
GNAi2/gip2-Regulated Transcriptome and Its Therapeutic Significance in Ovarian Cancer

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Transcriptomic Analysis
2.3. Bioinformatic Analysis
2.4. RT-qPCR Analysis
2.5. Immunoblot Analysis
2.6. Statistics
3. Results
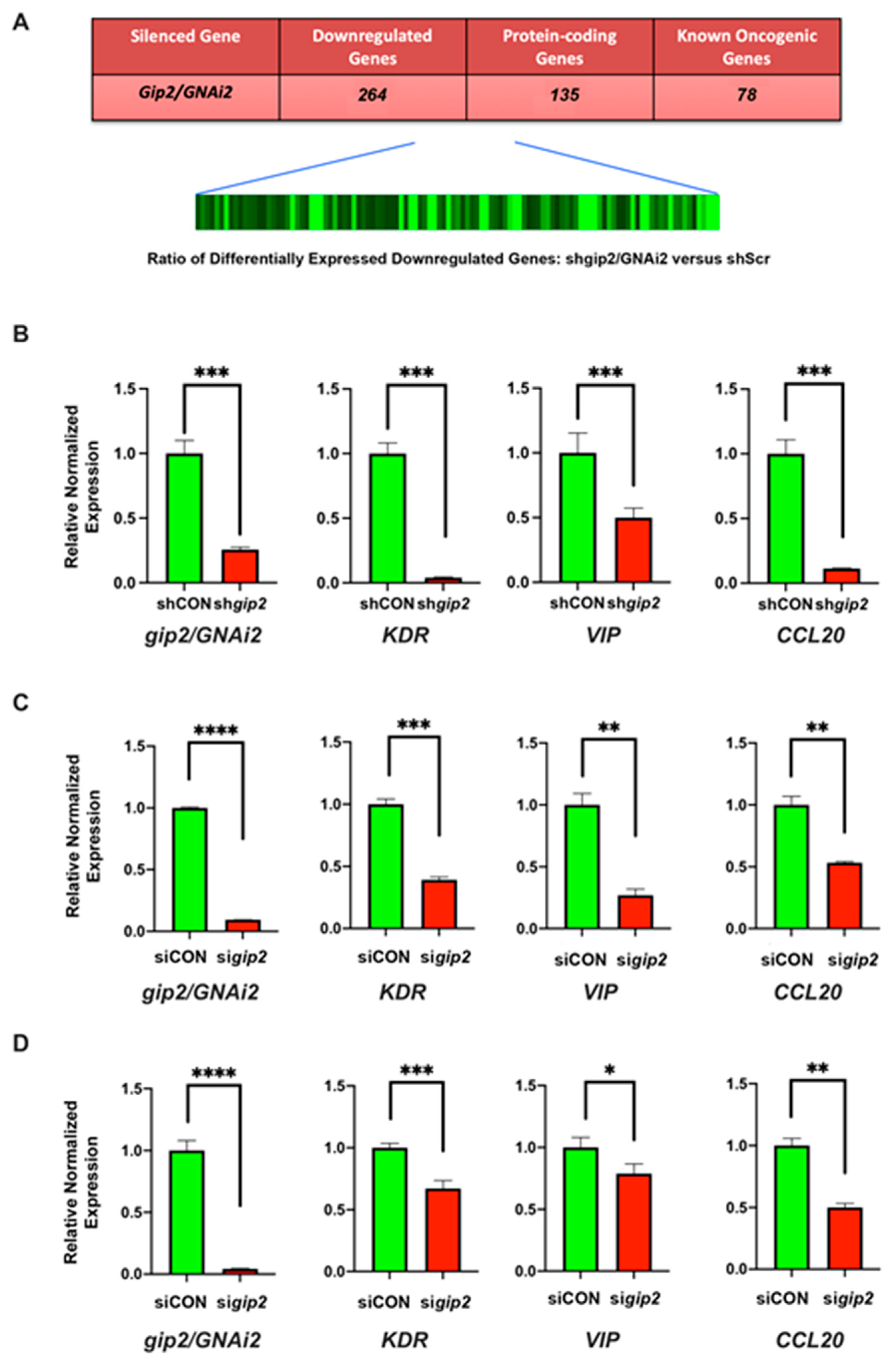
3.1. Characterization of GNAi2/gip2-Dependent Transcriptome
3.2. Gene Ontology Enrichment Analysis of gip2-Dependent Genes
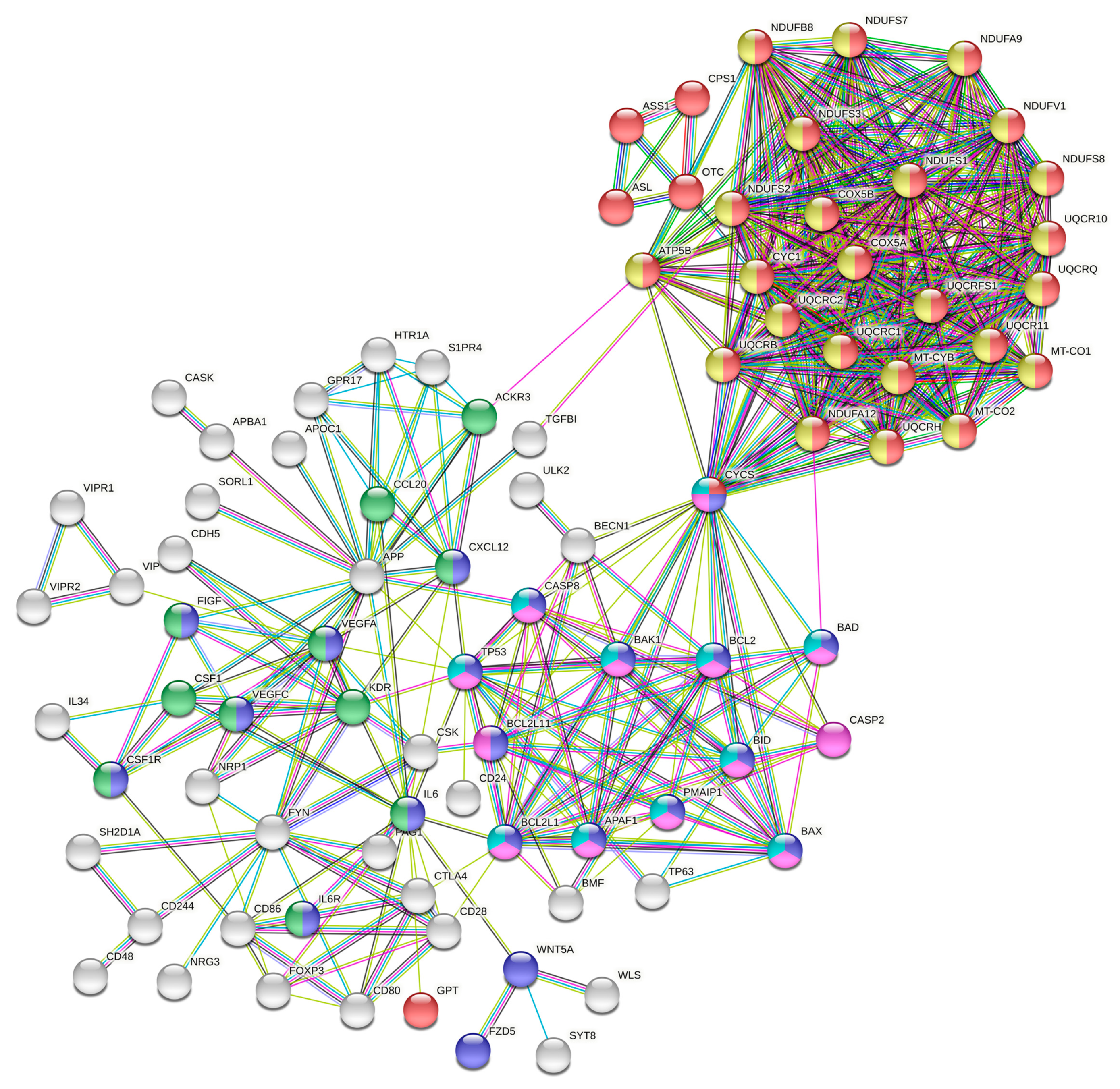
3.3. Analysis of PPI Networks and Pathways
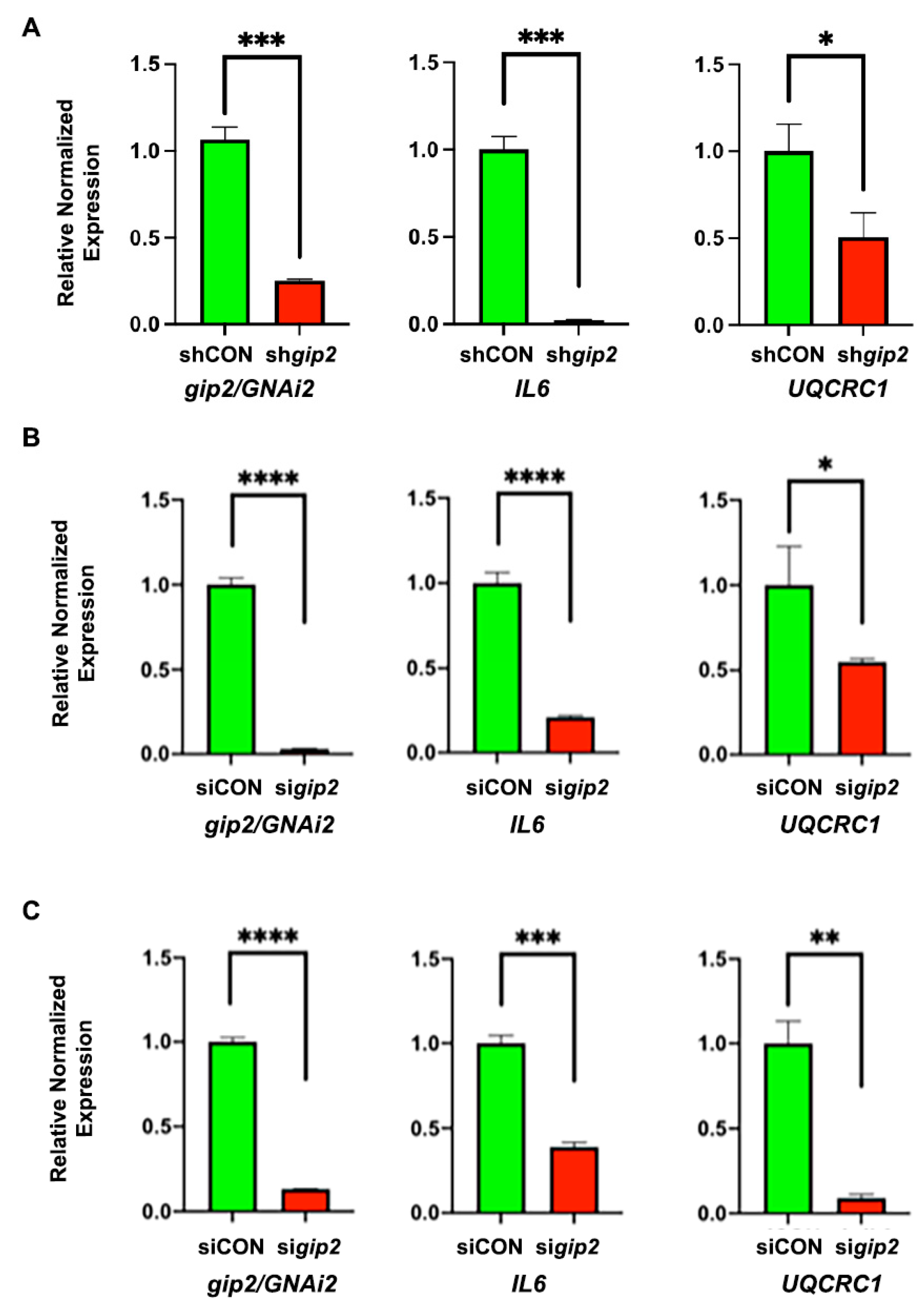
3.4. Identification of the Hub and Bottleneck Nodes
3.5. Significance of the Hub and Bottleneck Nodes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Epidemiology Working Group Steering Committee, Ovarian Cancer Association Consortium Members of the EWG SC; Doherty, J.A.; Jensen, A.; Kelemen, L.E.; Pearce, C.L.; Poole, E.; Schildkraut, J.M.; Terry, K.L.; Tworoger, S.S.; Webb, P.M.; et al. Current gaps in ovarian cancer epidemiology: The need for new population-based research. J. Natl. Cancer Inst. 2017, 109, djx144. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Lu, Z.; Han, C.Y.; Lu, K.H.; Anderson, K.S.; Drescher, C.W.; Skates, S.J. Biomarkers and strategies for early detection of ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Peng, H.; Qi, X.; Wu, M.; Zhao, X. Targeted therapies in gynecological cancers: A comprehensive review of clinical evidence. Signal Transduct. Target. Ther. 2020, 5, 137. [Google Scholar] [CrossRef] [PubMed]
- Willmott, L.J.; Fruehauf, J.P. Targeted therapy in ovarian cancer. J. Oncol. 2010, 2010, 740472. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A convergence-based framework for cancer drug resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Raymond, J.R., Jr.; Appleton, K.M.; Pierce, J.Y.; Peterson, Y.K. Suppression of GNAI2 message in ovarian cancer. J. Ovarian Res. 2014, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.H.; Radhakrishnan, R.; Jayaraman, M.; Yan, M.; Ward, J.D.; Fung, K.M.; Moxley, K.; Sood, A.K.; Isidoro, C.; Mukherjee, P.; et