
Introduction to Single-Cell DNA Methylation Profiling Methods


Abstract
:1. Introduction
2. Experimental Methodologies
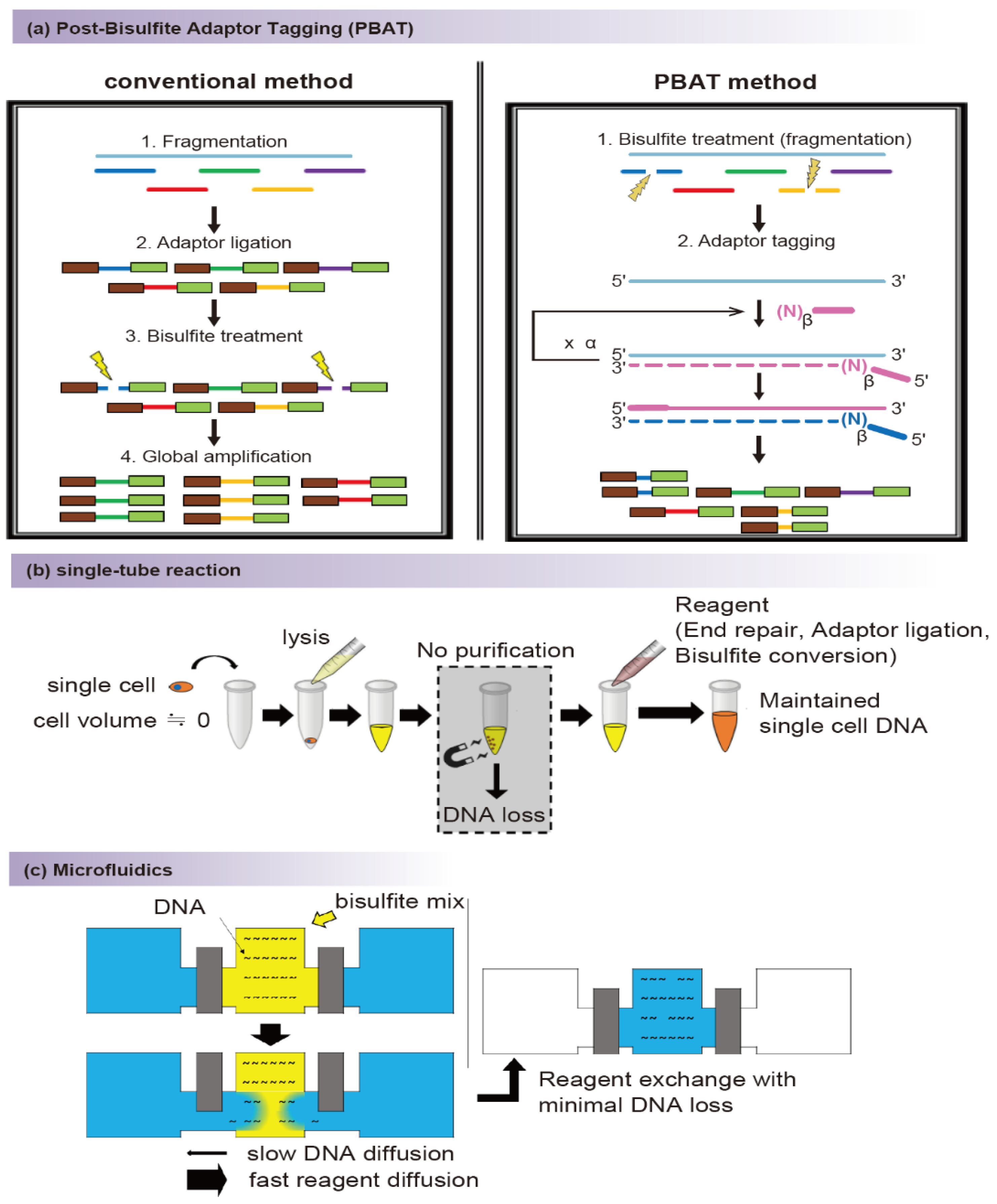
2.1. Bisulfite Conversion
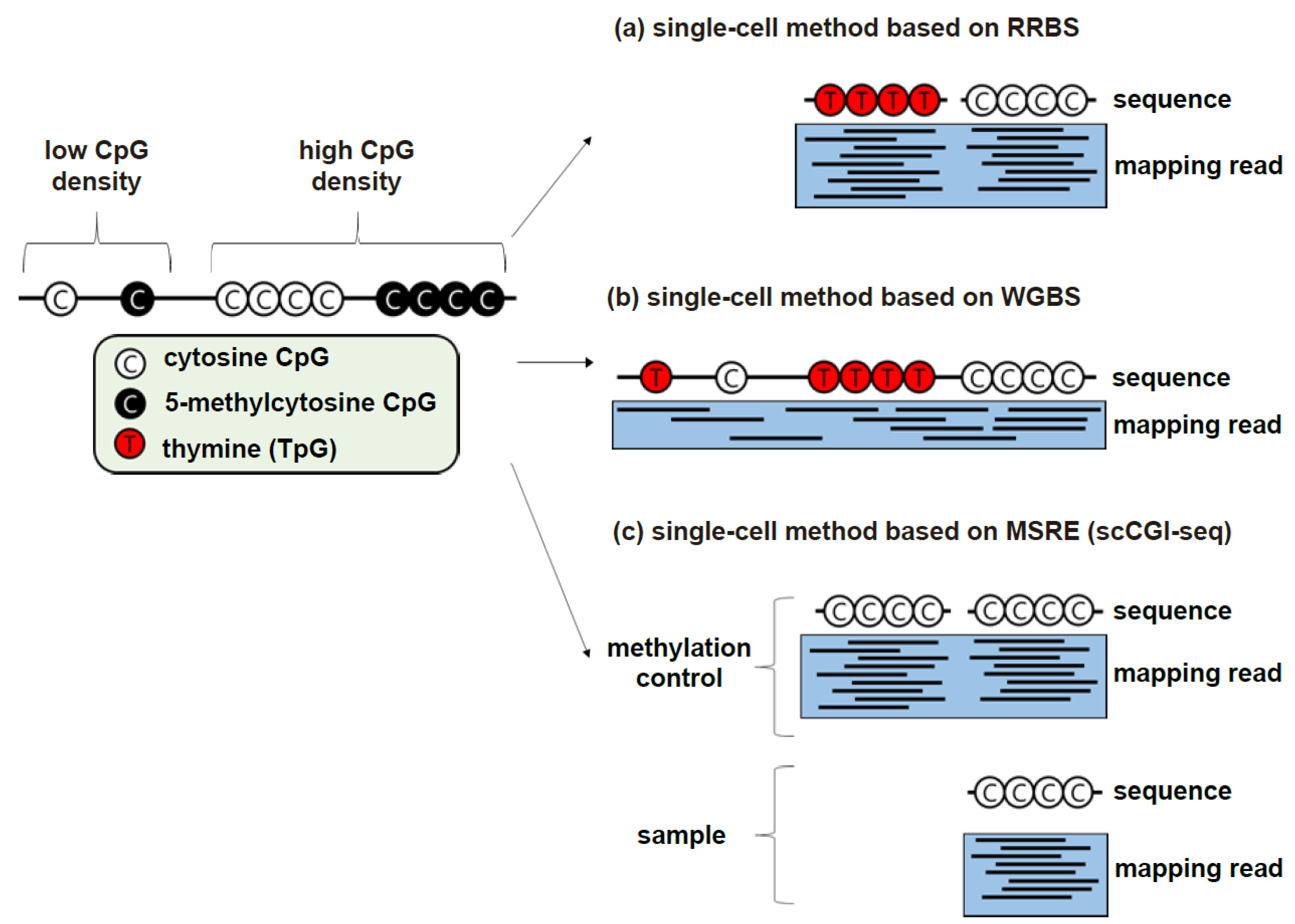
