
Ligands of Adrenergic Receptors: A Structural Point of View

Abstract
:1. Introduction
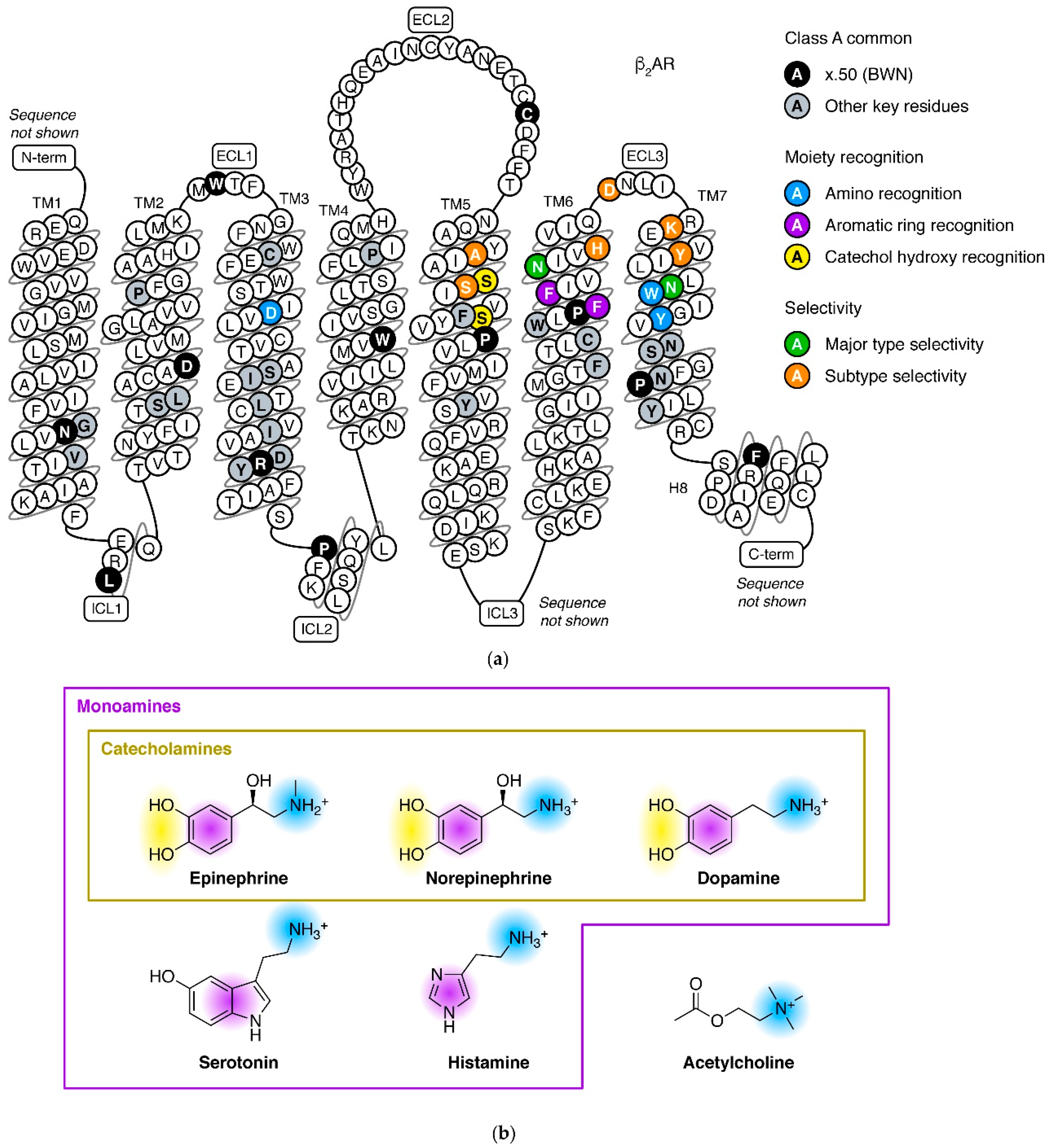
2. Adrenergic Receptors in the Aminergic Receptor Subfamily
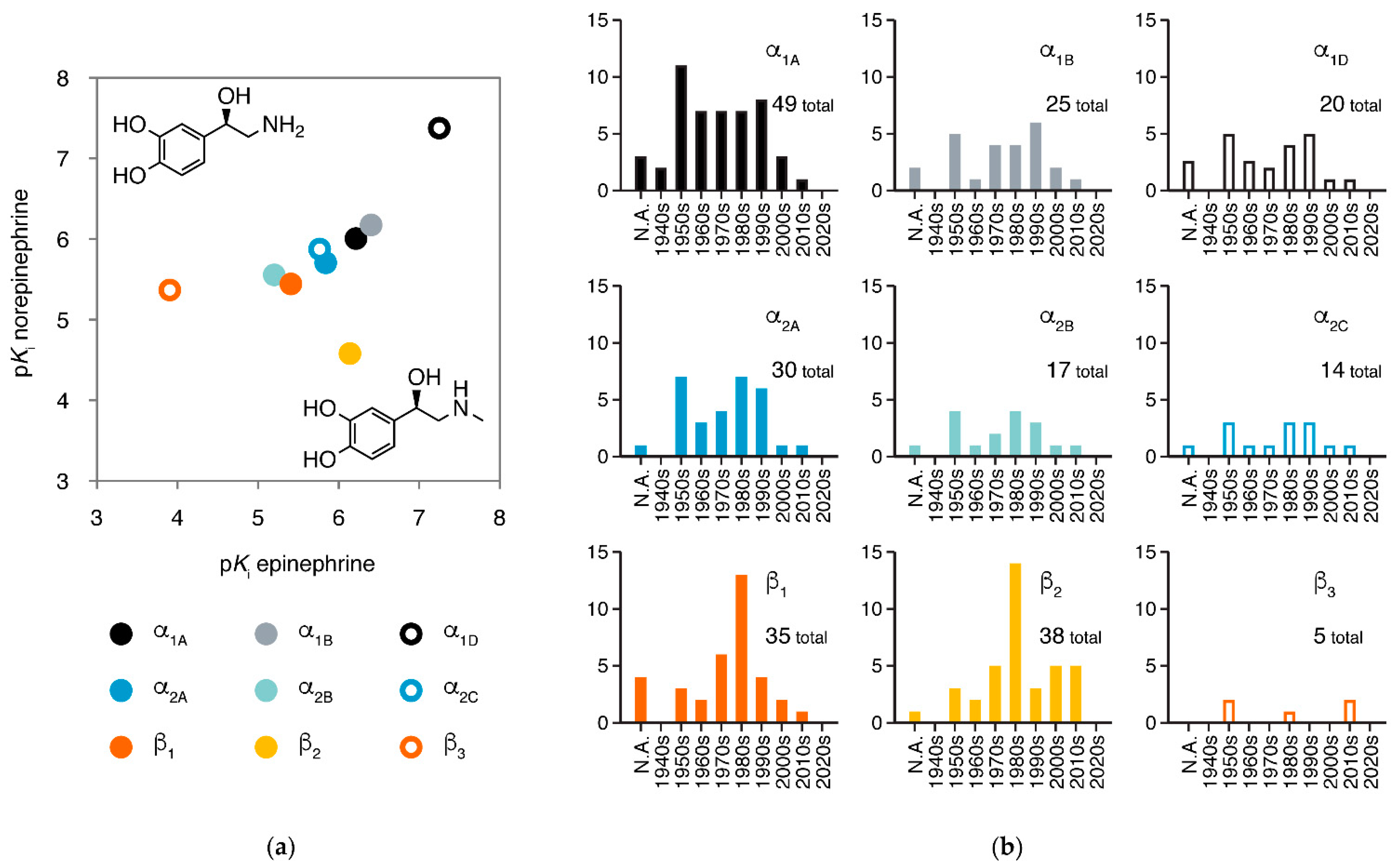
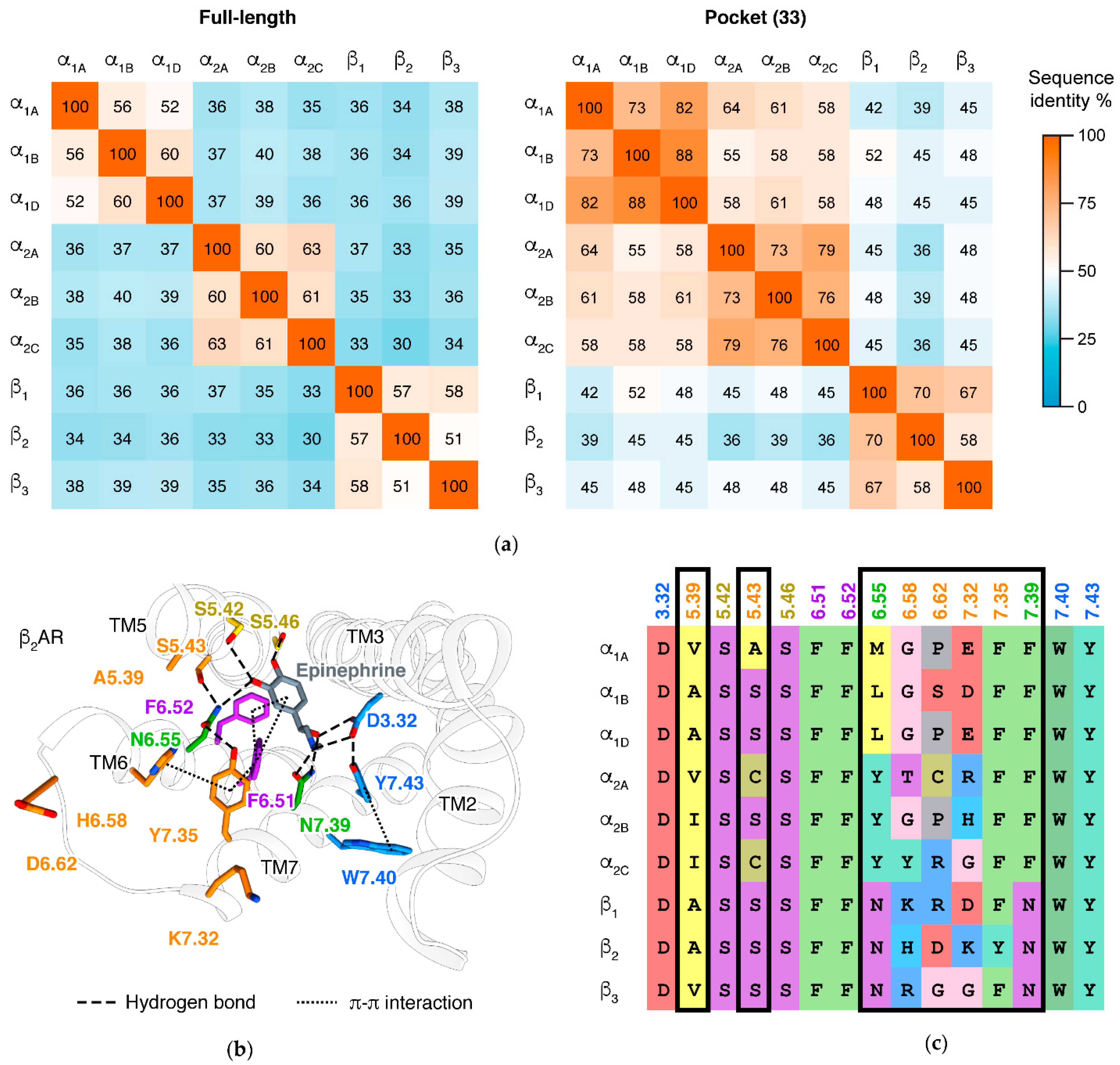
3. Major Types and Subtypes of Adrenergic Receptors
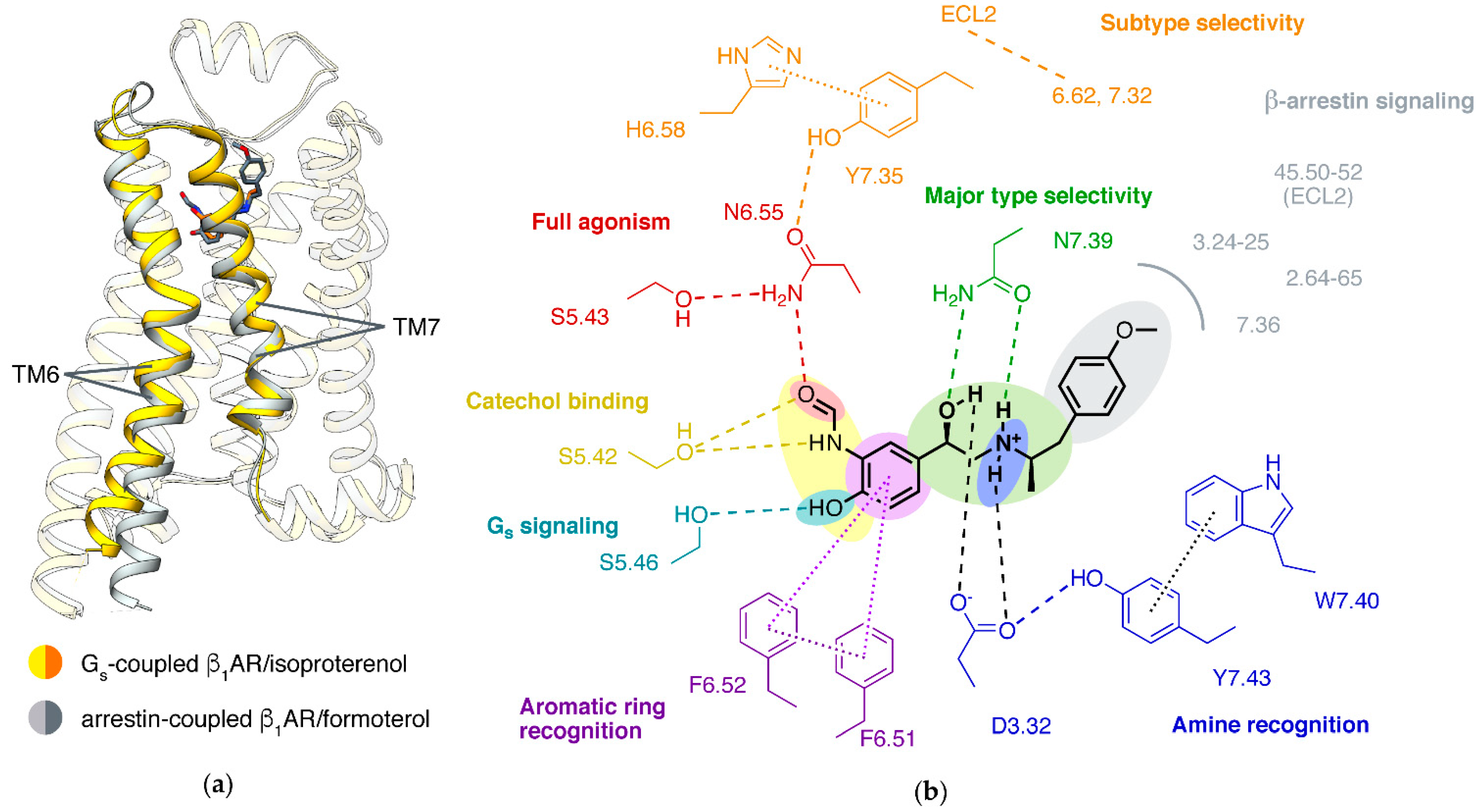
4. Mechanisms of β Adrenergic Receptors
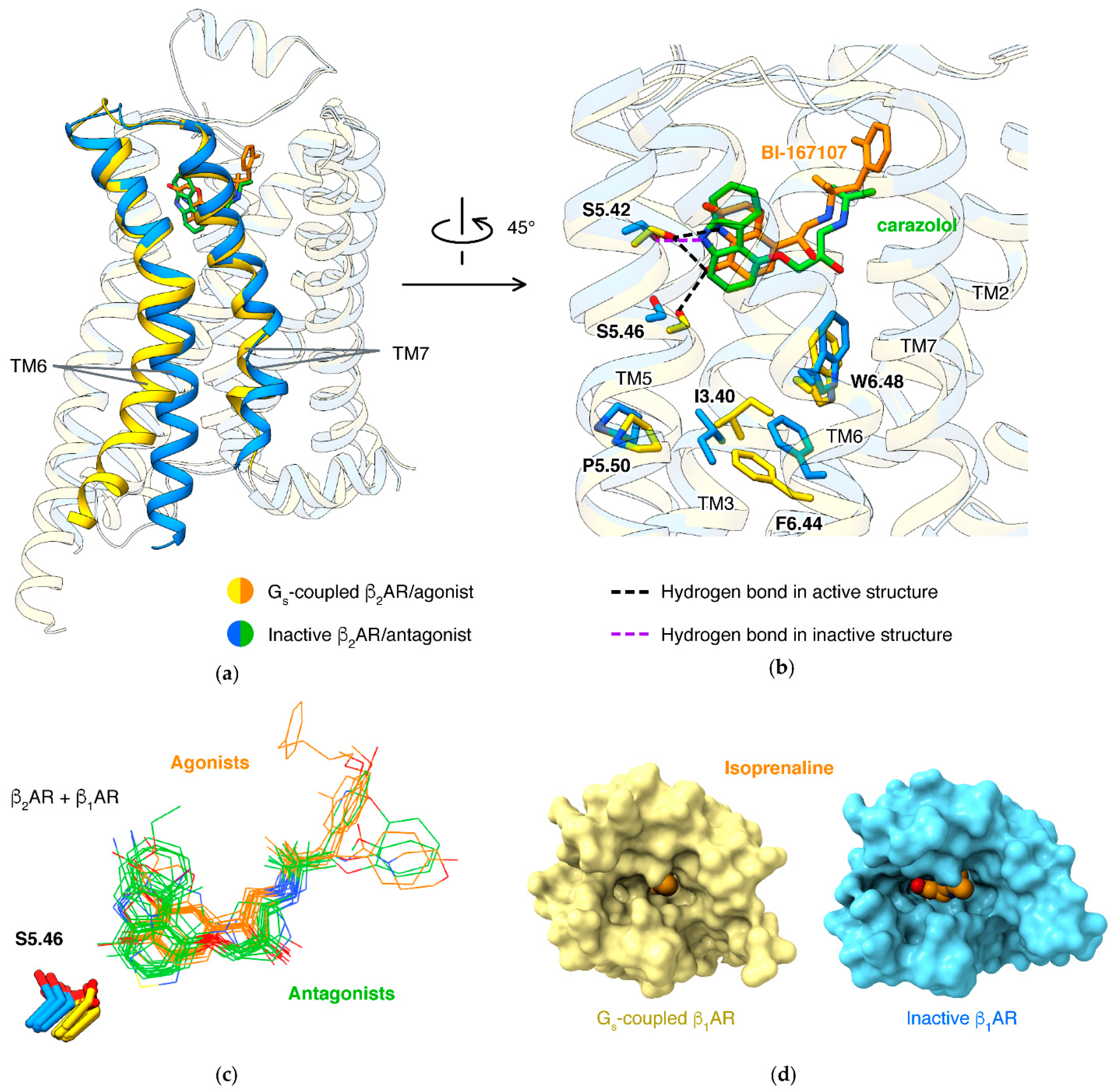
4.1. Activation Mechanism
4.2. Biased Signaling
4.3. Partial Agonism
4.4. β1/β2 Subtype Selectivity
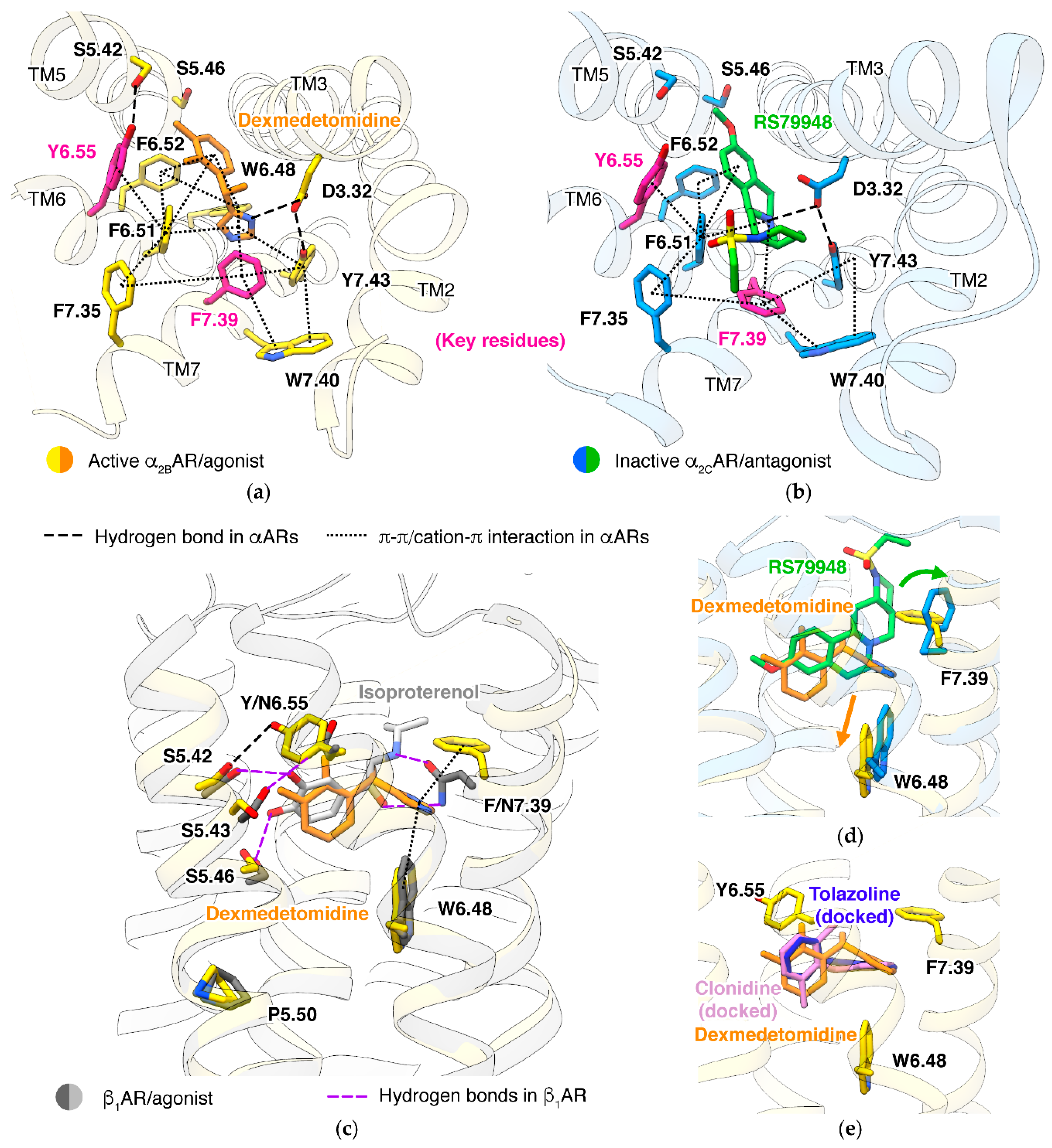
5. Mechanisms of α2 Receptors
5.1. Ligand Binding
5.2. α/β Selectivity
5.3. Alternative Activation Mechanism
5.4. Pathway Selectivity
5.5. α2A/α2C Subtype Selectivity
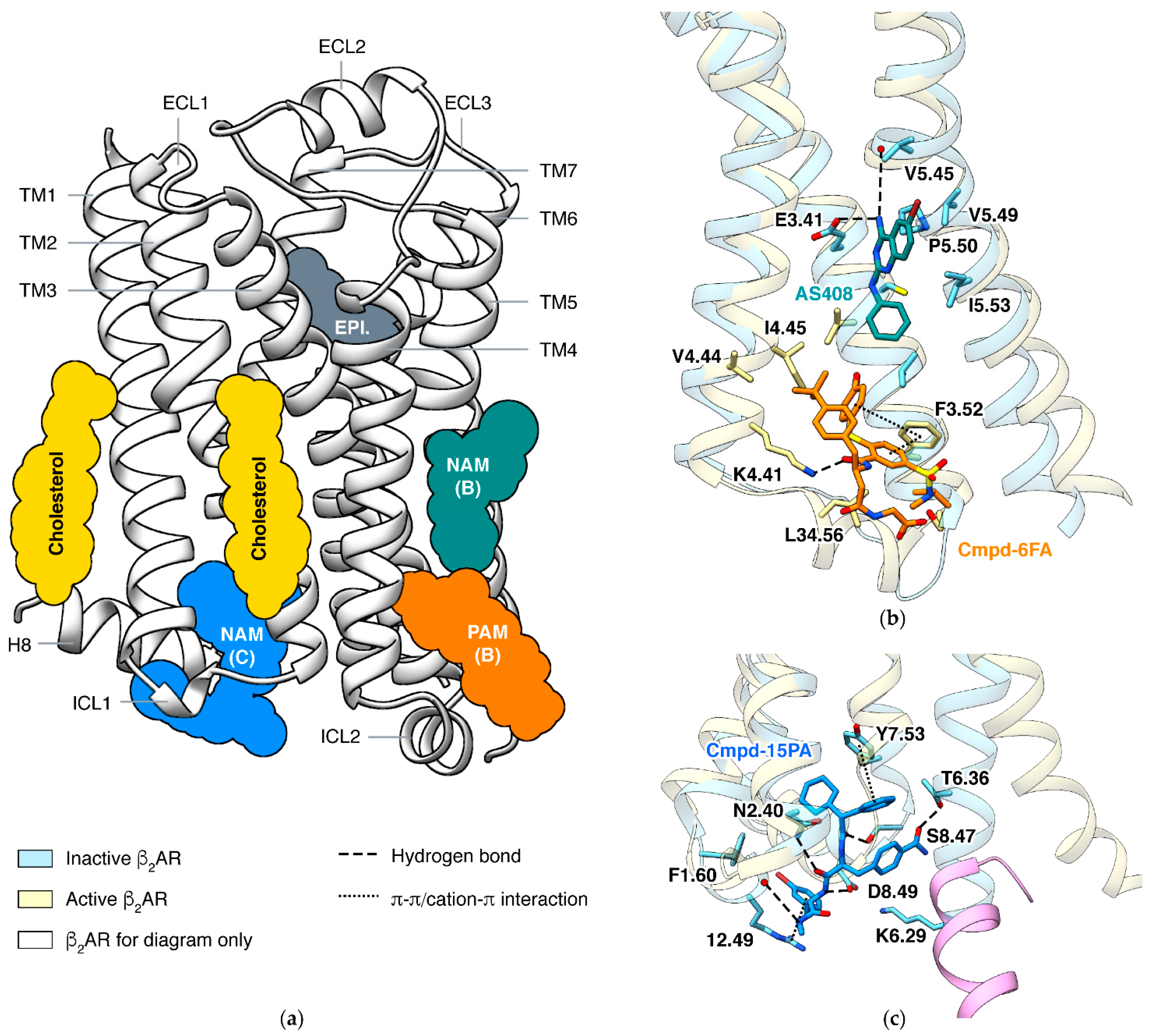
6. Allosteric Modulations of β2 Adrenergic Receptor
6.1. PAM and NAM at Lipid Interface of TM3/4/5
6.2. NAM at Cytoplasmic Surface
7. Structure-Based Ligand Discovery of Adrenergic Receptors
7.1. Virtual Screening with Molecular Docking
7.2. Rational Design
7.3. Predicting Actions of Ligands
7.4. Accelerated Structure Determination
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bylund, D.B.; Eikenberg, D.C.; Hieble, J.P.; Langer, S.Z.; Lefkowitz, R.J.; Minneman, K.P.; Molinoff, P.B.; Ruffolo, R.R.; Trendelenburg, U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 1994, 46, 121–136. [Google Scholar]
- Ciccarelli, M.; Santulli, G.; Pascale, V.; Trimarco, B.; Iaccarino, G. Adrenergic receptors and metabolism: Role in develop-ment of cardiovascular disease. Front Physiol. 2013, 4, 265. [Google Scholar] [CrossRef] [Green Version]
- Bylund, D.B. Adrenergic Receptors. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 57–60. [Google Scholar]
- Farzam, K.; Kidron, A.; Lakhkar, A.D. Adrenergic Drugs; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kobilka, B.K.; Dixon, R.A.; Frielle, T.; Dohlman, H.G.; Bolanowski, M.A.; Sigal, I.S.; Yang-Feng, T.L.; Francke, U.; Caron, M.G.; Lefkowitz, R.J. cDNA for the human beta 2-adrenergic receptor: A protein with multiple membrane-spanning do-mains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc. Natl. Acad. Sci. USA 1987, 84, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. beta-Arrestin: A protein that regulates beta-adrenergic re-ceptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Attramadal, H.; Arriza, J.; Aoki, C.; Dawson, T.; Codina, J.; Kwatra, M.; Snyder, S.; Caron, M.; Lefkowitz, R. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar] [CrossRef]
- Lee, Y.; Warne, T.; Nehme, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; Garcia-Nafria, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Hone, K.; Obika, K.; Foglar, R.; Tsujimoto, G. Selectivity of the imidazoline α-adrenoceptor agonists (oxymetazoline and cirazoline) for human cloned α1-adrenoceptor subtypes. Br. J. Pharmacol. 1995, 116, 1611–1618. [Google Scholar] [CrossRef]
- Jasper, J.R.; Lesnick, J.D.; Chang, L.K.; Yamanishi, S.S.; Chang, T.K.; Hsu, S.A.; Daunt, D.A.; Bonhaus, D.W.; Eglen, R.M. Lig-and efficacy and potency at recombinant alpha2 adrenergic receptors: Agonist-mediated [35S]GTPgammaS binding. Biochem. Pharmacol. 1998, 55, 1035–1043. [Google Scholar] [CrossRef]
- Hoffmann, C.; Leitz, M.R.; Oberdorf-Maass, S.; Lohse, M.J.; Klotz, K.N. Comparative pharmacology of human be-ta-adrenergic receptor subtypes--characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 369, 151–159. [Google Scholar] [CrossRef]
- Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Southan, C.; Sharman, J.L.; Campo, B.; Cavanagh, D.R.; Alexander, S.P.H.; Davenport, A.P.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 2019, 48, D1006–D1021. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, E.D. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Surgand, J.-S.; Rodrigo, J.; Kellenberger, E.; Rognan, D. A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins Struct. Funct. Bioinform. 2005, 62, 509–538. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flock, T.; Hauser, A.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR–G-protein binding. Nat. Cell Biol. 2017, 545, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Gloriam, D.E.; Foord, S.M.; Blaney, F.E.; Garland, S. Definition of the G Protein-Coupled Receptor Transmembrane Bundle Binding Pocket and Calculation of Receptor Similarities for Drug Design. J. Med. Chem. 2009, 52, 4429–4442. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences; Sealfon, S.C., Ed.; Academic Press: Waltham, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Peng, Y.; McCorvy, J.D.; Harpsøe, K.; Lansu, K.; Yuan, S.; Popov, P.; Qu, L.; Pu, M.; Che, T.; Nikolajsen, L.F.; et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172, 719–730.e14. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood dis-orders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Butini, S.; Nikolic, K.; Kassel, S.; Brückmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Baker, J.G. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br. J. Pharmacol. 2010, 160, 1048–1061. [Google Scholar] [CrossRef] [Green Version]
- Lei, S. Cross interaction of dopaminergic and adrenergic systems in neural modulation. Int. J. Physiol. Pathophysiol. Pharmacol. 2014, 6, 137–142. [Google Scholar]
- Lanau, F.; Zenner, M.T.; Civelli, O.; Hartman, D.S. Epinephrine and norepinephrine act as potent agonists at the recombi-nant human dopamine D4 receptor. J. Neurochem. 1997, 68, 804–812. [Google Scholar] [CrossRef]
- Sánchez-Soto, M.; Bonifazi, A.; Cai, N.S.; Ellenberger, M.P.; Newman, A.H.; Ferré, S.; Yano, H. Evidence for Noncanonical Neurotransmitter Activation: Norepinephrine as a Dopamine D2-Like Receptor Agonist. Mol. Pharmacol. 2016, 89, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insel, P.A. Adrenergic Receptors — Evolving Concepts and Clinical Implications. N. Engl. J. Med. 1996, 334, 580–585. [Google Scholar] [CrossRef]
- Pooput, C.; Rosemond, E.; Karpiak, J.; Deflorian, F.; Vilar, S.; Costanzi, S.; Wess, J.; Kirk, K.L. Structural basis of the selectiv-ity of the beta(2)-adrenergic receptor for fluorinated catecholamines. Bioorg. Med. Chem. 2009, 17, 7987–7992. [Google Scholar] [CrossRef] [Green Version]
- Suryanarayana, S.; Daunt, D.; Von Zastrow, M.; Kobilka, B. A point mutation in the seventh hydrophobic domain of the alpha 2 adrenergic receptor increases its affinity for a family of beta receptor antagonists. J. Biol. Chem. 1991, 266, 15488–15492. [Google Scholar] [CrossRef]
- Hieble, J.P. Adrenergic Receptors. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, MS, USA, 2009; pp. 135–139. [Google Scholar]
- Morales, D.R.; Jackson, C.; Lipworth, B.J.; Donnan, P.T.; Guthrie, B. Adverse respiratory effect of acute beta-blocker exposure in asthma: A systematic review and meta-analysis of randomized controlled trials. Chest 2014, 145, 779–786. [Google Scholar] [CrossRef]