al. LPA Induces metabolic reprogramming in ovarian cancer via a pseudohypoxic response. Cancer Res. 2018, 78, 1923–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.H.; Gomathinayagam, R.; Yan, M.; Jayaraman, M.; Ramesh, R.; Dhanasekaran, D.N. Determinant role for the gep oncogenes, Galpha12/13, in ovarian cancer cell proliferation and xenograft tumor growth. Genes Cancer 2015, 6, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. Cytohubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Meng, J.; Li, X.; Zhu, F.; Liu, T.; Wu, G.; Zhang, L. Identification of hub genes and key pathways associated with two subtypes of diffuse large B-cell lymphoma based on gene expression profiling via integrated bioinformatics. BioMed Res. Int. 2018, 2018, 3574534. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.D.; Ha, J.H.; Jayaraman, M.; Dhanasekaran, D.N. LPA-mediated migration of ovarian cancer cells involves translocalization of Galphai2 to invadopodia and association with Src and beta-pix. Cancer Lett. 2015, 356, 382–391. [Google Scholar] [CrossRef]
- Ward, J.D.; Dhanasekaran, D.N. LPA stimulates the phosphorylation of p130Cas via Galphai2 in ovarian cancer cells. Genes Cancer 2012, 3, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Ward, J.D.; Radhakrishnan, R.; Jayaraman, M.; Song, Y.S.; Dhanasekaran, D.N. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Galphai2, Src, and HIF1alpha signaling nexus. Oncotarget 2016, 7, 37664–37679. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, K.V.; Xiao, N.; Wang, W.; Gangi, L.; Greene, J.; Powell, J.I.; Dickson, R.; Furth, P.; Hunter, K.; Kucherlapati, R.; et al. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc. Natl. Acad. Sci. USA 2002, 99, 6967–6972. [Google Scholar] [CrossRef] [Green Version]
