14
4.2.12. Bcl-2/Bax
4.2.13. YY1/AKT
4.2.14. CD40/CD40L
4.3. Inhibitors of Protein Polymerization
4.4. Targeting PPIs Involving Viral and Human Proteins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peng, X.; Wang, J.; Peng, W.; Wu, F.-X.; Pan, Y. Protein-Protein Interactions: Detection, Reliability Assessment and Applications. Brief. Bioinform. 2017, 18, 798–819. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of Protein-Protein Interactions: What Are the Preferred Ways for Proteins to Interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef]
- Lite, T.-L.V.; Grant, R.A.; Nocedal, I.; Littlehale, M.L.; Guo, M.S.; Laub, M.T. Uncovering the Basis of Protein-Protein Interaction Specificity with a Combinatorially Complete Library. Elife 2020, 9, e60924. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A Reference Map of the Human Binary Protein Interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Ershov, P.V.; Yablokov, E.; Zgoda, V.; Mezentsev, Y.; Gnedenko, O.; Kaluzhskiy, L.; Svirid, A.; Gilep, A.; Usanov, S.A.; Ivanov, A. A New Insight into Subinteractomes of Functional Antagonists: Thromboxane (CYP5A1) and Prostacyclin (CYP8A1) Synthases. Cell Biol. Int. 2021, 45, 1175–1182. [Google Scholar] [CrossRef]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Gulfidan, G.; Turanli, B.; Beklen, H.; Sinha, R.; Arga, K.Y. Pan-Cancer Mapping of Differential Protein-Protein Interactions. Sci. Rep. 2020, 10, 3272. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Zeng, T.; Chen, L.; Huang, T.; Cai, Y.-D. Determining Protein-Protein Functional Associations by Functional Rules Based on Gene Ontology and KEGG Pathway. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140621. [Google Scholar] [CrossRef]
- Perez-Lopez, Á.R.; Szalay, K.Z.; Türei, D.; Módos, D.; Lenti, K.; Korcsmáros, T.; Csermely, P. Targets of Drugs Are Generally, and Targets of Drugs Having Side Effects Are Specifically Good Spreaders of Human Interactome Perturbations. Sci. Rep. 2015, 5, 10182. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, Q.; Wang, T. Drug Target Protein-Protein Interaction Networks: A Systematic Perspective. BioMed Res. Int. 2017, 2017, 1289259. [Google Scholar] [CrossRef] [Green Version]
- Thabault, L.; Liberelle, M.; Frédérick, R. Targeting Protein Self-Association in Drug Design. Drug Discov. Today 2021, 26, 1148–1163. [Google Scholar] [CrossRef]
- Hu, K. Summary and Conclusion. In Development of In-Tether Carbon Chiral Center-Induced Helical Peptide: Methodology and Applications; Hu, K., Ed.; Springer Theses; Springer: Singapore, 2021; pp. 101–102. ISBN 978-981-336-613-8. [Google Scholar]
- Loregian, A.; Palù, G. Disruption of Protein-Protein Interactions: Towards New Targets for Chemotherapy. J. Cell. Physiol. 2005, 204, 750–762. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein-Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Chang, C.-K.; Lin, S.-M.; Satange, R.; Lin, S.-C.; Sun, S.-C.; Wu, H.-Y.; Kehn-Hall, K.; Hou, M.-H. Targeting Protein-Protein Interaction Interfaces in COVID-19 Drug Discovery. Comput. Struct. Biotechnol. J. 2021, 19, 2246–2255. [Google Scholar] [CrossRef]
- Brautigan, D.L.; Farrington, C.; Narla, G. Targeting Protein Phosphatase PP2A for Cancer Therapy: Development of Allosteric Pharmaceutical Agents. Clin. Sci. 2021, 135, 1545–1556. [Google Scholar] [CrossRef]
- Song, T.; Guo, Y.; Xue, Z.; Guo, Z.; Wang, Z.; Lin, D.; Zhang, H.; Pan, H.; Zhang, X.; Yin, F.; et al. Small-Molecule Inhibitor Targeting the Hsp70-Bim Protein-Protein Interaction in CML Cells Overcomes BCR-ABL-Independent TKI Resistance. Leukemia 2021, 35, 2862–2874. [Google Scholar] [CrossRef]
- Lin, C.-M.; Jiang, Z.; Gao, Z.; Arancillo, M.; Burgess, K. Small Molecules Targeting the NEDD8·NAE Protein-Protein Interaction. Chem. Sci. 2020, 12, 1535–1543. [Google Scholar] [CrossRef]
- Siddiqui, F.A.; Parkkola, H.; Vukic, V.; Oetken-Lindholm, C.; Jaiswal, A.; Kiriazis, A.; Pavic, K.; Aittokallio, T.; Salminen, T.A.; Abankwa, D. Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling. Cancers 2021, 13, 927. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein-Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef]
- Engin, H.B.; Kreisberg, J.F.; Carter, H. Structure-Based Analysis Reveals Cancer Missense Mutations Target Protein Interaction Interfaces. PLoS ONE 2016, 11, e0152929. [Google Scholar] [CrossRef]