- Arch, J.R. Challenges in beta(3)-Adrenoceptor Agonist Drug Development. Ther. Adv. Endocrinol. Metab. 2011, 2, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Schwinn, D.A.; Price, D.T.; Narayan, P. α1-Adrenoceptor Subtype Selectivity and Lower Urinary Tract Symptoms. Mayo Clin. Proc. 2004, 79, 1423–1434. [Google Scholar] [CrossRef] [Green Version]
- Uys, M.M.; Shahid, M.; Harvey, B.H. Therapeutic Potential of Selectively Targeting the α2C-Adrenoceptor in Cognition, Depression, and Schizophrenia—New Developments and Future Perspective. Front. Psychiatry 2017, 8, 144. [Google Scholar] [CrossRef] [Green Version]
- Hwa, J.; Graham, R.M.; Perez, D.M. Identification of Critical Determinants of α1-Adrenergic Receptor Subtype Selective Agonist Binding. J. Biol. Chem. 1995, 270, 23189–23195. [Google Scholar] [CrossRef] [Green Version]
- Laurila, J.M.; Xhaard, H.; Ruuskanen, J.O.; Rantanen, M.J.; Karlsson, H.K.; Johnson, M.S.; Scheinin, M. The second extracellu-lar loop of alpha2A-adrenoceptors contributes to the binding of yohimbine analogues. Br. J. Pharmacol. 2007, 151, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.G.; Proudman, R.G.; Hill, S.J. Salmeterol’s extreme beta2 selectivity is due to residues in both extracellular loops and transmembrane domains. Mol. Pharmacol. 2015, 87, 103–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hubner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human beta1AR over beta2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.Y.; Jozwiak, K.; Toll, L.; Tanga, M.J.; Kozocas, J.A.; Jimenez, L.; Huang, Y.; Song, Y.; Plazinska, A.; Pajak, K.; et al. Tyrosine 308 is necessary for ligand-directed Gs protein-biased signaling of beta2-adrenoceptor. J. Biol. Chem. 2014, 289, 19351–19363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What Can Crystal Structures of Aminergic Receptors Tell Us about Designing Subtype-Selective Ligands? Pharmacol. Rev. 2015, 67, 198–213. [Google Scholar] [CrossRef] [Green Version]
- Dean, I.J.; Reddivari, A.K.R. Alpha 1 Receptor Agonists; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Nachawati, D.; Patel, J. Alpha Blockers; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Giovannitti, J.A.; Thoms, S.M.; Crawford, J.J. Alpha-2 Adrenergic Receptor Agonists: A Review of Current Clinical Applications. Anesthesia Prog. 2015, 62, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Morales, A. Yohimbine in erectile dysfunction: The facts. Int. J. Impot. Res. 2000, 12, S70–S74. [Google Scholar] [CrossRef] [Green Version]
- García-Sevilla, A.J.; Ventayol, P.; Pérez, V.; Rubovszky, G.; Puigdemont, D.; Ferrer-Alcón, M.; Andreoli, A.; Guimón, J.; Alvarez, E. Regulation of Platelet α2A-Adrenoceptors, Gi Proteins and Receptor Kinases in Major Depression: Effects of Mirtazapine Treatment. Neuropsychopharmacology 2003, 29, 580–588. [Google Scholar] [CrossRef]
- Dezsi, C.A.; Szentes, V. The Real Role of beta-Blockers in Daily Cardiovascular Therapy. Am. J. Cardiovasc. Drugs 2017, 17, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Liu, Z.; Kaindl, J.; Maeda, S.; Zhao, J.; Sun, X.; Xu, J.; Gmeiner, P.; Wang, H.-W.; Kobilka, B.K. Activation of the α2B adrenoceptor by the sedative sympatholytic dexmedetomidine. Nat. Chem. Biol. 2020, 16, 507–512. [Google Scholar] [CrossRef] [PubMed]