2.1.1. RRBS-Based Methods
2.1.2. WGBS-Based Methods
2.2. Conversion-Free Methods
3. Data Analysis
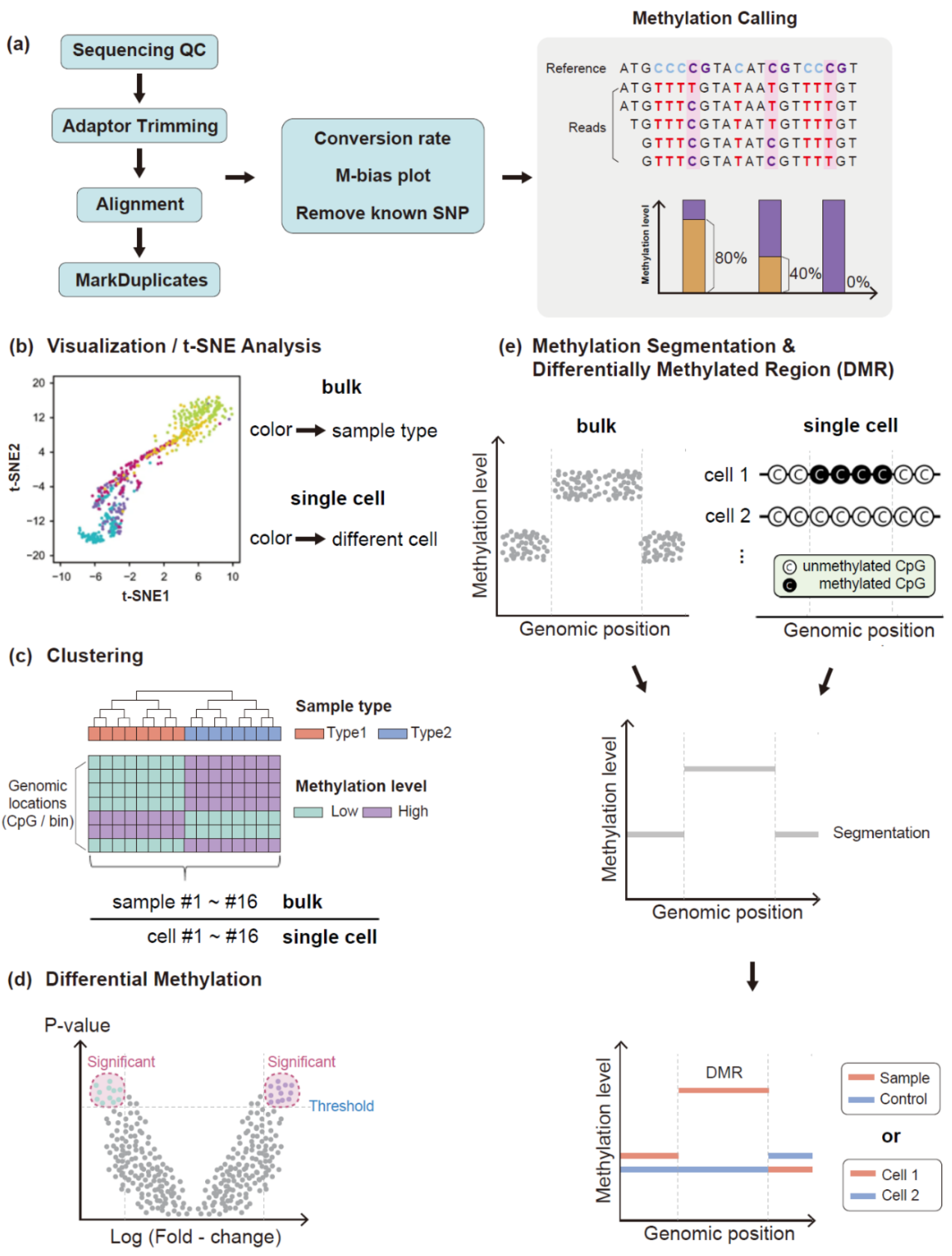
