
The Bigger Picture: Why Oral Mucosa Heals Better Than Skin

 , , ,
, , , 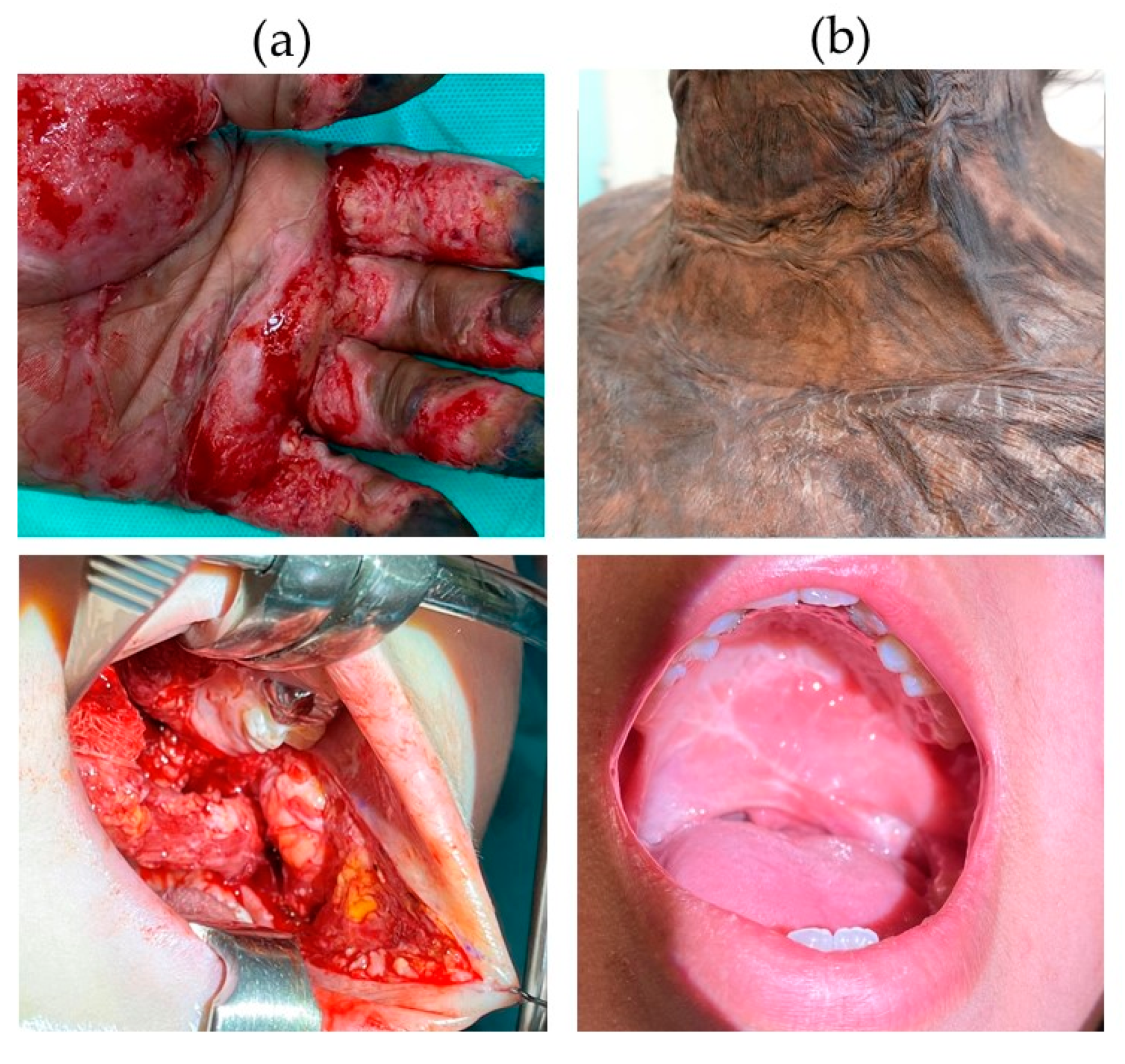{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
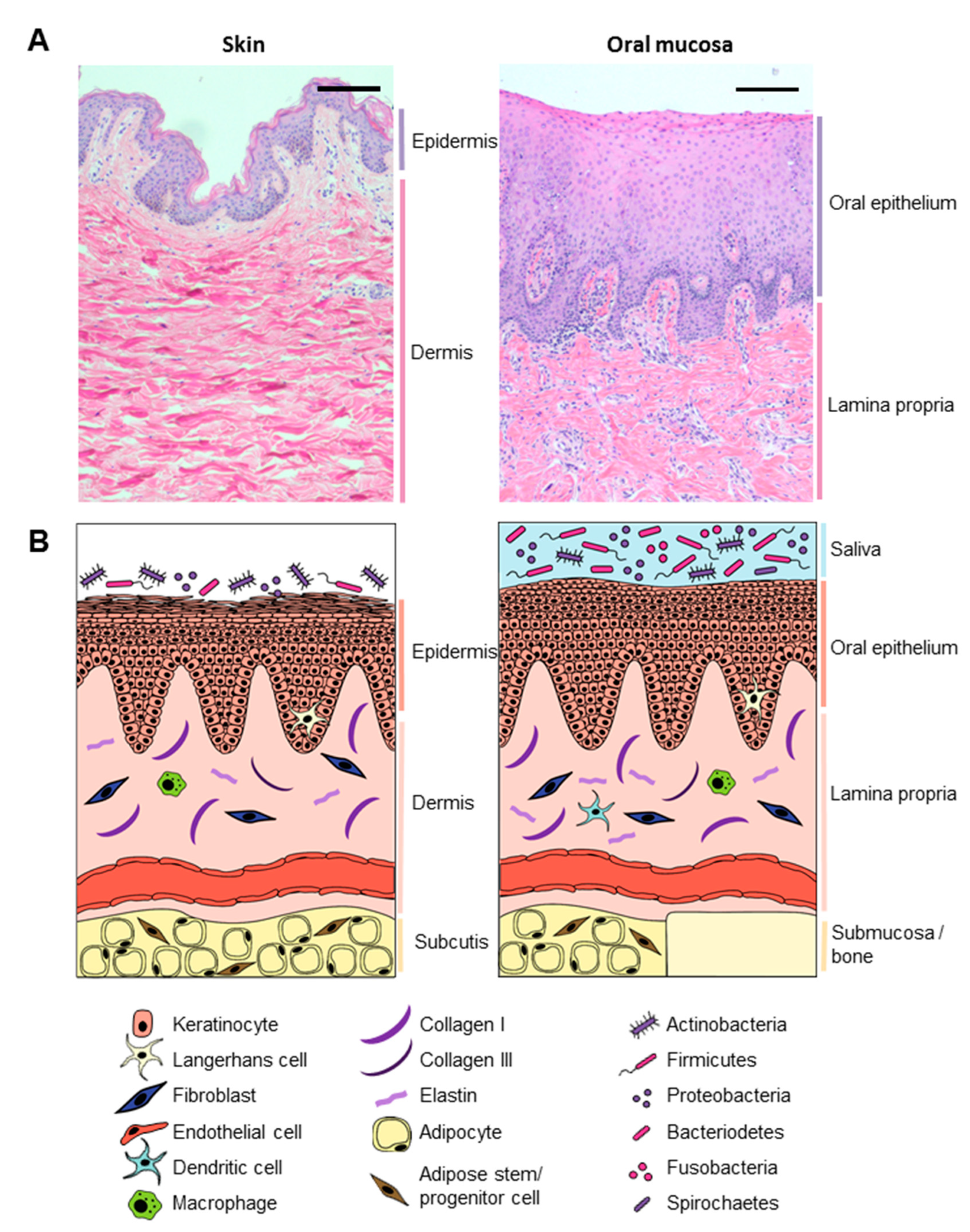
2. The Intrinsic Differences between Skin and Oral Mucosa
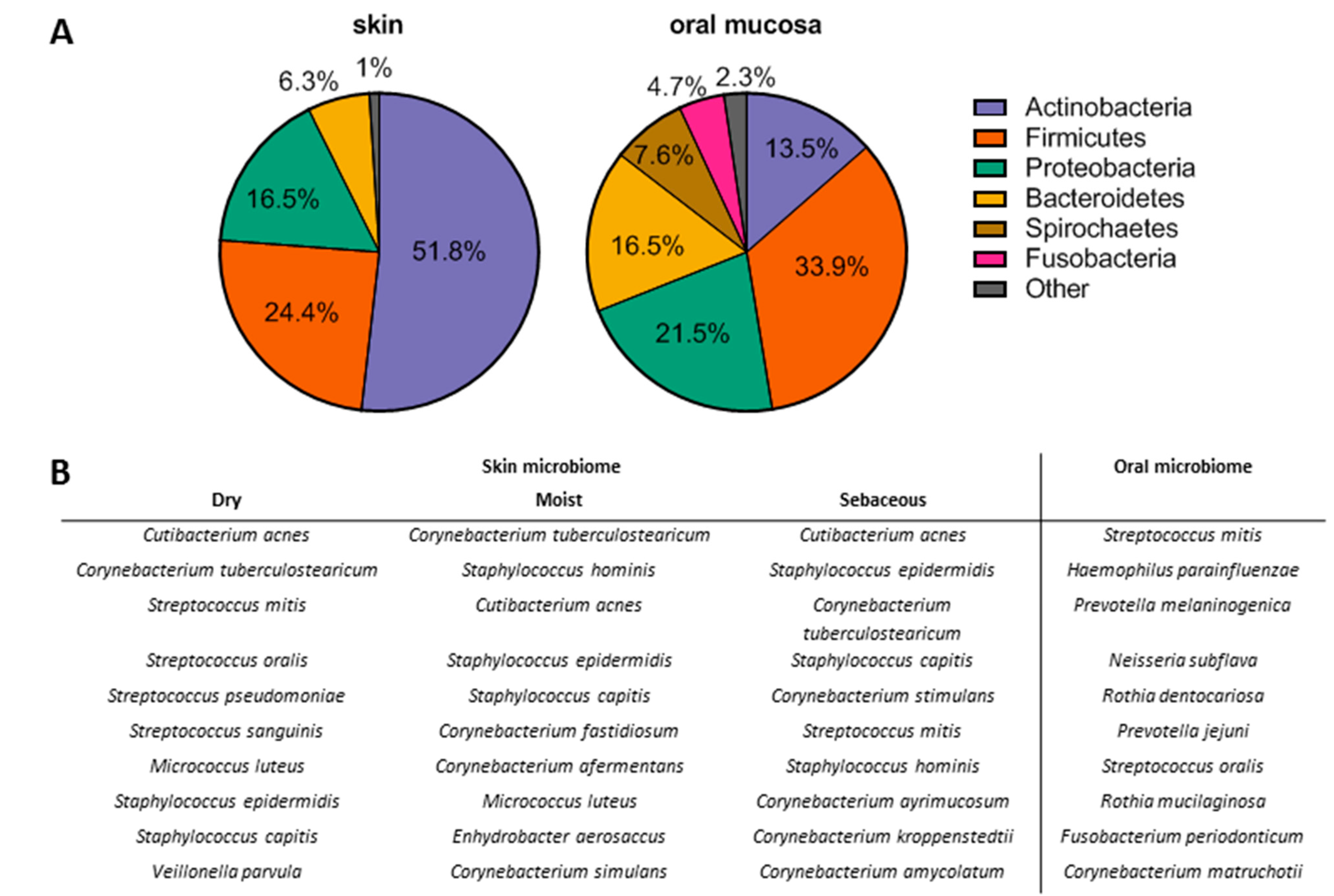
The Microenvironment, Microbiome, and Saliva
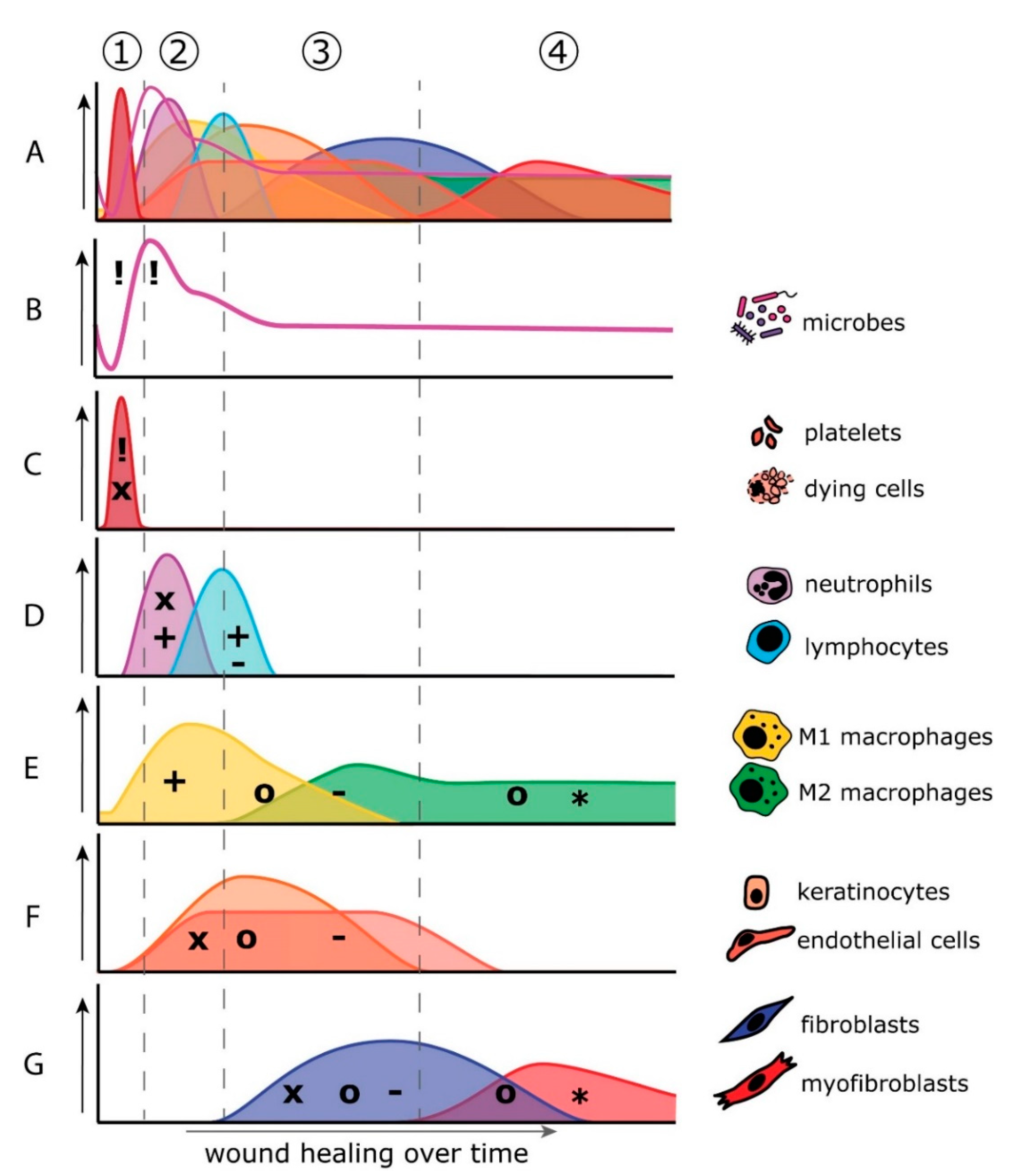
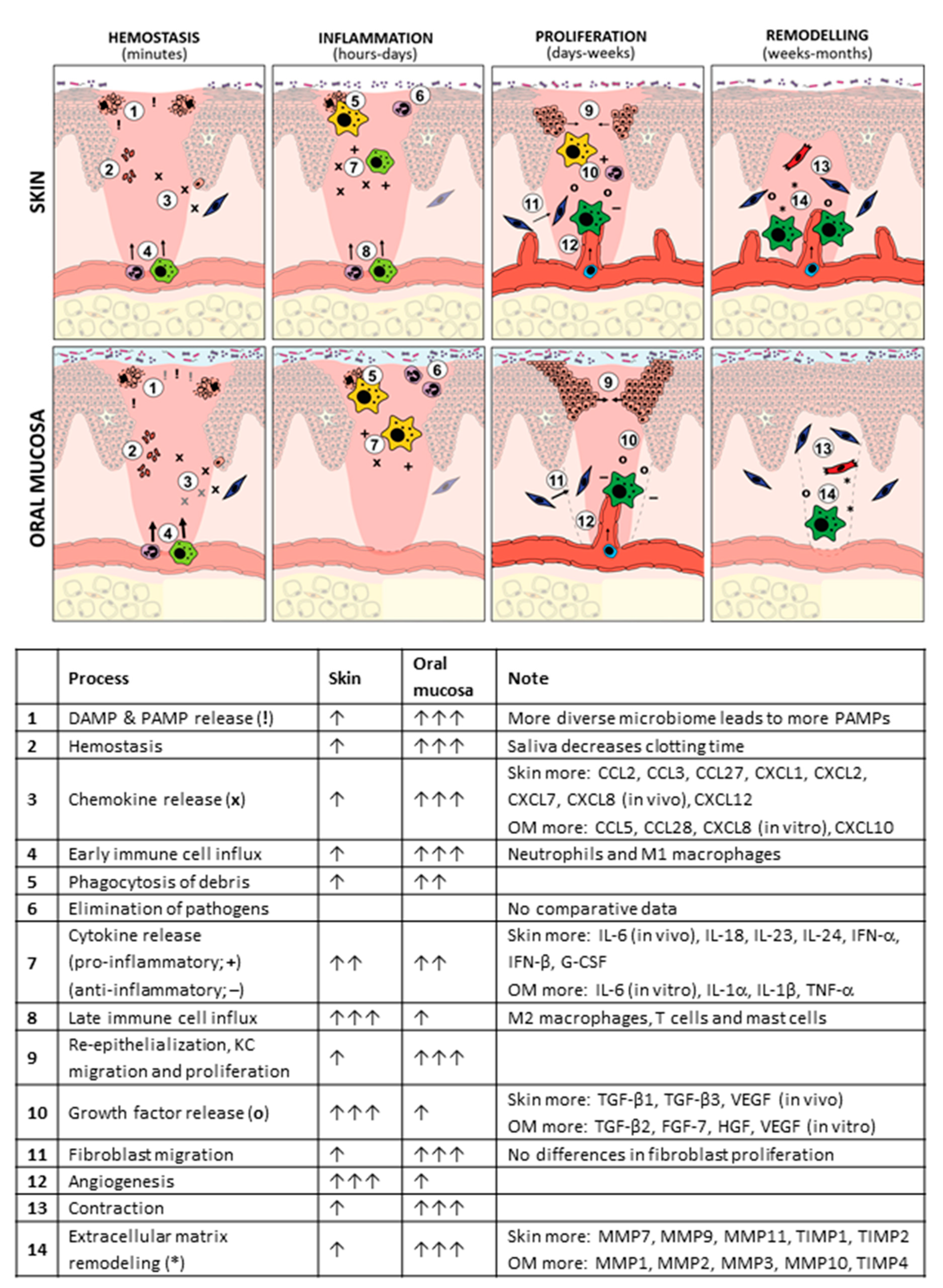
3. Differences between Skin and Oral Mucosa Wound Healing
3.1. Hemostasis Phase
3.2. Inflammation Phase
3.2.1. Phagocytosis
3.2.2. Neutrophil Infiltration
3.2.3. Macrophage Infiltration
3.2.4. T Cells
3.2.5. Mast Cells
3.2.6. Cytokines
3.3. Proliferation Phase
3.3.1. Angiogenesis
3.3.2. Re-Epithelialization
3.3.3. Granulation Tissue Formation
3.4. Remodeling Phase
4. The Bigger Picture: When Intrinsic, Local, Systemic, and External Factors Come Together
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Chiang, R.S.; Borovikova, A.A.; King, K.; Banyard, D.A.; Lalezari, S.; Toranto, J.D.; Paydar, K.Z.; Wirth, G.A.; Evans, G.R.; Widgerow, A.D. Current concepts related to hypertrophic scarring in burn injuries. Wound Repair Regen. 2016, 24, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Finnerty, C.C.; Jeschke, M.G.; Branski, L.K.; Barret, J.P.; Dziewulski, P.; Herndon, D.N. Hypertrophic scarring: The greatest unmet challenge after burn injury. Lancet 2016, 388, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Van den Broek, L.J.; van der Veer, W.M.; de Jong, E.H.; Gibbs, S.; Niessen, F.B. Suppressed inflammatory gene expression during human hypertrophic scar compared to normotrophic scar formation. Exp. Dermatol. 2015, 24, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, W.M.; Niessen, F.B.; Ferreira, J.A.; Zwiers, P.J.; de Jong, E.H.; Middelkoop, E.; Molema, G. Time course of the angiogenic response during normotrophic and hypertrophic scar formation in humans. Wound Repair Regen. 2011, 19, 292–301. [Google Scholar] [CrossRef]
- Linehan, J.L.; Harrison, O.J.; Han, S.J.; Byrd, A.L.; Vujkovic-Cvijin, I.; Villarino, A.V.; Sen, S.K.; Shaik, J.; Smelkinson, M.; Tamoutounour, S.; et al. Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 2018, 172, 784–796.e718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkaid, Y.; Segre, J.A. Dialogue between skin microbiota and immunity. Science 2014, 346, 954–959. [Google Scholar] [CrossRef]
- Lai, Y.; Di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.R.; Hooper, L.V.; Schmidt, R.R.; von Aulock, S.; et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Kido, D.; Mizutani, K.; Takeda, K.; Mikami, R.; Matsuura, T.; Iwasaki, K.; Izumi, Y. Impact of diabetes on gingival wound healing via oxidative stress. PLoS ONE 2017, 12, e0189601. [Google Scholar] [CrossRef]
- Evans, E.W. Treating Scars on the Oral Mucosa. Facial Plast. Surg. Clin. N. Am. 2017, 25, 89–97. [Google Scholar] [CrossRef]
- Semlali, A.; Chakir, J.; Goulet, J.P.; Chmielewski, W.; Rouabhia, M. Whole cigarette smoke promotes human gingival epithelial cell apoptosis and inhibits cell repair processes. J. Periodontal Res. 2011, 46, 533–541. [Google Scholar] [CrossRef]
- Presland, R.B.; Jurevic, R.J. Making sense of the epithelial barrier: What molecular biology and genetics tell us about the functions of oral mucosal and epidermal tissues. J. Dent. Educ. 2002, 66, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Steel, A.; Fazel, N. Oral mucosa biology and salivary biomarkers. Clin. Dermatol. 2017, 35, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, S.; Ponec, M. Intrinsic regulation of differentiation markers in human epidermis, hard palate and buccal mucosa. Arch. Oral Biol. 2000, 45, 149–158. [Google Scholar] [CrossRef]
- Turabelidze, A.; Guo, S.; Chung, A.Y.; Chen, L.; Dai, Y.; Marucha, P.T.; DiPietro, L.A. Intrinsic differences between oral and skin keratinocytes. PLoS ONE 2014, 9, e101480. [Google Scholar] [CrossRef]