- Tobacman, L.S.; Cammarato, A. Cardiomyopathic Troponin Mutations Predominantly Occur at Its Interface with Actin and Tropomyosin. J. Gen. Physiol. 2021, 153, e202012815. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot Spots in Protein-Protein Interfaces: Towards Drug Discovery. Prog. Biophys. Mol. Biol. 2014, 116, 165–173. [Google Scholar] [CrossRef]
- Fry, D.C. Targeting Protein-Protein Interactions for Drug Discovery. Methods Mol. Biol. 2015, 1278, 93–106. [Google Scholar] [CrossRef]
- Shin, W.-H.; Kumazawa, K.; Imai, K.; Hirokawa, T.; Kihara, D. Current Challenges and Opportunities in Designing Protein-Protein Interaction Targeted Drugs. Adv. Appl. Bioinform. Chem. 2020, 13, 11–25. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting Protein-Protein Interactions as an Anticancer Strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Sable, R.; Jois, S. Surfing the Protein-Protein Interaction Surface Using Docking Methods: Application to the Design of PPI Inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef] [Green Version]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. On the Specificity of Protein-Protein Interactions in the Context of Disorder. Biochem. J. 2021, 478, 2035–2050. [Google Scholar] [CrossRef] [PubMed]
- Erijman, A.; Rosenthal, E.; Shifman, J.M. How Structure Defines Affinity in Protein-Protein Interactions. PLoS ONE 2014, 9, e110085. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Genovese, F.; Ferrari, S.; Guaitoli, G.; Caselli, M.; Costi, M.P.; Ponterini, G. Dimer-Monomer Equilibrium of Human Thymidylate Synthase Monitored by Fluorescence Resonance Energy Transfer. Protein Sci. 2010, 19, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnez, R.; Thiroux, B.; Taront, S.; Segaoula, Z.; Quesnel, B.; Thuru, X. PD-1/PD-L1 Binding Studies Using Microscale Thermophoresis. Sci. Rep. 2017, 7, 17623. [Google Scholar] [CrossRef] [Green Version]
- Siligardi, G.; Panaretou, B.; Meyer, P.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Regulation of Hsp90 ATPase Activity by the Co-Chaperone Cdc37p/P50cdc37. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef] [Green Version]
- Perkins, J.R.; Diboun, I.; Dessailly, B.H.; Lees, J.G.; Orengo, C. Transient Protein-Protein Interactions: Structural, Functional, and Network Properties. Structure 2010, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, B.W.; Goncz, E.J.; Krist, D.T.; Statsyuk, A.V.; Nesvizhskii, A.I.; Weiss, E.L. Mapping Low-Affinity/High-Specificity Peptide-Protein Interactions Using Ligand-Footprinting Mass Spectrometry. Proc. Natl. Acad. Sci. USA 2019, 116, 21001–21011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small Heat Shock Proteins, Big Impact on Protein Aggregation in Neurodegenerative Disease. Front. Pharmacol. 2019, 10, 1047. [Google Scholar] [CrossRef] [PubMed]
- Chène, P. Drugs Targeting Protein-Protein Interactions. ChemMedChem 2006, 1, 400–411. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Chandana Epa, V.; Colman, P.M. Electrostatic Complementarity at Protein/Protein Interfaces. J. Mol. Biol. 1997, 268, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Sheinerman, F.B.; Honig, B. On the Role of Electrostatic Interactions in the Design of Protein-Protein Interfaces. J. Mol. Biol. 2002, 318, 161–177. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Colman, P.M. Shape Complementarity at Protein/Protein Interfaces. J. Mol. Biol. 1993, 234, 946–950. [Google Scholar] [CrossRef]
- Lanzarotti, E.; Defelipe, L.A.; Marti, M.A.; Turjanski, A.G. Aromatic Clusters in Protein-Protein and Protein-Drug Complexes. J. Cheminform. 2020, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.L.; Bonvin, A.M.J.J. On the Binding Affinity of Macromolecular Interactions: Daring to Ask Why Proteins Interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef]
- Xiao, Y.; Konermann, L. Protein Structural Dynamics at the Gas/Water Interface Examined by Hydrogen Exchange Mass Spectrometry. Protein Sci. 2015, 24, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Sorolla, A.; Wang, E.; Golden, E.; Duffy, C.; Henriques, S.T.; Redfern, A.D.; Blancafort, P. Precision Medicine by Designer Interference Peptides: Applications in Oncology and Molecular Therapeutics. Oncogene 2020, 39, 1167–1184. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Dawber, R.S.; Zhang, P.; Walko, M.; Wilson, A.J.; Wang, X. Peptide-Based Inhibitors of Protein-Protein Interactions: Biophysical, Structural and Cellular Consequences of Introducing a Constraint. Chem. Sci. 2021, 12, 5977–5993. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a Platform for Targeted Therapeutics for Cancer: Peptide-Drug Conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Bluntzer, M.T.J.; O’Connell, J.; Baker, T.S.; Michel, J.; Hulme, A.N. Designing Stapled Peptides to Inhibit Protein-Protein Interactions: An Analysis of Successes in a Rapidly Changing Field. Pept. Sci. 2021, 113, e24191. [Google Scholar] [CrossRef]