- DeVree, B.; Mahoney, J.P.; Vélez-Ruiz, G.A.; Rasmussen, S.G.F.; Kuszak, A.; Edwald, E.; Fung, J.-J.; Manglik, A.; Masureel, M.; Du, Y.; et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nat. Cell Biol. 2016, 535, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Warne, T.; Edwards, P.C.; Doré, A.S.; Leslie, A.G.W.; Tate, C.G. Molecular basis for high-affinity agonist binding in GPCRs. Science 2019, 364, 775–778. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos_Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef] [PubMed]
- Nojima, S.; Fujita, Y.; Kimura, K.T.; Nomura, N.; Suno, R.; Morimoto, K.; Yamamoto, M.; Noda, T.; Iwata, S.; Shigematsu, H.; et al. Cryo-EM Structure of the Prostaglandin E Receptor EP4 Coupled to G Protein. Structure 2021, 29, 252–260.e6. [Google Scholar] [CrossRef]
- Toyoda, Y.; Morimoto, K.; Suno, R.; Horita, S.; Yamashita, K.; Hirata, K.; Sekiguchi, Y.; Yasuda, S.; Shiroishi, M.; Shimizu, T.; et al. Ligand binding to human prostaglandin E receptor EP4 at the lipid-bilayer interface. Nat. Chem. Biol. 2019, 15, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Deluigi, M.; Klipp, A.; Klenk, C.; Merklinger, L.; Eberle, S.A.; Morstein, L.; Heine, P.; Mittl, P.R.E.; Ernst, P.; Kamenecka, T.M.; et al. Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial, and inverse agonism. Sci. Adv. 2021, 7, eabe5504. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of delta-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active delta-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.K.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of Human A 1 and A 2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef] [PubMed]
- Draper-Joyce, C.J.; Khoshouei, M.; Thal, D.M.; Liang, Y.-L.; Nguyen, A.T.N.; Furness, S.G.B.; Venugopal, H.; Baltos, J.-A.; Plitzko, J.M.; Danev, R.; et al. Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nat. Cell Biol. 2018, 558, 559–563. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, J.; Gao, Z.-G.; Zhang, D.; Zhu, L.; Han, G.W.; Moss, S.M.; Paoletta, S.; Kiselev, E.; Lu, W.; et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nat. Cell Biol. 2014, 509, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, K.; Gao, Z.-G.; Paoletta, S.; Zhang, D.; Han, G.W.; Li, T.; Ma, L.; Zhang, W.; Müller, C.E.; et al. Agonist-bound structure of the human P2Y12 receptor. Nat. Cell Biol. 2014, 509, 119–122. [Google Scholar] [CrossRef]
- Su, M.; Zhu, L.; Zhang, Y.; Paknejad, N.; Dey, R.; Huang, J.; Lee, M.Y.; Williams, D.; Jordan, K.D.; Eng, E.T.; et al. Structural Basis of the Activation of Heterotrimeric Gs-Protein by Isoproterenol-Bound beta1-Adrenergic Receptor. Mol. Cell 2020, 80, 59–71 e54. [Google Scholar] [CrossRef]
- Warne, T.; Moukhametzianov, R.; Baker, J.G.; Nehme, R.; Edwards, P.C.; Leslie, A.G.; Schertler, G.F.; Tate, C.G. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 2011, 469, 241–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yang, F.; Ling, S.; Lv, P.; Zhou, Y.; Fang, W.; Sun, W.; Zhang, L.; Shi, P.; Tian, C. Single-particle cryo-EM structural studies of the beta2AR-Gs complex bound with a full agonist formoterol. Cell Discov. 2020, 6, 45. [Google Scholar] [CrossRef]
- Yang, F.; Ling, S.; Zhou, Y.; Zhang, Y.; Lv, P.; Liu, S.; Fang, W.; Sun, W.; Hu, A.L.; Zhang, L.; et al. Different conformational responses of the β2-adrenergic receptor-Gs complex upon binding of the partial agonist salbutamol or the full agonist isoprenaline. Natl. Sci. Rev. 2020, nwaa284. [Google Scholar] [CrossRef]