- Winning, T.A.; Townsend, G.C. Oral mucosal embryology and histology. Clin. Dermatol. 2000, 18, 499–511. [Google Scholar] [CrossRef]
- Glim, J.E.; Beelen, R.H.; Niessen, F.B.; Everts, V.; Ulrich, M.M. The number of immune cells is lower in healthy oral mucosa compared to skin and does not increase after scarring. Arch. Oral Biol. 2015, 60, 272–281. [Google Scholar] [CrossRef]
- Szpaderska, A.M.; Walsh, C.G.; Steinberg, M.J.; DiPietro, L.A. Distinct patterns of angiogenesis in oral and skin wounds. J. Dent. Res. 2005, 84, 309–314. [Google Scholar] [CrossRef]
- Glim, J.E.; Everts, V.; Niessen, F.B.; Ulrich, M.M.; Beelen, R.H. Extracellular matrix components of oral mucosa differ from skin and resemble that of foetal skin. Arch. Oral Biol. 2014, 59, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.; Manji, A.; Gallant-Behm, C.; Wiebe, C.; Hart, D.A.; Larjava, H.; Hakkinen, L. Scarless healing of oral mucosa is characterized by faster resolution of inflammation and control of myofibroblast action compared to skin wounds in the red Duroc pig model. J. Dermatol. Sci. 2009, 56, 168–180. [Google Scholar] [CrossRef]
- Hsieh, P.C.; Jin, Y.T.; Chang, C.W.; Huang, C.C.; Liao, S.C.; Yuan, K. Elastin in oral connective tissue modulates the keratinization of overlying epithelium. J. Clin. Periodontol. 2010, 37, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Dyson, M.; Young, S.R.; Hart, J.; Lynch, J.A.; Lang, S. Comparison of the effects of moist and dry conditions on the process of angiogenesis during dermal repair. J. Investig. Dermatol. 1992, 99, 729–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junker, J.P.; Kamel, R.A.; Caterson, E.J.; Eriksson, E. Clinical Impact upon Wound Healing and Inflammation in Moist, Wet, and Dry Environments. Adv. Wound Care 2013, 2, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Svensjo, T.; Pomahac, B.; Yao, F.; Slama, J.; Eriksson, E. Accelerated healing of full-thickness skin wounds in a wet environment. Plast. Reconstr. Surg. 2000, 106, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.M.; Andree, C.; Breuing, K.; Liu, P.Y.; Slama, J.; Helo, G.; Eriksson, E. Dry, moist, and wet skin wound repair. Ann. Plast. Surg. 1995, 34, 493–499. [Google Scholar] [CrossRef]
- Brand, H.S.; Ligtenberg, A.J.; Veerman, E.C. Saliva and wound healing. Monogr. Oral Sci. 2014, 24, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Cheng, L.; Lin, H.; Pang, M.; Yao, Z.; Chen, C.; Forouzanfar, T.; Bikker, F.J.; Wu, G.; Cheng, B. Human Salivary Histatin-1 Is More Efficacious in Promoting Acute Skin Wound Healing Than Acellular Dermal Matrix Paste. Front. Bioeng. Biotechnol. 2020, 8, 999. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Sun, W.; Nazmi, K.; Veerman, E.C.I.; Bikker, F.J.; Jaspers, R.T.; Bolscher, J.G.M.; Wu, G. Salivary Histatin 1 and 2 Are Targeted to Mitochondria and Endoplasmic Reticulum in Human Cells. Cells 2020, 9, 795. [Google Scholar] [CrossRef] [Green Version]
- Bodner, L.; Knyszynski, A.; Adler-Kunin, S.; Danon, D. The effect of selective desalivation on wound healing in mice. Exp. Gerontol. 1991, 26, 357–363. [Google Scholar] [CrossRef]
- Bodner, L.; Dayan, D.; Pinto, Y.; Hammel, I. Characteristics of palatal wound healing in desalivated rats. Arch. Oral Biol. 1993, 38, 17–21. [Google Scholar] [CrossRef]
- Bodner, L.; Kaffe, I.; Cohen, Z.; Dayan, D. Long-term effect of desalivation on extraction wound healing: A densitometric study in rats. Dentomaxillofac. Radiol. 1993, 22, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Fabbri, C.; D’Accolti, M.; Soffritti, I.; Bassi, C.; Mazzacane, S.; Franchi, M. Defining the oral microbiome by whole-genome sequencing and resistome analysis: The complexity of the healthy picture. BMC Microbiol. 2020, 20, 120. [Google Scholar] [CrossRef]
- Deo, P.N.; Deshmukh, R. Oral microbiome: Unveiling the fundamentals. J. Oral Maxillofac. Pathol. 2019, 23, 122–128. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef]