- González-Muñiz, R.; Bonache, M.Á.; Pérez de Vega, M.J. Modulating Protein-Protein Interactions by Cyclic and Macrocyclic Peptides. Prominent Strategies and Examples. Molecules 2021, 26, 445. [Google Scholar] [CrossRef]
- Mihara, E.; Watanabe, S.; Bashiruddin, N.K.; Nakamura, N.; Matoba, K.; Sano, Y.; Maini, R.; Yin, Y.; Sakai, K.; Arimori, T.; et al. Lasso-Grafting of Macrocyclic Peptide Pharmacophores Yields Multi-Functional Proteins. Nat. Commun. 2021, 12, 1543. [Google Scholar] [CrossRef]
- Philippe, G.J.B.; Craik, D.J.; Henriques, S.T. Converting Peptides into Drugs Targeting Intracellular Protein-Protein Interactions. Drug Discov. Today 2021, 26, 1521–1531. [Google Scholar] [CrossRef]
- Morimoto, J.; Hosono, Y.; Sando, S. Isolation of a Peptide Containing D-Amino Acid Residues That Inhibits the α-Helix-Mediated P53-MDM2 Interaction from a One-Bead One-Compound Library. Bioorg. Med. Chem. Lett. 2018, 28, 231–234. [Google Scholar] [CrossRef]
- Orafaie, A.; Sadeghian, H.; Bahrami, A.R.; Rafatpanah, H.; Matin, M.M. Design, Synthesis and Evaluation of PD-L1 Peptide Antagonists as New Anticancer Agents for Immunotherapy. Bioorg. Med. Chem. 2021, 30, 115951. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Talapatra, S.K.; Gatliff, J.; Kozielski, F.; Wells, G. Modified Peptide Inhibitors of the Keap1-Nrf2 Protein-Protein Interaction Incorporating Unnatural Amino Acids. ChemBioChem 2018, 19, 1810–1816. [Google Scholar] [CrossRef]
- Li, X.; Tolbert, W.D.; Hu, H.-G.; Gohain, N.; Zou, Y.; Niu, F.; He, W.-X.; Yuan, W.; Su, J.-C.; Pazgier, M.; et al. Dithiocarbamate-Inspired Side Chain Stapling Chemistry for Peptide Drug Design. Chem. Sci. 2019, 10, 1522–1530. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Kaur, A.; Fowler, E.; Wiedmann, M.M.; Young, R.; Galloway, W.R.J.D.; Olsen, L.; Sore, H.F.; Chattopadhyay, A.; Kwan, T.T.-L.; et al. Toolbox of Diverse Linkers for Navigating the Cellular Efficacy Landscape of Stapled Peptides. ACS Chem. Biol. 2019, 14, 526–533. [Google Scholar] [CrossRef]
- Strizhak, A.V.; Babii, O.; Afonin, S.; Bakanovich, I.; Pantelejevs, T.; Xu, W.; Fowler, E.; Eapen, R.; Sharma, K.; Platonov, M.O.; et al. Diarylethene Moiety as an Enthalpy-Entropy Switch: Photoisomerizable Stapled Peptides for Modulating P53/MDM2 Interaction. Org. Biomol. Chem. 2020, 18, 5359–5369. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Wen, J.; Pan, X.; Koley, A.; Ren, J.-G.; Sahni, A.; Basu, R.; Salim, H.; Appiah Kubi, G.; Qian, Z.; et al. Enhancing the Cell Permeability of Stapled Peptides with a Cyclic Cell-Penetrating Peptide. J. Med. Chem. 2019, 62, 10098–10107. [Google Scholar] [CrossRef]
- Niu, F.; Yan, J.; Ma, B.; Li, S.; Shao, Y.; He, P.; Zhang, W.; He, W.; Ma, P.X.; Lu, W. Lanthanide-Doped Nanoparticles Conjugated with an Anti-CD33 Antibody and a P53-Activating Peptide for Acute Myeloid Leukemia Therapy. Biomaterials 2018, 167, 132–142. [Google Scholar] [CrossRef]
- Roscoe, I.; Parker, M.; Dong, D.; Li, X.; Li, Z. Human Serum Albumin and the P53-Derived Peptide Fusion Protein Promotes Cytotoxicity Irrespective of P53 Status in Cancer Cells. Mol. Pharm. 2018, 15, 5046–5057. [Google Scholar] [CrossRef]
- Liang, C.; Yan, X.; Zhang, R.; Xu, T.; Zheng, D.; Tan, Z.; Chen, Y.; Gao, Z.; Wang, L.; Li, X.; et al. Enhanced Cellular Uptake and Nuclear Accumulation of Drug-Peptide Nanomedicines Prepared by Enzyme-Instructed Self-Assembly. J. Control. Release 2020, 317, 109–117. [Google Scholar] [CrossRef]
- Miyafusa, T.; Hirota, K.; Honda, S. Generation of Ubiquitin-Based Binder with an Inserted Active Peptide. Biochem. Biophys. Res. Commun. 2018, 503, 3162–3166. [Google Scholar] [CrossRef]
- Philippe, G.J.-B.; Gaspar, D.; Sheng, C.; Huang, Y.-H.; Benfield, A.H.; Condon, N.D.; Weidmann, J.; Lawrence, N.; Löwer, A.; Castanho, M.A.R.B.; et al. Cell Membrane Composition Drives Selectivity and Toxicity of Designed Cyclic Helix-Loop-Helix Peptides with Cell Penetrating and Tumor Suppressor Properties. ACS Chem. Biol. 2019, 14, 2071–2087. [Google Scholar] [CrossRef]