- Masureel, M.; Zou, Y.; Picard, L.P.; van der Westhuizen, E.; Mahoney, J.P.; Rodrigues, J.; Mildorf, T.J.; Dror, R.O.; Shaw, D.E.; Bouvier, M.; et al. Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat. Chem. Biol. 2018, 14, 1059–1066. [Google Scholar] [CrossRef]
- Warne, T.; Serrano-Vega, M.J.; Baker, J.G.; Moukhametzianov, R.; Edwards, P.C.; Henderson, R.; Leslie, A.G.; Tate, C.G.; Schertler, G.F. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 2008, 454, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopher, J.A.; Brown, J.; Dore, A.S.; Errey, J.C.; Koglin, M.; Marshall, F.H.; Myszka, D.G.; Rich, R.L.; Tate, C.G.; Tehan, B.; et al. Biophysical fragment screening of the beta1-adrenergic receptor: Identification of high affinity arylpiperazine leads using structure-based drug design. J. Med. Chem. 2013, 56, 3446–3455. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.-P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, D.; Fenalti, G.; Brown, M.A.; Katritch, V.; Abagyan, R.; Cherezov, V.; Stevens, R.C. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Moukhametzianov, R.; Warne, T.; Edwards, P.C.; Serrano-Vega, M.J.; Leslie, A.G.; Tate, C.G.; Schertler, G.F. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 8228–8232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Baker, J.; Warne, T.; Brown, G.A.; Leslie, A.G.; Congreve, M.; Tate, C.G. Pharmacological Analysis and Structure Determination of 7-Methylcyanopindolol-Bound beta1-Adrenergic Receptor. Mol. Pharmacol. 2015, 88, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [Green Version]
- Mangmool, S.; Parichatikanond, W.; Kurose, H. Therapeutic Targets for Treatment of Heart Failure: Focus on GRKs and beta-Arrestins Affecting betaAR Signaling. Front. Pharmacol. 2018, 9, 1336. [Google Scholar] [CrossRef]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nat. Cell Biol. 2015, 523, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.-L.; De Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 2019, 29, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wuthrich, K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [Green Version]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of beta-blocker action: Carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [Green Version]
- Galandrin, S.; Oligny-Longpre, G.; Bonin, H.; Ogawa, K.; Gales, C.; Bouvier, M. Conformational rearrangements and signaling cascades involved in ligand-biased mitogen-activated protein kinase signaling through the beta1-adrenergic receptor. Mol. Pharmacol. 2008, 74, 162–172. [Google Scholar] [CrossRef]
- Rajagopal, S.; Ahn, S.; Rominger, D.H.; Gowen-MacDonald, W.; Lam, C.M.; DeWire, S.M.; Violin, J.D.; Lefkowitz, R.J. Quantifying Ligand Bias at Seven-Transmembrane Receptors. Mol. Pharmacol. 2011, 80, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Weiss, D.R.; Ahn, S.; Sassano, M.F.; Kleist, A.; Zhu, X.; Strachan, R.; Roth, B.L.; Lefkowitz, R.J.; Shoichet, B.K. Conformation guides molecular efficacy in docking screens of activated beta-2 adrenergic G protein coupled receptor. ACS Chem. Biol. 2013, 8, 1018–1026. [Google Scholar] [CrossRef]