- Krishnan, K.; Chen, T.; Paster, B.J. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. 2017, 23, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Deng, D.; Buskermolen, J.K.; Janus, M.M.; Krom, B.P.; Roffel, S.; Waaijman, T.; van Loveren, C.; Crielaard, W.; Gibbs, S. Multi-species oral biofilm promotes reconstructed human gingiva epithelial barrier function. Sci. Rep. 2018, 8, 16061. [Google Scholar] [CrossRef] [Green Version]
- Laheij, A.M.; de Soet, J.J.; Veerman, E.C.; Bolscher, J.G.; van Loveren, C. The influence of oral bacteria on epithelial cell migration in vitro. Mediators Inflamm. 2013, 2013, 154532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn wound infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van’t Hof, W.; Veerman, E.C.; Nieuw Amerongen, A.V.; Ligtenberg, A.J. Antimicrobial defense systems in saliva. Monogr. Oral Sci. 2014, 24, 40–51. [Google Scholar] [CrossRef]
- Amerongen, A.V.; Veerman, E.C. Saliva—The defender of the oral cavity. Oral Dis. 2002, 8, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Bikker, F.J.; End, C.; Ligtenberg, A.J.M.; Blaich, S.; Lyer, S.; Renner, M.; Wittig, R.; Nazmi, K.; van Nieuw Amerongen, A.; Poustka, A.; et al. The scavenging capacity of DMBT1 is impaired by germline deletions. Immunogenetics 2017, 69, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenink, J.; Ligtenberg, A.J.; Veerman, E.C.; Bolscher, J.G.; Nieuw Amerongen, A.V. Interaction of the salivary low-molecular-weight mucin (MG2) with Actinobacillus actinomycetemcomitans. Antonie Leeuwenhoek 1996, 70, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, T.J.; Bikker, F.J.; Groenink, J.; Tornoe, I.; Leth-Larsen, R.; Veerman, E.C.; Nieuw Amerongen, A.V.; Holmskov, U. Human salivary agglutinin binds to lung surfactant protein-D and is identical with scavenger receptor protein gp-340. Biochem. J. 2001, 359, 243–248. [Google Scholar] [CrossRef]
- Murray, P.A.; Prakobphol, A.; Lee, T.; Hoover, C.I.; Fisher, S.J. Adherence of oral streptococci to salivary glycoproteins. Infect. Immun. 1992, 60, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, P.D.; Do, T.; Beighton, D.; Devine, D.A. Influence of saliva on the oral microbiota. Periodontol. 2000 2016, 70, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Molhoek, E.M.; den Hertog, A.L.; de Vries, A.M.; Nazmi, K.; Veerman, E.C.; Hartgers, F.C.; Yazdanbakhsh, M.; Bikker, F.J.; van der Kleij, D. Structure-function relationship of the human antimicrobial peptide LL-37 and LL-37 fragments in the modulation of TLR responses. Biol. Chem. 2009, 390, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Krom, B.P.; Oskam, J. Microbial biofilms and wound healing: An ecological hypothesis. Phlebology 2014, 29, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Abusleme, L.; Hoare, A.; Hong, B.Y.; Diaz, P.I. Microbial signatures of health, gingivitis, and periodontitis. Periodontology 2000 2021, 86, 57–78. [Google Scholar] [CrossRef]
- Glazko, A.J.; Greenberg, D.M. The mechanism of the action of saliva in blood coagulation. Am. J. Physiol. 1938, 125, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Berckmans, R.J.; Sturk, A.; van Tienen, L.M.; Schaap, M.C.; Nieuwland, R. Cell-derived vesicles exposing coagulant tissue factor in saliva. Blood 2011, 117, 3172–3180. [Google Scholar] [CrossRef]
- Ridiandries, A.; Tan, J.T.M.; Bursill, C.A. The Role of Chemokines in Wound Healing. Int. J. Mol. Sci. 2018, 19, 3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger, F.; Patzelt, J.; Langer, H.F. The Platelet Response to Tissue Injury. Front. Med. 2018, 5, 317. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Fu, J.; Shao, K.; Wang, L.; Lan, X.; Shi, J. Biomimetic hydrogel for rapid and scar-free healing of skin wounds inspired by the healing process of oral mucosa. Acta Biomater. 2019, 100, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A. Inflammation as an orchestrator of cutaneous scar formation: A review of the literature. Plast. Aesthet. Res. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Farthing, P.M.; Ireland, G.W.; Thornhill, M.H. IL-1 alpha and IL-6 production by oral and skin keratinocytes: Similarities and differences in response to cytokine treatment in vitro. J. Oral Pathol. Med. 1996, 25, 157–162. [Google Scholar] [CrossRef]