- Ma, B.; Niu, F.; Qu, X.; He, W.; Feng, C.; Wang, S.; Ouyang, Z.; Yan, J.; Wen, Y.; Xu, D.; et al. A Tetrameric Protein Scaffold as a Nano-Carrier of Antitumor Peptides for Cancer Therapy. Biomaterials 2019, 204, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Partridge, A.W.; Kaan, H.Y.K.; Juang, Y.-C.; Sadruddin, A.; Lim, S.; Brown, C.J.; Ng, S.; Thean, D.; Ferrer, F.; Johannes, C.; et al. Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties. Molecules 2019, 24, 2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Ando, N.; Ishihara, M.; Sato, M. Development of Novel Heparin/Protamine Nanoparticles Useful for Delivery of Exogenous Proteins In Vitro and In Vivo. Nanomaterials 2020, 10, 1584. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Lee, M.W.; Vishnubhotla, P.; Perez, J.M.; Khaled, A.R. The Use of Therapeutic Peptides to Target and to Kill Cancer Cells. Curr. Med. Chem. 2012, 19, 3794–3804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, M.; Cao, N.; Qin, W.; Zhao, M.; Wu, J.; Lin, D. Construction of a Tumor Microenvironment PH-Responsive Cleavable PEGylated Hyaluronic Acid Nano-Drug Delivery System for Colorectal Cancer Treatment. Biomater. Sci. 2020, 8, 1885–1896. [Google Scholar] [CrossRef]
- da Silva, A.L.; Cruz, F.F.; Rocco, P.R.M.; Morales, M.M. New Perspectives in Nanotherapeutics for Chronic Respiratory Diseases. Biophys. Rev. 2017, 9, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Gayle, S.; Aiello, R.; Leelatian, N.; Beckta, J.M.; Bechtold, J.; Bourassa, P.; Csengery, J.; Maguire, R.J.; Marshall, D.; Sundaram, R.K.; et al. Tumor-Selective, Antigen-Independent Delivery of a PH Sensitive Peptide-Topoisomerase Inhibitor Conjugate Suppresses Tumor Growth without Systemic Toxicity. NAR Cancer 2021, 3, zcab021. [Google Scholar] [CrossRef]
- Sakurai, H.; Nishimura, K.; Yamamoto, S.; Maruyama, T.; Tamura, A. Molecular Design of PH-Responsive Helix Peptides That Can Damage Tumor Cells Selectively. ACS Appl. Biol. Mater. 2021, 4, 2442–2452. [Google Scholar] [CrossRef]
- Srivastava, D.; Santiso, E.; Gubbins, K.; Barroso da Silva, F.L. Computationally Mapping PKa Shifts Due to the Presence of a Polyelectrolyte Chain around Whey Proteins. Langmuir 2017, 33, 11417–11428. [Google Scholar] [CrossRef]
- Caetano, D.L.Z.; Metzler, R.; Cherstvy, A.G.; de Carvalho, S.J. Adsorption of Lysozyme into a Charged Confining Pore. Phys. Chem. Chem. Phys. 2021, 23, 27195–27206. [Google Scholar] [CrossRef]
- Lunkad, R.; Murmiliuk, A.; Tošner, Z.; Štěpánek, M.; Košovan, P. Role of PKA in Charge Regulation and Conformation of Various Peptide Sequences. Polymers 2021, 13, 214. [Google Scholar] [CrossRef]
- Nadal-Bufi, F.; Mason, J.M.; Chan, L.Y.; Craik, D.J.; Kaas, Q.; Troeira Henriques, S. Designed β-Hairpins Inhibit LDH5 Oligomerization and Enzymatic Activity. J. Med. Chem. 2021, 64, 3767–3779. [Google Scholar] [CrossRef] [PubMed]
- Marverti, G.; Marraccini, C.; Martello, A.; D’Arca, D.; Pacifico, S.; Guerrini, R.; Spyrakis, F.; Gozzi, G.; Lauriola, A.; Santucci, M.; et al. Folic Acid-Peptide Conjugates Combine Selective Cancer Cell Internalization with Thymidylate Synthase Dimer Interface Targeting. J. Med. Chem. 2021, 64, 3204–3221. [Google Scholar] [CrossRef] [PubMed]
- Cen, L.; Xing, F.; Xu, L.; Cao, Y. Potential Role of Gene Regulator NFAT5 in the Pathogenesis of Diabetes Mellitus. J. Diabetes Res. 2020, 2020, 6927429. [Google Scholar] [CrossRef]
- Lee, N.; Kim, D.; Kim, W.-U. Role of NFAT5 in the Immune System and Pathogenesis of Autoimmune Diseases. Front. Immunol. 2019, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Timucin, A.C. Structure Based Peptide Design, Molecular Dynamics and MM-PBSA Studies for Targeting C Terminal Dimerization of NFAT5 DNA Binding Domain. J. Mol. Graph. Model. 2021, 103, 107804. [Google Scholar] [CrossRef]
- Lee, J.-U.; Kim, L.-K.; Choi, J.-M. Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front. Immunol. 2018, 9, 2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, D.; Zhang, Y.; Zheng, J. Regulation of P53: A Collaboration between Mdm2 and Mdmx. Oncotarget 2012, 3, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gohain, N.; Chen, S.; Li, Y.; Zhao, X.; Li, B.; Tolbert, W.D.; He, W.; Pazgier, M.; Hu, H.; et al. Design of Ultrahigh-Affinity and Dual-Specificity Peptide Antagonists of MDM2 and MDMX for P53 Activation and Tumor Suppression. Acta Pharm. Sin. B 2021, 11, 2655–2669. [Google Scholar] [CrossRef] [PubMed]
- Philippe, G.J.-B.; Mittermeier, A.; Lawrence, N.; Huang, Y.-H.; Condon, N.D.; Loewer, A.; Craik, D.J.; Henriques, S.T. Angler Peptides: Macrocyclic Conjugates Inhibit P53:MDM2/X Interactions and Activate Apoptosis in Cancer Cells. ACS Chem. Biol. 2021, 16, 414–428. [Google Scholar] [CrossRef]