- Smolle, E.; Taucher, V.; Pichler, M.; Petru, E.; Lax, S.; Haybaeck, J. Targeting signaling pathways in epithelial ovarian cancer. Int. J. Mol. Sci. 2013, 14, 9536–9555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentzensen, N.; Poole, E.M.; Trabert, B.; White, E.; Arslan, A.A.; Patel, A.V.; Setiawan, V.W.; Visvanathan, K.; Weiderpass, E.; Adami, H.O.; et al. Ovarian cancer risk factors by histologic subtype: An analysis from the ovarian cancer cohort consortium. J. Clin. Oncol. 2016, 34, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP inhibitors in ovarian cancer. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-angiogenic agents in ovarian cancer: Past, present, and future. Ann. Oncol. 2016, 27, i33–i39. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Yamulla, R.J.; Nalubola, S.; Flesken-Nikitin, A.; Nikitin, A.Y.; Schimenti, J.C. Most commonly mutated genes in high-grade serous ovarian carcinoma are nonessential for ovarian surface epithelial stem cell transformation. Cell Rep. 2020, 32, 108086. [Google Scholar] [CrossRef]
- Cybulski, M.; Jarosz, B.; Nowakowski, A.; Jeleniewicz, W.; Seroczynski, P.; Mazurek-Kociubowska, M. Cyclin I correlates with VEGFR-2 and cell proliferation in human epithelial ovarian cancer. Gynecol. Oncol. 2012, 127, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cheng, X.; Tu, W.; Qi, Z.; Li, H.; Liu, F.; Yang, Y.; Zhang, Z.; Wang, Z. Apatinib inhibits glycolysis by suppressing the VEGFR2/AKT1/SOX5/GLUT4 signaling pathway in ovarian cancer cells. Cell. Oncol. 2019, 42, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Moody, T.W.; Hill, J.M.; Jensen, R.T. VIP as a trophic factor in the CNS and cancer cells. Peptides 2003, 24, 163–177. [Google Scholar] [CrossRef]
- Liu, W.; Wang, W.; Zhang, N.; Di, W. The role of CCL20-CCR6 axis in ovarian cancer metastasis. OncoTargets Ther. 2020, 13, 12739–12750. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Sun, X.; Zhang, Q.; Zhang, Z.; Chen, J. CCL20 Promotes ovarian cancer chemotherapy resistance by regulating ABCB1 expression. Cell Struct. Funct. 2019, 44, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.J.; Choi, H.J.; Bischoff, F.Z.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Au Yeung, C.L.