- Butzelaar, L.; Schooneman, D.P.; Soykan, E.A.; Talhout, W.; Ulrich, M.M.; van den Broek, L.J.; Gibbs, S.; Beelen, R.H.; Mink van der Molen, A.B.; Niessen, F.B. Inhibited early immunologic response is associated with hypertrophic scarring. Exp. Dermatol. 2016, 25, 797–804. [Google Scholar] [CrossRef] [PubMed]
- De Bakker, E.; van der Putten, M.A.M.; Heymans, M.W.; Spiekstra, S.W.; Waaijman, T.; Butzelaar, L.; Negenborn, V.L.; Beekman, V.K.; Akpinar, E.O.; Rustemeyer, T.; et al. Prognostic tools for hypertrophic scar formation based on fundamental differences in systemic immunity. Exp. Dermatol. 2021, 30, 169–178. [Google Scholar] [CrossRef]
- Bootun, R. Effects of immunosuppressive therapy on wound healing. Int. Wound J. 2013, 10, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.; Sheoran, S.; Maruyama, K.; Martino, M.M. Immune Regulation of Skin Wound Healing: Mechanisms and Novel Therapeutic Targets. Adv. Wound Care 2018, 7, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.A.; Zhang, L.J.; Williams, M.R.; Gangoiti, J.A.; Huang, C.M.; Gallo, R.L. Inhibition of HDAC8 and HDAC9 by microbial short-chain fatty acids breaks immune tolerance of the epidermis to TLR ligands. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Sciubba, J.J.; Waterhouse, J.P.; Meyer, J. A fine structural comparison of the healing of incisional wounds of mucosa and skin. J. Oral Pathol. 1978, 7, 214–227. [Google Scholar] [CrossRef]
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Szpaderska, A.M.; Zuckerman, J.D.; DiPietro, L.A. Differential injury responses in oral mucosal and cutaneous wounds. J. Dent. Res. 2003, 82, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Breger, J.; Baeva, L.; Agrawal, A.; Shindell, E.; Godar, D.E. UVB-induced inflammatory cytokine release, DNA damage and apoptosis of human oral compared with skin tissue equivalents. Photochem. Photobiol. 2013, 89, 665–670. [Google Scholar] [CrossRef]
- Kosten, I.J.; Buskermolen, J.K.; Spiekstra, S.W.; de Gruijl, T.D.; Gibbs, S. Gingiva Equivalents Secrete Negligible Amounts of Key Chemokines Involved in Langerhans Cell Migration Compared to Skin Equivalents. J. Immunol. Res. 2015, 2015, 627125. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ireland, G.W.; Farthing, P.M.; Thornhill, M.H. Epidermal and oral keratinocytes are induced to produce RANTES and IL-8 by cytokine stimulation. J. Investig. Dermatol. 1996, 106, 661–666. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Arbieva, Z.H.; Guo, S.; Marucha, P.T.; Mustoe, T.A.; DiPietro, L.A. Positional differences in the wound transcriptome of skin and oral mucosa. BMC Genom. 2010, 11, 471. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mahiouz, D.L.; Farthing, P.M.; Haskard, D.O.; Thornhill, M.H. Heterogeneity of ICAM-1 expression, and cytokine regulation of ICAM-1 expression, in skin and oral keratinocytes. J. Oral Pathol. Med. 1996, 25, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef]
- Li, J.; Farthing, P.M.; Thornhill, M.H. Oral and skin keratinocytes are stimulated to secrete monocyte chemoattractant protein-1 by tumour necrosis factor-alpha and interferon-gamma. J. Oral Pathol. Med. 2000, 29, 438–444. [Google Scholar] [CrossRef]