- Shi, Y.; Sang, P.; Lu, J.; Higbee, P.; Chen, L.; Yang, L.; Odom, T.; Daughdrill, G.; Chen, J.; Cai, J. Rational Design of Right-Handed Heterogeneous Peptidomimetics as Inhibitors of Protein-Protein Interactions. J. Med. Chem. 2020, 63, 13187–13196. [Google Scholar] [CrossRef]
- He, W.; Yan, J.; Li, Y.; Yan, S.; Wang, S.; Hou, P.; Lu, W. Resurrecting a P53 Peptide Activator—An Enabling Nanoengineering Strategy for Peptide Therapeutics. J. Control. Release 2020, 325, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Lundsten, S.; Hernández, V.A.; Gedda, L.; Sarén, T.; Brown, C.J.; Lane, D.P.; Edwards, K.; Nestor, M. Tumor-Targeted Delivery of the P53-Activating Peptide VIP116 with PEG-Stabilized Lipodisks. Nanomaterials 2020, 10, 783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.-Y.; Jiang, X.-M.; Wang, B.-L.; Sun, Y.; Lu, J.-J. Combination Therapy with PD-1/PD-L1 Blockade in Non-Small Cell Lung Cancer: Strategies and Mechanisms. Pharmacol. Ther. 2021, 219, 107694. [Google Scholar] [CrossRef]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 Immune Checkpoint Inhibitors in Glioblastoma: Clinical Studies, Challenges and Potential. Hum. Vaccines Immunother. 2021, 17, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Ma, B.B.Y. Targeting the PD-1/ PD-L1 Interaction in Nasopharyngeal Carcinoma. Oral Oncol. 2021, 113, 105127. [Google Scholar] [CrossRef]
- Zhai, W.; Zhou, X.; Zhai, M.; Li, W.; Ran, Y.; Sun, Y.; Du, J.; Zhao, W.; Xing, L.; Qi, Y.; et al. Blocking of the PD-1/PD-L1 Interaction by a Novel Cyclic Peptide Inhibitor for Cancer Immunotherapy. Sci. China Life Sci. 2021, 64, 548–562. [Google Scholar] [CrossRef]
- Zhang, P.; Li, C.; Ji, X.; Gao, M.; Lyu, S.; Dai, X.; Du, J. In Silico Screening and Surface Plasma Resonance-Based Verification of Programmed Death 1-Targeted Peptides. Chem. Biol. Drug Des. 2020, 95, 332–342. [Google Scholar] [CrossRef]
- Niu, B.; Appleby, T.C.; Wang, R.; Morar, M.; Voight, J.; Villaseñor, A.G.; Clancy, S.; Wise, S.; Belzile, J.-P.; Papalia, G.; et al. Protein Footprinting and X-Ray Crystallography Reveal the Interaction of PD-L1 and a Macrocyclic Peptide. Biochemistry 2020, 59, 541–551. [Google Scholar] [CrossRef]
- Meng, X.; Wang, J.; Zhou, J.; Tian, Q.; Qie, B.; Zhou, G.; Duan, W.; Zhu, Y. Tumor Cell Membrane-Based Peptide Delivery System Targeting the Tumor Microenvironment for Cancer Immunotherapy and Diagnosis. Acta Biomater. 2021, 127, 266–275. [Google Scholar] [CrossRef]
- Zou, S.; Liu, J.; Sun, Z.; Feng, X.; Wang, Z.; Jin, Y.; Yang, Z. Discovery of HPRDX5-Based Peptide Inhibitors Blocking PD-1/PD-L1 Interaction through in Silico Proteolysis and Rational Design. Cancer Chemother. Pharmacol. 2020, 85, 185–193. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Z.; Zhang, L.; Li, Y.; Jain, A.; Barve, A.; Jin, W.; Liu, Y.; Fetse, J.; Cheng, K. Discovery of Low-Molecular Weight Anti-PD-L1 Peptides for Cancer Immunotherapy. J. Immunother. Cancer 2019, 7, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, W.-J.; Bu, J.; Han, Y.; Drelich, A.J.; Nair, A.; Král, P.; Hong, S. Nanoparticle Conjugation Stabilizes and Multimerizes β-Hairpin Peptides To Effectively Target PD-1/PD-L1 β-Sheet-Rich Interfaces. J. Am. Chem. Soc. 2020, 142, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Song, Y.; Su, Y.; Liang, Y.; Wang, L. Effect of the Hairpin Structure of Peptide Inhibitors on the Blockade of PD-1/PD-L1 Axis. Biochem. Biophys. Res. Commun. 2020, 527, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Boohaker, R.J.; Sambandam, V.; Segura, I.; Miller, J.; Suto, M.; Xu, B. Rational Design and Development of a Peptide Inhibitor for the PD-1/PD-L1 Interaction. Cancer Lett. 2018, 434, 11–21. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhu, X.; Zhou, X.; Wang, X.; Zhai, W.; Li, B.; Du, J.; Li, G.; Sui, X.; Wu, Y.; et al. An Orally Available PD-1/PD-L1 Blocking Peptide OPBP-1-Loaded Trimethyl Chitosan Hydrogel for Cancer Immunotherapy. J. Control. Release 2021, 334, 376–388. [Google Scholar] [CrossRef]
- Luo, L.; Zhu, C.; Yin, H.; Jiang, M.; Zhang, J.; Qin, B.; Luo, Z.; Yuan, X.; Yang, J.; Li, W.; et al. Laser Immunotherapy in Combination with Perdurable PD-1 Blocking for the Treatment of Metastatic Tumors. ACS Nano 2018, 12, 7647–7662. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 Transcription Factors--Similar but Not Identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [Google Scholar] [CrossRef]