- Warne, T.; Edwards, P.C.; Leslie, A.G.; Tate, C.G. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 2012, 20, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, M.T.; Violin, J.D.; Whalen, E.J.; Wisler, J.W.; Shenoy, S.K.; Lefkowitz, R.J. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J. Biol. Chem. 2008, 283, 5669–5676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukasheva, V.; Devost, D.; Le Gouill, C.; Namkung, Y.; Martin, R.D.; Longpre, J.M.; Amraei, M.; Shinjo, Y.; Hogue, M.; Lagace, M.; et al. Signal profiling of the beta1AR reveals coupling to novel signalling pathways and distinct phenotypic responses mediated by beta1AR and beta2AR. Sci. Rep. 2020, 10, 8779. [Google Scholar] [CrossRef]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Yokomizo, T.; Kofuku, Y.; Shiraishi, Y.; Ueda, T.; Shimada, I. Structural equilibrium underlying ligand-dependent activation of beta2-adrenoreceptor. Nat. Chem. Biol. 2020, 16, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Lerch, M.T.; Matt, R.A.; Masureel, M.; Elgeti, M.; Kumar, K.K.; Hilger, D.; Foys, B.; Kobilka, B.K.; Hubbell, W.L. Viewing rare conformations of the beta2 adrenergic receptor with pressure-resolved DEER spectroscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 31824–31831. [Google Scholar] [CrossRef]
- Gregorio, G.G.; Masureel, M.; Hilger, D.; Terry, D.S.; Juette, M.; Zhao, H.; Zhou, Z.; Perez-Aguilar, J.M.; Hauge, M.; Mathiasen, S.; et al. Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 2017, 547, 68–73. [Google Scholar] [CrossRef]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Maeda, M.; Tsujishita, H.; Shimada, I. Efficacy of the beta(2)-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 2012, 3, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Zhou, Q.; Xu, Y.; Guo, Y.; Chen, X.; Yao, D.; Han, G.W.; Liu, Z.-J.; Stevens, R.C.; Zhong, G.; et al. Structural Basis of the Diversity of Adrenergic Receptors. Cell Rep. 2019, 29, 2929–2935.e4. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, Y.; Qu, L.; Wu, L.; Han, G.W.; Guo, Y.; Wu, Y.; Zhou, Q.; Sun, Q.; Chu, C.; et al. Molecular Mechanism for Ligand Recognition and Subtype Selectivity of α2C Adrenergic Receptor. Cell Rep. 2019, 29, 2936–2943.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffolo, R.R.; Bondinell, W.; Hieble, J.P. .alpha.- and .beta.-Adrenoceptors: From the Gene to the Clinic. 2. Structure-Activity Relationships and Therapeutic Applications. J. Med. Chem. 1995, 38, 3681–3716. [Google Scholar] [CrossRef]
- Ruffolo, R.R., Jr.; Spradlin, T.A.; Pollock, G.D.; Waddell, J.E.; Murphy, P.J. Alpha and beta adrenergic effects of the stereoi-somers of dobutamine. J. Pharmacol. Exp. Ther. 1981, 219, 447–452. [Google Scholar] [PubMed]
- Pauwels, P.J.; Colpaert, F.C. Disparate ligand-mediated Ca(2+) responses by wild-type, mutant Ser(200)Ala and Ser(204)Ala alpha(2A)-adrenoceptor: G(alpha15) fusion proteins: Evidence for multiple ligand-activation binding sites. Br. J. Pharmacol. 2000, 130, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- Sallinen, J.; Hoglund, I.; Engstrom, M.; Lehtimaki, J.; Virtanen, R.; Sirvio, J.; Wurster, S.; Savola, J.M.; Haapalinna, A. Phar-macological characterization and CNS effects of a novel highly selective alpha2C-adrenoceptor antagonist JP-1302. Br. J. Pharmacol. 2007, 150, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Lockyer, S.; Li, J.; Chen, R.; Yoshitake, M.; Kambayashi, J.I. OPC-28326, a selective femoral vasodilator, is an al-pha2C-adrenoceptor-selective antagonist. J. Pharmacol. Exp. Ther. 2001, 299, 652–658. [Google Scholar]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nat. Cell Biol. 2018, 559, 45–53. [Google Scholar] [CrossRef]