- Boothby, I.C.; Cohen, J.N.; Rosenblum, M.D. Regulatory T cells in skin injury: At the crossroads of tolerance and tissue repair. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, A.S.; Mansbridge, J.N. The Innate Immune System in Acute and Chronic Wounds. Adv. Wound Care 2016, 5, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strbo, N.; Yin, N.; Stojadinovic, O. Innate and Adaptive Immune Responses in Wound Epithelialization. Adv. Wound Care 2014, 3, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazza, M.; Mor, A. Consider the chemokines: A review of the interplay between chemokines and T cell subset function. Discov. Med. 2017, 24, 31–39. [Google Scholar] [PubMed]
- Viola, A.; Contento, R.L.; Molon, B. T cells and their partners: The chemokine dating agency. Trends Immunol. 2006, 27, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Mah, W.; Jiang, G.; Olver, D.; Cheung, G.; Kim, B.; Larjava, H.; Hakkinen, L. Human gingival fibroblasts display a non-fibrotic phenotype distinct from skin fibroblasts in three-dimensional cultures. PLoS ONE 2014, 9, e90715. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Bouladoux, N.; Wilhelm, C.; Molloy, M.J.; Salcedo, R.; Kastenmuller, W.; Deming, C.; Quinones, M.; Koo, L.; Conlan, S.; et al. Compartmentalized control of skin immunity by resident commensals. Science 2012, 337, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Bouladoux, N.; Linehan, J.L.; Han, S.J.; Harrison, O.J.; Wilhelm, C.; Conlan, S.; Himmelfarb, S.; Byrd, A.L.; Deming, C.; et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015, 520, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.F. The role of mast cells in wound healing. Int. Wound J. 2010, 7, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Do, D.V.; Ong, C.T.; Khoo, Y.T.; Carbone, A.; Lim, C.P.; Wang, S.; Mukhopadhyay, A.; Cao, X.; Cho, D.H.; Wei, X.Q.; et al. Interleukin-18 system plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. Br. J. Dermatol. 2012, 166, 1275–1288. [Google Scholar] [CrossRef]
- Lee, J.; Rodero, M.P.; Patel, J.; Moi, D.; Mazzieri, R.; Khosrotehrani, K. Interleukin-23 regulates interleukin-17 expression in wounds, and its inhibition accelerates diabetic wound healing through the alteration of macrophage polarization. FASEB J. 2018, 32, 2086–2094. [Google Scholar] [CrossRef] [Green Version]
- Matias, M.A.; Saunus, J.M.; Ivanovski, S.; Walsh, L.J.; Farah, C.S. Accelerated wound healing phenotype in Interleukin 12/23 deficient mice. J. Inflamm. 2011, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, W.; Jinnin, M.; Tomizawa, Y.; Nakamura, K.; Kudo, H.; Inoue, K.; Makino, K.; Honda, N.; Kajihara, I.; Fukushima, S.; et al. Dysregulated interleukin-23 signalling contributes to the increased collagen production in scleroderma fibroblasts via balancing microRNA expression. Rheumatology 2017, 56, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Poindexter, N.J.; Williams, R.R.; Powis, G.; Jen, E.; Caudle, A.S.; Chada, S.; Grimm, E.A. IL-24 is expressed during wound repair and inhibits TGFalpha-induced migration and proliferation of keratinocytes. Exp. Dermatol. 2010, 19, 714–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, L.Z.; Wang, Y.; Zhang, L.; Wu, G.; Zhang, L.; Wang, F.X.; Chen, L.M.; Sun, F.; Jia, S.; Zhang, S.; et al. IL-24 deficiency protects mice against bleomycin-induced pulmonary fibrosis by repressing IL-4-induced M2 program in macrophages. Cell Death Differ. 2021, 28, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.J.; Gresser, I.; Thompson, W.D. Inhibition of wound healing in mice by local interferon alpha/beta injection. Int. J. Exp. Pathol. 1993, 74, 79–85. [Google Scholar]
- Wong, J.W.; Gallant-Behm, C.; Wiebe, C.; Mak, K.; Hart, D.A.; Larjava, H.; Hakkinen, L. Wound healing in oral mucosa results in reduced scar formation as compared with skin: Evidence from the red Duroc pig model and humans. Wound Repair Regen. 2009, 17, 717–729. [Google Scholar] [CrossRef]