- Semenza, G.L. Physiology Meets Biophysics: Visualizing the Interaction of Hypoxia-Inducible Factor 1 Alpha with P300 and CBP. Proc. Natl. Acad. Sci. USA 2002, 99, 11570–11572. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Kyle, H.F.; Nelson, A.; Edwards, T.A.; Wilson, A.J. Hypoxia Inducible Factor (HIF) as a Model for Studying Inhibition of Protein-Protein Interactions. Chem. Sci. 2017, 8, 4188–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colarusso, S.; De Simone, D.; Frattarelli, T.; Andreini, M.; Cerretani, M.; Missineo, A.; Moretti, D.; Tambone, S.; Kempf, G.; Augustin, M.; et al. Optimization of Linear and Cyclic Peptide Inhibitors of KEAP1-NRF2 Protein-Protein Interaction. Bioorg. Med. Chem. 2020, 28, 115738. [Google Scholar] [CrossRef]
- Lu, M.-C.; Jiao, Q.; Liu, T.; Tan, S.-J.; Zhou, H.-S.; You, Q.-D.; Jiang, Z.-Y. Discovery of a Head-to-Tail Cyclic Peptide as the Keap1-Nrf2 Protein-Protein Interaction Inhibitor with High Cell Potency. Eur. J. Med. Chem. 2018, 143, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Steel, R.J.; O’Connell, M.A.; Searcey, M. Perfluoroarene-Based Peptide Macrocycles That Inhibit the Nrf2/Keap1 Interaction. Bioorg. Med. Chem. Lett. 2018, 28, 2728–2731. [Google Scholar] [CrossRef] [Green Version]
- Salim, H.; Song, J.; Sahni, A.; Pei, D. Development of a Cell-Permeable Cyclic Peptidyl Inhibitor against the Keap1-Nrf2 Interaction. J. Org. Chem. 2020, 85, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Huang, Y.-H.; Best, S.A.; Sutherland, K.D.; Craik, D.J.; Wang, C.K. An Integrated Molecular Grafting Approach for the Design of Keap1-Targeted Peptide Inhibitors. ACS Chem. Biol. 2021, 16, 1276–1287. [Google Scholar] [CrossRef]
- Camarero, J.A. Cyclotides, a Versatile Ultrastable Micro-Protein Scaffold for Biotechnological Applications. Bioorg. Med. Chem. Lett. 2017, 27, 5089–5099. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.; Hommen, P.; Noisier, A.; Krzyzanowski, A.; Schüler, D.; Porfetye, A.T.; Akbarzadeh, M.; Vetter, I.R.; Adihou, H.; Waldmann, H. Structure Based Design of Bicyclic Peptide Inhibitors of RbAp48. Angew. Chem. Int. Ed. Engl. 2021, 60, 1813–1820. [Google Scholar] [CrossRef]
- Li, Q.; Zeng, C.; Liu, H.; Yung, K.W.Y.; Chen, C.; Xie, Q.; Zhang, Y.; Wan, S.W.C.; Mak, B.S.W.; Xia, J.; et al. Protein-Protein Interaction Inhibitor of SRPKs Alters the Splicing Isoforms of VEGF and Inhibits Angiogenesis. iScience 2021, 24, 102423. [Google Scholar] [CrossRef] [PubMed]
- Jafari, A.; Rezaei-Tavirani, M.; Farhadihosseinabadi, B.; Taranejoo, S.; Zali, H. HSP90 and Co-Chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Investig. 2020, 38, 310–328. [Google Scholar] [CrossRef] [PubMed]
- D’Annessa, I.; Hurwitz, N.; Pirota, V.; Beretta, G.L.; Tinelli, S.; Woodford, M.; Freccero, M.; Mollapour, M.; Zaffaroni, N.; Wolfson, H.; et al. Design of Disruptors of the Hsp90-Cdc37 Interface. Molecules 2020, 25, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Bao, Q.-C.; Xu, X.-L.; Jiang, F.; Gu, K.; Jiang, Z.-Y.; Zhang, X.-J.; Guo, X.-K.; You, Q.-D.; Sun, H.-P. Discovery and Identification of Cdc37-Derived Peptides Targeting the Hsp90–Cdc37 Protein–Protein Interaction. RSC Adv. 2015, 5, 96138–96145. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Fu, W.-T.; Jiang, Z.-Y.; You, Q.-D.; Xu, X.-L. Optimization and Bioevaluation of Cdc37-Derived Peptides: An Insight into Hsp90-Cdc37 Protein-Protein Interaction Modulators. Bioorg. Med. Chem. 2017, 25, 233–240. [Google Scholar] [CrossRef]
- Knauer, S.K.; Mann, W.; Stauber, R.H. Survivin’s Dual Role: An Export’s View. Cell Cycle 2007, 6, 518–521. [Google Scholar] [CrossRef]
- Fokkens, M.; Schrader, T.; Klärner, F.-G. A Molecular Tweezer for Lysine and Arginine. J. Am. Chem. Soc. 2005, 127, 14415–14421. [Google Scholar] [CrossRef]
- Meiners, A.; Bäcker, S.; Hadrović, I.; Heid, C.; Beuck, C.; Ruiz-Blanco, Y.B.; Mieres-Perez, J.; Pörschke, M.; Grad, J.-N.; Vallet, C.; et al. Specific Inhibition of the Survivin-CRM1 Interaction by Peptide-Modified Molecular Tweezers. Nat. Commun. 2021, 12, 1505. [Google Scholar] [CrossRef]
- Dohi, T.; Okada, K.; Xia, F.; Wilford, C.E.; Samuel, T.; Welsh, K.; Marusawa, H.; Zou, H.; Armstrong, R.; Matsuzawa, S.; et al. An IAP-IAP Complex Inhibits Apoptosis. J. Biol. Chem. 2004, 279, 34087–34090. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Che, X.; Li, G.; Wang, A.; Wang, Y.; Shi, X.; Hou, K.; Zhang, X.; Qu, X.; Liu, Y. Sur-X, a Novel Peptide, Kills Colorectal Cancer Cells by Targeting Survivin-XIAP Complex. J. Exp. Clin. Cancer Res. 2020, 39, 82. [Google Scholar] [CrossRef]