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Darb-Esfahani, S.; Richter, R.; Taube, E.T.; Ruscito, I.; Mahner, S.; Woelber, L.; Prieske, K.; Concin, N.; Vergote, I.; et al. Vascular endothelial growth factor receptor 2 (VEGFR2) correlates with long-term survival in patients with advanced high-grade serous ovarian cancer (HGSOC): A study from the tumor bank ovarian cancer (TOC) consortium. J. Cancer Res. Clin. Oncol. 2019, 145, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Sopo, M.; Anttila, M.; Hamalainen, K.; Kivela, A.; Yla-Herttuala, S.; Kosma, V.M.; Keski-Nisula, L.; Sallinen, H. Expression profiles of VEGF-A, VEGF-D and VEGFR1 are higher in distant metastases than in matched primary high grade epithelial ovarian cancer. BMC Cancer 2019, 19, 584. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hu, Z.; Shi, H.; Wang, C.; Lei, J.; Cheng, Y. Inhibition of VEGFA increases the sensitivity of ovarian cancer cells to chemotherapy by suppressing VEGFA-mediated autophagy. OncoTargets Ther. 2020, 13, 8161–8171. [Google Scholar] [CrossRef]
- Wang, Y.; Zong, X.; Mitra, S.; Mitra, A.K.; Matei, D.; Nephew, K.P. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight 2018, 3, e122360. [Google Scholar] [CrossRef] [Green Version]
- Azar, W.J.; Christie, E.L.; Mitchell, C.; Liu, D.S.; Au-Yeung, G.; Bowtell, D.D.L. Noncanonical IL6 signaling-mediated activation of YAP regulates cell migration and invasion in ovarian clear cell cancer. Cancer Res. 2020, 80, 4960–4971. [Google Scholar] [CrossRef]
- Neves, M.; Fumagalli, A.; van den Bor, J.; Marin, P.; Smit, M.J.; Mayor, F. The role of ACKR3 in breast, lung, and brain cancer. Mol. Pharmacol. 2019, 96, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.J.; Schlecht-Louf, G.; Neves, M.; van den Bor, J.; Penela, P.; Siderius, M.; Bachelerie, F.; Mayor, F., Jr. The CXCL12/CXCR4/ACKR3 axis in the tumor microenvironment: Signaling, crosstalk, and therapeutic targeting. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 541–563. [Google Scholar] [CrossRef]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A novel molecular target in cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Yoo, K.C.; An, Y.; Lee, H.J.; Lee, M.; Uddin, N.; Kim, M.J.; Kim, I.G.; Suh, Y.; Lee, S.J. FYN promotes mesenchymal phenotypes of basal type breast cancer cells through STAT5/NOTCH2 signaling node. Oncogene 2018, 37, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Xu, L.; Chen, L.; Wang, Y.; Jiang, H.; Wang, Y.; Yan, Y.; Luo, S.; Zhai, Z. FYN is required for ARHGEF16 to promote proliferation and migration in colon cancer cells. Cell Death Dis. 2020, 11, 652. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, M.; Gan, Y.; Jiang, S.; Qiao, J.; Zhang, W.; Fan, Y.; Shen, Y.; Song, Y.; Meng, Z.; et al. Mitochondrial protein UQCRC1 is oncogenic and a potential therapeutic target for pancreatic cancer. Theranostics 2020, 10, 2141–2157. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.J.; Gerasimova, T.; Smith, S.T.; Lloyd, K.O.; Suzumori, K.; Young, S.R. CA125 and UQCRFS1 FISH studies of ovarian carcinoma. Gynecol. Oncol. 2003, 90, 29–36. [Google Scholar] [CrossRef]
- Ohashi, Y.; Kaneko, S.J.; Cupples, T.E.; Young, S.R. Ubiquinol cytochrome c reductase (UQCRFS1) gene amplification in primary breast cancer core biopsy samples. Gynecol. Oncol. 2004, 93, 54–58. [Google Scholar] [CrossRef]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS complex III in breast cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, K.H.; Kim, S.Y.; Yoon, J.H.; Song, J.H.; Park, W.S. Amplification of the UQCRFS1 gene in gastric cancers. J. Gastric Cancer 2012, 12, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.P.; Sun, H.F.; Fu, W.Y.; Li, L.D.; Zhao, Y.; Chen, M.T.; Jin, W. High expression of COX5B is associated with poor prognosis in breast cancer. Future Oncol. 2017, 13, 1711–1719. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lin, W.R.; Lin, Y.H.; Kuo, W.H.; Tseng, C.J.; Lim, S.N.; Huang, Y.L.; Huang, S.C.; Wu, T.J.; Lin, K.H.; et al. COX5B-mediated bioenergetic alteration regulates tumor growth and migration by modulating AMPK-UHMK1-ERK cascade in hepatoma. Cancers 2020, 12, 1646. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2011, 1807, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, A.P.; Kapur, A.; Barroilhet, L.; Patankar, M.S. Oxidative phosphorylation: A target for novel therapeutic strategies against ovarian cancer. Cancers 2018, 10, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zirlik, K.; Duyster, J. Anti-angiogenics: Current situation and future perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | Description | Gene Count | p-Value |
|---|---|---|---|---|
| GO: BP | GO:0050896 | Response to stimulus | 67 | 2.9E−2 |
| GO:0009605 | Intracellular signal transduction | 25 | 7.1E−2 | |
| GO:0042127 | Regulation of cell proliferation | 19 | 1.3E−1 | |
| GO:0007155 | Cell adhesion | 17 | 9.5E−2 | |
| GO:0016477 | Cell migration | 14 | 5.2E−2 | |
| GO:CC | GO:0005886 | Plasma membrane | 46 | 1.7E−2 |
| GO:0071944 | Cell periphery | 46 | 2.5E−2 | |
| GO:0005576 | Extracellular region | 41 | 3.1E−2 | |
| GO:0042995 | Extracellular space | 20 | 2.8E−3 | |
| GO:0098862 | Membrane region | 6 | 8.5E−2 | |
| GO:MF | GO:0060089 | Molecular transducer activity | 18 | 3.8E−2 |
| GO:0038023 | Signaling receptor activity | 18 | 3.8E−2 | |
| GO:0004888 | Transmembrane signaling receptor activity | 15 | 5.2E−2 | |
| GO:0003982 | Lipid binding | 10 | 2.5E−2 | |
| GO:0001228 | Protein tyrosine kinase binding | 3 | 5.5E−2 |
| Pathway | Term | Description | Gene Count | False Discovery Rate |
|---|---|---|---|---|
| KEGG Pathway | hsa01100 | Metabolic pathways | 36 | 5.40e−08 |
| hsa00190 | Oxidative phosphorylation | 24 | 6.13e−21 | |
| hsa05200 | Pathways in cancer | 21 | 5.83e−07 | |
| hsa01524 | Platinum drug resistance | 11 | 5.45e−09 | |
| hsa01521 | EGFR tyrosine kinase inhibitor resistance | 9 | 1.75e−06 | |
| Reactome Pathway | HSA-1428517 | TCA Cycle and Respiratory electron transport | 21 | 7.61e−16 |
| HSA-109581 | Apoptosis | 14 | 1.62e−08 | |
| HSA-109582 | Hemostasis | 14 | 1.67e−02 | |
| HSA-1268020 | Mitochondrial protein import | 4 | 2.76e−02 | |
| HSA-1059683 | Interleukin-6 signaling | 2 | 3.69e−02 |
| Genes. | Nodes | Function | References |