- Schrementi, M.E.; Ferreira, A.M.; Zender, C.; Di Pietro, L.A. Site-specific production of TGF-beta in oral mucosal and cutaneous wounds. Wound Repair Regen. 2008, 16, 80–86. [Google Scholar] [CrossRef]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramirez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [Green Version]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Khan, A. Detection and quantitation of forty eight cytokines, chemokines, growth factors and nine acute phase proteins in healthy human plasma, saliva and urine. J. Proteom. 2012, 75, 4802–4819. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gajendrareddy, P.K.; DiPietro, L.A. Differential expression of HIF-1alpha in skin and mucosal wounds. J. Dent. Res. 2012, 91, 871–876. [Google Scholar] [CrossRef] [Green Version]
- Wietecha, M.S.; Cerny, W.L.; DiPietro, L.A. Mechanisms of vessel regression: Toward an understanding of the resolution of angiogenesis. Curr. Top. Microbiol. Immunol. 2013, 367, 3–32. [Google Scholar] [CrossRef]
- Xue, M.; Jackson, C.J. Extracellular Matrix Reorganization during Wound Healing and Its Impact on Abnormal Scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, M.A.; Paller, A.S. The Roles of Growth Factors in Keratinocyte Migration. Adv. Wound Care 2015, 4, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, D.B.; McKeown, S.T.; Lundy, F.T.; Irwin, C.R. Phenotypic differences between oral and skin fibroblasts in wound contraction and growth factor expression. Wound Repair Regen. 2006, 14, 172–178. [Google Scholar] [CrossRef]
- Jahovic, N.; Guzel, E.; Arbak, S.; Yegen, B.C. The healing-promoting effect of saliva on skin burn is mediated by epidermal growth factor (EGF): Role of the neutrophils. Burns 2004, 30, 531–538. [Google Scholar] [CrossRef]
- Oudhoff, M.J.; Bolscher, J.G.; Nazmi, K.; Kalay, H.; van’t Hof, W.; Amerongen, A.V.; Veerman, E.C. Histatins are the major wound-closure stimulating factors in human saliva as identified in a cell culture assay. FASEB J. 2008, 22, 3805–3812. [Google Scholar] [CrossRef] [Green Version]
- Zelles, T.; Purushotham, K.R.; Macauley, S.P.; Oxford, G.E.; Humphreys-Beher, M.G. Saliva and growth factors: The fountain of youth resides in us all. J. Dent. Res. 1995, 74, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Neves, C.; Buskermolen, J.; Roffel, S.; Waaijman, T.; Thon, M.; Veerman, E.; Gibbs, S. Human saliva stimulates skin and oral wound healing in vitro. J. Tissue Eng. Regen. Med. 2019, 13, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Oudhoff, M.J.; Kroeze, K.L.; Nazmi, K.; van den Keijbus, P.A.; van’t Hof, W.; Fernandez-Borja, M.; Hordijk, P.L.; Gibbs, S.; Bolscher, J.G.; Veerman, E.C. Structure-activity analysis of histatin, a potent wound healing peptide from human saliva: Cyclization of histatin potentiates molar activity 1000-fold. FASEB J. 2009, 23, 3928–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudhoff, M.J.; van den Keijbus, P.A.; Kroeze, K.L.; Nazmi, K.; Gibbs, S.; Bolscher, J.G.; Veerman, E.C. Histatins enhance wound closure with oral and non-oral cells. J. Dent. Res. 2009, 88, 846–850. [Google Scholar] [CrossRef]
- Haverman, T.M.; Laheij, A.; de Soet, J.J.; de Lange, J.; Rozema, F.R. Candida and Porphyromonas gingivalis: The effect on wound closure in vitro. J. Oral Microbiol. 2017, 9, 1328266. [Google Scholar] [CrossRef] [Green Version]
- De Ryck, T.; Vanlancker, E.; Grootaert, C.; Roman, B.I.; De Coen, L.M.; Vandenberghe, I.; Stevens, C.V.; Bracke, M.; Van de Wiele, T.; Vanhoecke, B. Microbial inhibition of oral epithelial wound recovery: Potential role for quorum sensing molecules? AMB Express 2015, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maquart, F.X.; Monboisse, J.C. Extracellular matrix and wound healing. Pathol. Biol. 2014, 62, 91–95. [Google Scholar] [CrossRef]