- Wu, D.; Luo, L.; Yang, Z.; Chen, Y.; Quan, Y.; Min, Z. Targeting Human Hippo TEAD Binding Interface with YAP/TAZ-Derived, Flexibility-Reduced Peptides in Gastric Cancer. Int. J. Pept. Res. Ther. 2021, 27, 119–128. [Google Scholar] [CrossRef]
- Crawford, J.J.; Bronner, S.M.; Zbieg, J.R. Hippo Pathway Inhibition by Blocking the YAP/TAZ-TEAD Interface: A Patent Review. Expert Opin. Ther. Pat. 2018, 28, 867–873. [Google Scholar] [CrossRef]
- Saunders, J.T.; Holmes, B.; Benavides-Serrato, A.; Kumar, S.; Nishimura, R.N.; Gera, J. Targeting the YAP-TEAD Interaction Interface for Therapeutic Intervention in Glioblastoma. J. Neurooncol. 2021, 152, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Badia-Villanueva, M.; Defaus, S.; Foj, R.; Andreu, D.; Oliva, B.; Sierra, A.; Fernandez-Fuentes, N. Evaluation of Computationally Designed Peptides against TWEAK, a Cytokine of the Tumour Necrosis Factor Ligand Family. Int. J. Mol. Sci. 2021, 22, 1066. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, R.; Yan, H.; Zhao, Z.; Zhang, J.; You, W. Assembling BH3-Mimic Peptide into a Nanocluster to Target Intracellular Bcl2 towards the Apoptosis Induction of Cancer Cell. Nanotechnology 2021, 33, 085103. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, T.; Chen, L.; Zhang, Q.; Wang, W.; Han, X.; Li, D.; Shi, J.; Sui, G. Characterization of YY1 OPB Peptide for Its Anticancer Activity. Curr. Cancer Drug Targets 2019, 19, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Bojadzic, D.; Chen, J.; Alcazar, O.; Buchwald, P. Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40−CD154 Costimulatory Protein-Protein Interaction. Molecules 2018, 23, 1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojadzic, D.; Buchwald, P. CD40-Targeting KGYY15 Peptides Do Not Efficiently Block the CD40-CD40L Interaction. Diabetologia 2019, 62, 2158–2160. [Google Scholar] [CrossRef]
- Bartling, C.R.O.; Jensen, T.M.T.; Henry, S.M.; Colliander, A.L.; Sereikaite, V.; Wenzler, M.; Jain, P.; Maric, H.M.; Harpsøe, K.; Pedersen, S.W.; et al. Targeting the APP-Mint2 Protein-Protein Interaction with a Peptide-Based Inhibitor Reduces Amyloid-β Formation. J. Am. Chem. Soc. 2021, 143, 891–901. [Google Scholar] [CrossRef]
- Razzaghi-Asl, N.; Ebadi, A. In Silico Design of Peptide Inhibitors of Tubulin: Amyloid-β as a Lead Compound. J. Biomol. Struct. Dyn. 2021, 39, 2189–2198. [Google Scholar] [CrossRef]
- Ulysse, L.G.; Chmielewski, J. Restricting the Flexibility of Crosslinked, Interfacial Peptide Inhibitors of HIV-1 Protease. Bioorg. Med. Chem. Lett. 1998, 8, 3281–3286. [Google Scholar] [CrossRef]
- Srivastava, S.; Sluis-Cremer, N.; Tachedjian, G. Dimerization of Human Immunodeficiency Virus Type 1 Reverse Transcriptase as an Antiviral Target. Curr. Pharm. Des. 2006, 12, 1879–1894. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Liu, Z.-P. Recent Developments of Peptidomimetic HIV-1 Protease Inhibitors. Curr. Med. Chem. 2011, 18, 4513–4537. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, K.; Kimura, T.; Sankaranarayanan, R.; Wang, J.; McDaniel, K.F.; Kempf, D.J.; Kameoka, M.; Adachi, M.; Kuroki, R.; Nguyen, J.-T.; et al. Identification of Highly Potent Human Immunodeficiency Virus Type-1 Protease Inhibitors against Lopinavir and Darunavir Resistant Viruses from Allophenylnorstatine-Based Peptidomimetics with P2 Tetrahydrofuranylglycine. J. Med. Chem. 2018, 61, 5138–5153. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, Y.; Huang, S.; Zhao, N.; Cheng, M.; Zhang, X. Computational Exploration of Natural Peptides Targeting ACE2. J. Biomol. Struct. Dyn. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Odolczyk, N.; Marzec, E.; Winiewska-Szajewska, M.; Poznański, J.; Zielenkiewicz, P. Native Structure-Based Peptides as Potential Protein-Protein Interaction Inhibitors of SARS-CoV-2 Spike Protein and Human ACE2 Receptor. Molecules 2021, 26, 2157. [Google Scholar] [CrossRef] [PubMed]
- Alagumuthu, M.; Rajpoot, S.; Baig, M.S. Structure-Based Design of Novel Peptidomimetics Targeting the SARS-CoV-2 Spike Protein. Cell. Mol. Bioeng. 2020, 14, 177–185. [Google Scholar] [CrossRef]
- Maas, M.N.; Hintzen, J.C.J.; Löffler, P.M.G.; Mecinović, J. Targeting SARS-CoV-2 Spike Protein by Stapled HACE2 Peptides. Chem. Commun. 2021, 57, 3283–3286. [Google Scholar] [CrossRef]