- Wu, Y.; Tong, J.; Ding, K.; Zhou, Q.; Zhao, S. GPCR Allosteric Modulator Discovery. Adv. Exp. Med. Biol. 2019, 1163, 225–251. [Google Scholar] [CrossRef]
- Sarkar, P.; Chattopadhyay, A. Cholesterol interaction motifs in G protein-coupled receptors: Slippery hot spots? Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1481. [Google Scholar] [CrossRef]
- Oswald, C.; Rappas, M.; Kean, J.; Doré, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; et al. Intracellular allosteric antagonism of the CCR9 receptor. Nat. Cell Biol. 2016, 540, 462–465. [Google Scholar] [CrossRef]
- Liu, X.; Masoudi, A.; Kahsai, A.W.; Huang, L.Y.; Pani, B.; Staus, D.P.; Shim, P.J.; Hirata, K.; Simhal, R.K.; Schwalb, A.M.; et al. Mechanism of beta2AR regulation by an intracellular positive allosteric modulator. Science 2019, 364, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kaindl, J.; Korczynska, M.; Stossel, A.; Dengler, D.; Stanek, M.; Hubner, H.; Clark, M.J.; Mahoney, J.; Matt, R.A.; et al. An allosteric modulator binds to a conformational hub in the beta2 adrenergic receptor. Nat. Chem. Biol. 2020, 16, 749–755. [Google Scholar] [CrossRef]
- Lu, J.; Byrne, N.; Wang, J.; Bricogne, G.; Brown, F.K.; Chobanian, H.R.; Colletti, S.L.; Di Salvo, J.; Thomas-Fowlkes, B.; Guo, Y.; et al. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat. Struct. Mol. Biol. 2017, 24, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.D.; Chau, B.; Rodgers, L.; Lu, F.; Wilbur, K.L.; Otto, K.A.; Chen, Y.; Song, M.; Riley, J.P.; Yang, H.-C.; et al. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Y.; Krumm, B.; Zhang, H.; Zhou, X.E.; Wang, Y.; Huang, X.P.; Liu, Y.; Cheng, X.; Jiang, Y.; Jiang, H.; et al. Mecha-nism of dopamine binding and allosteric modulation of the human D1 dopamine receptor. Cell Res. 2021, 31, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.; Rappas, M.; Doré, A.S.; Brown, J.; Bottegoni, G.; Koglin, M.; Cansfield, J.; Jazayeri, A.; Cooke, R.M.; Marshall, F.H. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nat. Cell Biol. 2018, 553, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kim, H.R.; Deepak, R.N.V.K.; Wang, L.; Chung, K.Y.; Fan, H.; Wei, Z.; Zhang, C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018, 25, 472–481. [Google Scholar] [CrossRef]
- Zheng, Y.; Qin, L.; Zacarías, N.V.O.; De Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nat. Cell Biol. 2016, 540, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, K.; Brünle, S.; Weinert, T.; Guba, W.; Muehle, J.; Miyazaki, T.; Weber, M.; Furrer, A.; Haenggi, N.; Tetaz, T.; et al. Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell 2019, 178, 1222–1230.e10. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nat. Cell Biol. 2020, 585, 1–9. [Google Scholar] [CrossRef]
- Liu, X.; Ahn, S.; Kahsai, A.W.; Meng, K.C.; Latorraca, N.R.; Pani, B.; Venkatakrishnan, A.J.; Masoudi, A.; Weis, W.I.; Dror, R.O.; et al. Mechanism of intracellular allosteric beta2AR antagonist revealed by X-ray crystal structure. Nature 2017, 548, 480–484. [Google Scholar] [CrossRef]
- Ahn, S.; Kahsai, A.W.; Pani, B.; Wang, Q.T.; Zhao, S.; Wall, A.L.; Strachan, R.T.; Staus, D.P.; Wingler, L.M.; Sun, L.D.; et al. Al-losteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. USA 2017, 114, 1708–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoichet, B.K.; Kobilka, B.K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G pro-tein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Topiol, S.; Sabio, M. Use of the X-ray structure of the Beta2-adrenergic receptor for drug discovery. Bioorg. Med. Chem. Lett. 2008, 18, 1598–1602. [Google Scholar] [CrossRef]