|---|---|---|---|
| CYC5 | Hub and Bottleneck | Increased oxidative phosphorylation and pro-survival cellular events | Huttemann et al., 2011 |
| VEGFA | Hub and Bottleneck | Overexpression in ovarian cancer patients; tumor angiogenesis, associated with distant metastasis and resistance to chemotherapy | Guan et al., 2019; Sopo et al., 2019; Li et al., 2020 |
| IL6 | Hub and Bottleneck | Ovarian Cancer Growth, stemness, and therapy resistance | Wang et al., 2018; Azar et al., 2020 |
| UQCRFS1 | Hub | Oncogenic reprogramming of metabolism role in Pancreatic cancer | Kaneko et al., 2003; Ohashi et al., 2004; Owens et al., 2011; Jun et al., 2012 |
| UQCRC1 | Hub | Oncogenic reprogramming of metabolism role in Pancreatic cancer | Wang et al., 2020 |
| COX5B | Hub | COX5B-mediated metabolic reprogramming is Associated with poor prognosis in many cancers | Gao et al., 2017; Chu et al., 2020 |
| ACKR3/CXCR7 | Bottleneck | Chemokine receptor activated in many cancers to promote invasive metastasis. | Neves et al., 2019; Smit et al., 2020 |
| FYN | Bottleneck | Mediates oncogenic cell proliferation, migration, EMT, and therapy resistance in many cancers. | Saito et al., 2010; Lee et al., 2018; Yu et al., 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, J.H.; Jayaraman, M.; Yan, M.; Dhanasekaran, P.; Isidoro, C.; Song, Y.S.; Dhanasekaran, D.N. GNAi2/gip2-Regulated Transcriptome and Its Therapeutic Significance in Ovarian Cancer. Biomolecules 2021, 11, 1211. https://doi.org/10.3390/biom11081211
Ha JH, Jayaraman M, Yan M, Dhanasekaran P, Isidoro C, Song YS, Dhanasekaran DN. GNAi2/gip2-Regulated Transcriptome and Its Therapeutic Significance in Ovarian Cancer. Biomolecules. 2021; 11(8):1211. https://doi.org/10.3390/biom11081211
Chicago/Turabian StyleHa, Ji Hee, Muralidharan Jayaraman, Mingda Yan, Padmaja Dhanasekaran, Ciro Isidoro, Yong Sang Song, and Danny N. Dhanasekaran. 2021. "GNAi2/gip2-Regulated Transcriptome and Its Therapeutic Significance in Ovarian Cancer" Biomolecules 11, no. 8: 1211. https://doi.org/10.3390/biom11081211