- Hara-Saito, Y.; Kato, H.; Saito, N.; Shiomi, A.; Uenoyama, A.; Takagi, R.; Izumi, K. Distinct differences in hypoxic responses between human oral mucosa and skin fibroblasts in a 3D collagen matrix. In Vitro Cell. Dev. Biol. Anim. 2020, 56, 452–479. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, P.; Montmasson, M.; Garnier, C. Extracellular matrix contribution to skin wound re-epithelialization. Matrix Biol. 2019, 75–76, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Lee, J.; Kim, E.H.; Shin, D. Plasticity of oral mucosal cell sheets for accelerated and scarless skin wound healing. Oral Oncol. 2017, 75, 81–88. [Google Scholar] [CrossRef]
- Boink, M.A.; van den Broek, L.J.; Roffel, S.; Nazmi, K.; Bolscher, J.G.; Gefen, A.; Veerman, E.C.; Gibbs, S. Different wound healing properties of dermis, adipose, and gingiva mesenchymal stromal cells. Wound Repair Regen. 2016, 24, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Lygoe, K.A.; Wall, I.; Stephens, P.; Lewis, M.P. Role of vitronectin and fibronectin receptors in oral mucosal and dermal myofibroblast differentiation. Biol. Cell 2007, 99, 601–614. [Google Scholar] [CrossRef]
- Stephens, P.; Davies, K.J.; al-Khateeb, T.; Shepherd, J.P.; Thomas, D.W. A comparison of the ability of intra-oral and extra-oral fibroblasts to stimulate extracellular matrix reorganization in a model of wound contraction. J. Dent. Res. 1996, 75, 1358–1364. [Google Scholar] [CrossRef]
- Van Wyk, C.W.; Olivier, A.; Hoal-van Helden, E.G.; Grobler-Rabie, A.F. Growth of oral and skin fibroblasts from patients with oral submucous fibrosis. J. Oral. Pathol. Med. 1995, 24, 349–353. [Google Scholar] [CrossRef]
- Palaiologou, A.A.; Yukna, R.A.; Moses, R.; Lallier, T.E. Gingival, dermal, and periodontal ligament fibroblasts express different extracellular matrix receptors. J. Periodontol. 2001, 72, 798–807. [Google Scholar] [CrossRef]
- Guo, F.; Carter, D.E.; Mukhopadhyay, A.; Leask, A. Gingival fibroblasts display reduced adhesion and spreading on extracellular matrix: A possible basis for scarless tissue repair? PLoS ONE 2011, 6, e27097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-beta family in wound healing, burns and scarring: A review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar]
- Eslami, A.; Gallant-Behm, C.L.; Hart, D.A.; Wiebe, C.; Honardoust, D.; Gardner, H.; Hakkinen, L.; Larjava, H.S. Expression of integrin alphavbeta6 and TGF-beta in scarless vs scar-forming wound healing. J. Histochem. Cytochem. 2009, 57, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.G.; Eun, H.C. Differences between fibroblasts cultured from oral mucosa and normal skin: Implication to wound healing. J. Dermatol. Sci. 1999, 21, 176–182. [Google Scholar] [CrossRef]
- Meran, S.; Luo, D.D.; Simpson, R.; Martin, J.; Wells, A.; Steadman, R.; Phillips, A.O. Hyaluronan facilitates transforming growth factor-beta1-dependent proliferation via CD44 and epidermal growth factor receptor interaction. J. Biol. Chem. 2011, 286, 17618–17630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aplin, A.C.; Zhu, W.H.; Fogel, E.; Nicosia, R.F. Vascular regression and survival are differentially regulated by MT1-MMP and TIMPs in the aortic ring model of angiogenesis. Am. J. Physiol. Cell Physiol. 2009, 297, C471–C480. [Google Scholar] [CrossRef] [Green Version]
- McKeown, S.T.; Barnes, J.J.; Hyland, P.L.; Lundy, F.T.; Fray, M.J.; Irwin, C.R. Matrix metalloproteinase-3 differences in oral and skin fibroblasts. J. Dent. Res. 2007, 86, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.; Davies, K.J.; Occleston, N.; Pleass, R.D.; Kon, C.; Daniels, J.; Khaw, P.T.; Thomas, D.W. Skin and oral fibroblasts exhibit phenotypic differences in extracellular matrix reorganization and matrix metalloproteinase activity. Br. J. Dermatol. 2001, 144, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.; Peake, M.A.; Wall, I.; Davies, L.; Farrier, J.; Giles, P.; Kipling, D.; Price, P.; Moseley, R.; Thomas, D.; et al. ’Young’ oral fibroblasts are geno/phenotypically distinct. J. Dent. Res. 2010, 89, 1407–1413. [Google Scholar] [CrossRef]