- Irwin, J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, P.; Rosenbaum, D.M.; Irwin, J.J.; Fung, J.J.; Kobilka, B.K.; Shoichet, B.K. Structure-based discovery of beta2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 6843–6848. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.; Choi, H.J.; Devree, B.T.; Suna-hara, R.K.; et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, D.; Kruse, A.C.; Manglik, A.; Hiller, C.; Zhang, C.; Hübner, H.; Kobilka, B.K.; Gmeiner, P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2014, 111, 10744–10748. [Google Scholar] [CrossRef] [Green Version]
- McCorvy, J.D.; Butler, K.V.; Kelly, B.; Rechsteiner, K.; Karpiak, J.; Betz, R.M.; Kormos, B.L.; Shoichet, B.K.; Dror, R.O.; Jin, J.; et al. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol. 2018, 14, 126–134. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, C.; Rognan, D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic re-ceptor. J. Med. Chem. 2008, 51, 4978–4985. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Leurs, R.; de Esch, I.J.; de Graaf, C. Structure-Based Prediction of G-Protein-Coupled Receptor Ligand Func-tion: A beta-Adrenoceptor Case Study. J. Chem. Inf. Model. 2015, 55, 1045–1061. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Vischer, H.F.; McNaught-Flores, D.; Leurs, R.; de Esch, I.; De Graaf, C. Function-specific virtual screening for GPCR ligands using a combined scoring method. Sci. Rep. 2016, 6, 28288. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.P.; et al. Structural in-sights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942 e918. [Google Scholar] [CrossRef]
- Xiao, P.; Yan, W.; Gou, L.; Zhong, Y.-N.; Kong, L.; Wu, C.; Wen, X.; Yuan, Y.; Cao, S.; Qu, C.; et al. Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes. Cell 2021, 184, 943–956.e18. [Google Scholar] [CrossRef]
- Sun, B.; Feng, D.; Chu, M.L.; Fish, I.; Kelm, S.; Lebon, F.; Lovera, S.; Valade, A.; Wood, M.; Ceska, T.; et al. PDB 7JOZ. Available online: https://www.rcsb.org/structure/7JOZ (accessed on 24 May 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Type | Primary Pathway | Main Effects | Indications of Agonists | Indications of Antagonists |
|---|---|---|---|---|
| α1 | Gq/11 | smooth muscle contraction | vasodilatory shock, hypotension [46] | hypertension, benign prostatic hyperplasia [47] |
| α2 | Gi/o | inhibition of norepinephrine release | hypertension, pain and panic disorders [48] | erectile dysfunction [49], depression [50] |
| β | Gs | β1: cardiac stimulation β2: bronchodilation β3: increased lipolysis | cardiogenic shock, heart failure, asthma, overactive bladder [4] | heart failure, arrhythmias, hypertension [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zeng, L.; Zhao, S. Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules 2021, 11, 936. https://doi.org/10.3390/biom11070936
Wu Y, Zeng L, Zhao S. Ligands of Adrenergic Receptors: A Structural Point of View. Biomolecules. 2021; 11(7):936. https://doi.org/10.3390/biom11070936
Chicago/Turabian StyleWu, Yiran, Liting Zeng, and Suwen Zhao. 2021. "Ligands of Adrenergic Receptors: A Structural Point of View" Biomolecules 11, no. 7: 936. https://doi.org/10.3390/biom11070936