- Chitsike, L.; Duerksen-Hughes, P.J. PPI Modulators of E6 as Potential Targeted Therapeutics for Cervical Cancer: Progress and Challenges in Targeting E6. Molecules 2021, 26, 3004. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for Gene List Enrichment Analysis and Candidate Gene Prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Cabri, W.; Cantelmi, P.; Corbisiero, D.; Fantoni, T.; Ferrazzano, L.; Martelli, G.; Mattellone, A.; Tolomelli, A. Therapeutic Peptides Targeting PPI in Clinical Development: Overview, Mechanism of Action and Perspectives. Front. Mol. Biosci. 2021, 8, 697586. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-Type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- Pakhrin, S.C.; Shrestha, B.; Adhikari, B.; Kc, D.B. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci. 2021, 22, 5553. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Zhang, N.; Bersch, B.; Fidelis, K.; Inouye, M.; Ishida, Y.; Kryshtafovych, A.; Kobayashi, N.; Kuroda, Y.; Liu, G.; et al. Assessment of Prediction Methods for Protein Structures Determined by NMR in CASP14: Impact of AlphaFold2. Proteins 2021, 89, 1959–1976. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Design Concepts | References |
|---|---|
| Alteration of linear peptide amino acid composition | |
| Substitution of l- for d-amino acids | [51,52] |
| Inclusion of non-proteinogenic amino acids | [53] |
| Macrocyclization (stapled peptides) | |
| Stapling via a dithiocarbamate linker | [54] |
| Stapling via dialkynyl linker | [55] |
| Stapling via photoisomerizable diarylethene residue | [56] |
| Peptides conjugates | |
| Stapled peptide, conjugated with a cell-penetrating peptide | [57] |
| Lanthanum oxyfluoride nanoparticles with the affine peptide and monoclonal antibody | [58] |
| A conjugate of affine peptide and serum albumin | [59] |
| Self-assembling nanostructures based on lipopeptide conjugates | [60] |
| Peptide grafting | |
| Peptide grafting into ubiquitin scaffold | [61] |
| Peptide grafting into scaffold based on cyclic motif helix-loop-helix | [62] |
| Peptide grafting into scaffold based on N-terminal domain of chimeric oncoprotein Bcr/Abl | [63] |
| Combined approaches | |
| Alpha-helical cyclic peptide with inclusion of D- and N-methylated amino acids | [64] |
| Parameters/ Complexes | Interface Area of a Complex, Å 2 | Percent of Protein SASA 1 Occupied by a PPI Inhibitor | SASA of an Inhibitor, Å 2 | A Number of Hydrogen Bonds |
|---|---|---|---|---|
| Protein/peptide 2 | 560 | 8.3 | 1332 | 3 |
| Protein/compound 3 | 389 | 5.1 | 720 | 1 |
| Ratio | 1.43 | 1.62 | 1.85 | 3 |
| Subcategory Name (FDR B & Y Value) | Proteins | Enrichment Ratio, % |
|---|---|---|
| Protein domain specific binding (3.868 × 10−4) | Bax, Bcl-2, KEAP1, CRM1, Mdm2, NRF2, p53, Hsp90, CD40 | 36 |
| Response to cytokine (1.680 × 10−9) | Fn14, TYMS, Cdc37, BIRC5, AKT, HIF-1, Bcl-2. KEAP1, NFAT5, TWEAK, YY1. NRF2. YAP, p53, Hsp90, CD40 | 64 |
| Transcription regulator complex (2.621 × 10−3) | MTA1, NFAT5, YY1, RbAp48, NRF2, YAP, p53 | 24 |
| Hematological neoplasm (8.940 × 10−3) | Bax, XIAP, AKT, Bcl-2, CRM1, Mdm2, TWEAK, NRF2, YAP, p53 | 40 |
| Platinum drug resistance (3.236 × 10−7), Apoptosis (5.256 × 10−7) Pathways in cancer (3.750 × 10−5) | Bax, XIAP, BIRC5, AKT, Bcl-2, Mdm2, p53, Hsp90 | 32 |
| Doxorubicin (6.303 × 10−11) | Fn14, TYMS, Bax, XIAP, BIRC5, PDCD1, AKT Bcl-2, KEAP1, Mdm2, LDH5, NFAT5, RbAp48, NRF2, YAP, p53, Hsp90, CD40 | 72 |
| Triple Negative Breast Neoplasms (1.822 × 10−13) | Fn14, TYMS, Cdc37, Bax, XIAP, BIRC5, PDCD1, AKT, HIF-1, Bcl-2, MTA1, KEAP1, CRM1, Mdm2, LDH5, TWEAK, NRF2, YAP, p53, Hsp90 | 80 |
| Diffuse Large B-Cell Lymphoma (9.284 × 10−14) | TYMS, Bax, XIAP, BIRC5, PDCD1, AKT, HIF-1 Bcl-2, MTA1, KEAP1, CRM1, Mdm2, YY1, NRF2, YAP, p53, Hsp90, CD40 | 72 |
| Renal Cell Carcinoma (1.087 × 10−12) | Fn14, TYMS, Bax, XIAP, BIRC5, SRPK1, PDCD1, AKT, HIF-1, Bcl-2, KEAP1, CRM1, Mdm2, LDH5, TWEAK, YY1, NRF2, YAP, p53, CD40 | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ershov, P.V.; Mezentsev, Y.V.; Ivanov, A.S. Interfacial Peptides as Affinity Modulating Agents of Protein-Protein Interactions. Biomolecules 2022, 12, 106. https://doi.org/10.3390/biom12010106
Ershov PV, Mezentsev YV, Ivanov AS. Interfacial Peptides as Affinity Modulating Agents of Protein-Protein Interactions. Biomolecules. 2022; 12(1):106. https://doi.org/10.3390/biom12010106
Chicago/Turabian StyleErshov, Pavel V., Yuri V. Mezentsev, and Alexis S. Ivanov. 2022. "Interfacial Peptides as Affinity Modulating Agents of Protein-Protein Interactions" Biomolecules 12, no. 1: 106. https://doi.org/10.3390/biom12010106