- Jansen, R.G.; van Kuppevelt, T.H.; Daamen, W.F.; Kuijpers-Jagtman, A.M.; Von den Hoff, J.W. Tissue reactions to collagen scaffolds in the oral mucosa and skin of rats: Environmental and mechanical factors. Arch. Oral Biol. 2008, 53, 376–387. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-beta Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Le, N.T.; Jackson, C.J. Targeting matrix metalloproteases to improve cutaneous wound healing. Expert Opin. Ther. Targets 2006, 10, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.G.; Parks, W.C. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015, 44–46, 113–121. [Google Scholar] [CrossRef]
- Chinnathambi, S.; Bickenbach, J.R. Human skin and gingival keratinocytes show differential regulation of matrix metalloproteinases when combined with fibroblasts in 3-dimensional cultures. J. Periodontol. 2005, 76, 1072–1083. [Google Scholar] [CrossRef]
- Caley, M.P.; Martins, V.L.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnaswamy, V.R.; Mintz, D.; Sagi, I. Matrix metalloproteinases: The sculptors of chronic cutaneous wounds. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2220–2227. [Google Scholar] [CrossRef]
- Letra, A.; Ghaneh, G.; Zhao, M.; Ray, H., Jr.; Francisconi, C.F.; Garlet, G.P.; Silva, R.M. MMP-7 and TIMP-1, new targets in predicting poor wound healing in apical periodontitis. J. Endod. 2013, 39, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Reiss, M.J.; Han, Y.P.; Garcia, E.; Goldberg, M.; Yu, H.; Garner, W.L. Matrix metalloproteinase-9 delays wound healing in a murine wound model. Surgery 2010, 147, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Kosten, I.J.; van de Ven, R.; Thon, M.; Gibbs, S.; de Gruijl, T.D. Comparative phenotypic and functional analysis of migratory dendritic cell subsets from human oral mucosa and skin. PLoS ONE 2017, 12, e0180333. [Google Scholar] [CrossRef] [PubMed]
- Buskermolen, J.K.; Janus, M.M.; Roffel, S.; Krom, B.P.; Gibbs, S. Saliva-Derived Commensal and Pathogenic Biofilms in a Human Gingiva Model. J. Dent. Res. 2018, 97, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Haverman, T.M.; Laheij, A.; Nie, M.; Deng, D.M.; Raber-Durlacher, J.E.; de Soet, J.J.; Rozema, F.R. Exploring the role of oral microorganisms in the pathogenesis of mucositis by assessing their impact on metabolic activity and reproductive capacity of epithelial cells in vitro. Support. Care Cancer 2020, 28, 4729–4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laheij, A.M.; de Soet, J.J.; von dem Borne, P.A.; Kuijper, E.J.; Kraneveld, E.A.; van Loveren, C.; Raber-Durlacher, J.E. Oral bacteria and yeasts in relationship to oral ulcerations in hematopoietic stem cell transplant recipients. Support. Care Cancer 2012, 20, 3231–3240. [Google Scholar] [CrossRef] [Green Version]
- Loomis, K.H.; Wu, S.K.; Ernlund, A.; Zudock, K.; Reno, A.; Blount, K.; Karig, D.K. A mixed community of skin microbiome representatives influences cutaneous processes more than individual members. Microbiome 2021, 9, 22. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Veer, W.M.; Bloemen, M.C.; Ulrich, M.M.; Molema, G.; van Zuijlen, P.P.; Middelkoop, E.; Niessen, F.B. Potential cellular and molecular causes of hypertrophic scar formation. Burns 2009, 35, 15–29. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waasdorp, M.; Krom, B.P.; Bikker, F.J.; van Zuijlen, P.P.M.; Niessen, F.B.; Gibbs, S. The Bigger Picture: Why Oral Mucosa Heals Better Than Skin. Biomolecules 2021, 11, 1165. https://doi.org/10.3390/biom11081165
Waasdorp M, Krom BP, Bikker FJ, van Zuijlen PPM, Niessen FB, Gibbs S. The Bigger Picture: Why Oral Mucosa Heals Better Than Skin. Biomolecules. 2021; 11(8):1165. https://doi.org/10.3390/biom11081165
Chicago/Turabian StyleWaasdorp, Maaike, Bastiaan P. Krom, Floris J. Bikker, Paul P. M. van Zuijlen, Frank B. Niessen, and Susan Gibbs. 2021. "The Bigger Picture: Why Oral Mucosa Heals Better Than Skin" Biomolecules 11, no. 8: 1165. https://doi.org/10.3390/biom11081165