3.1. Data Quality Assessment
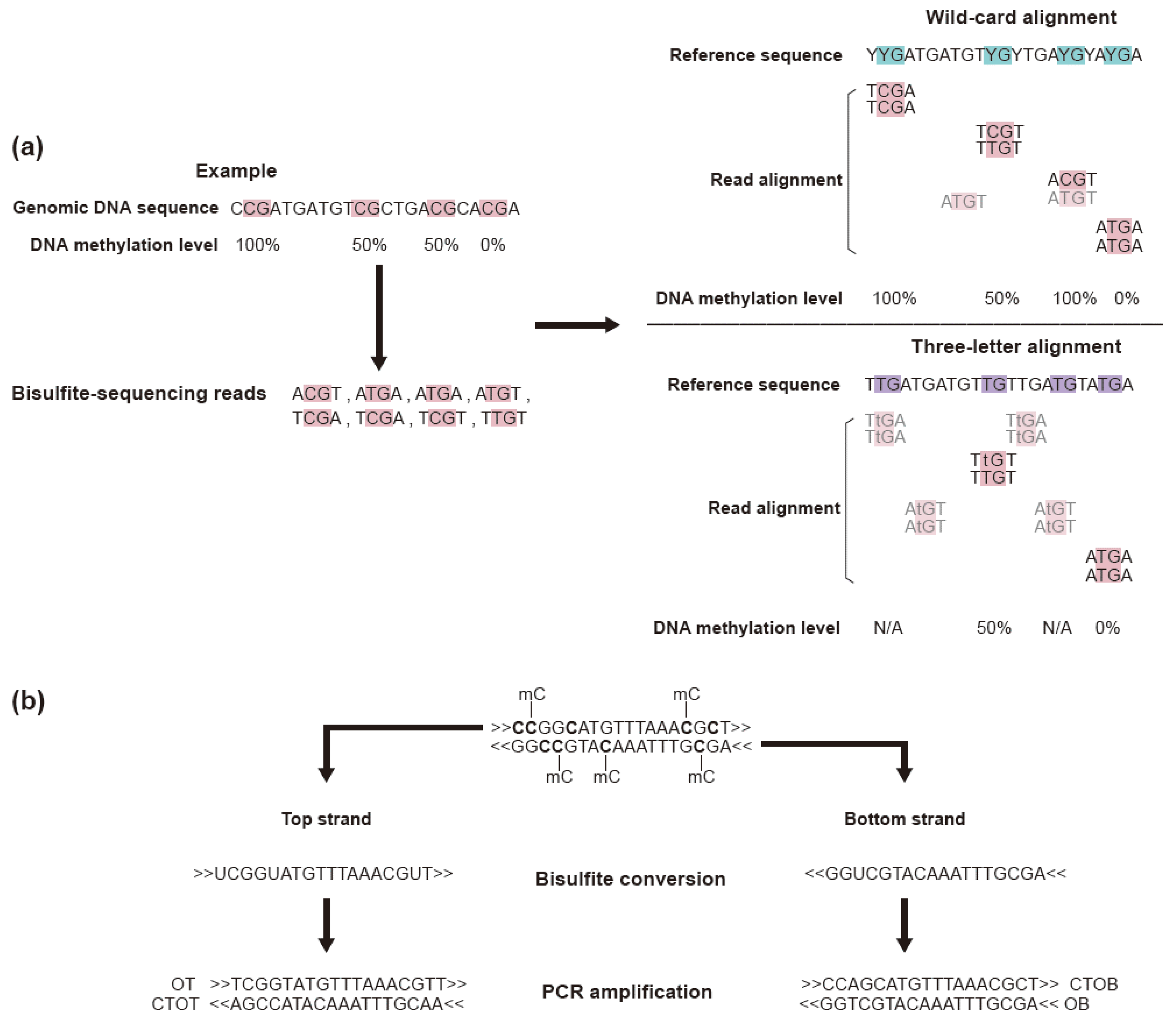
3.2. Read Trimming and Sequence Alignment
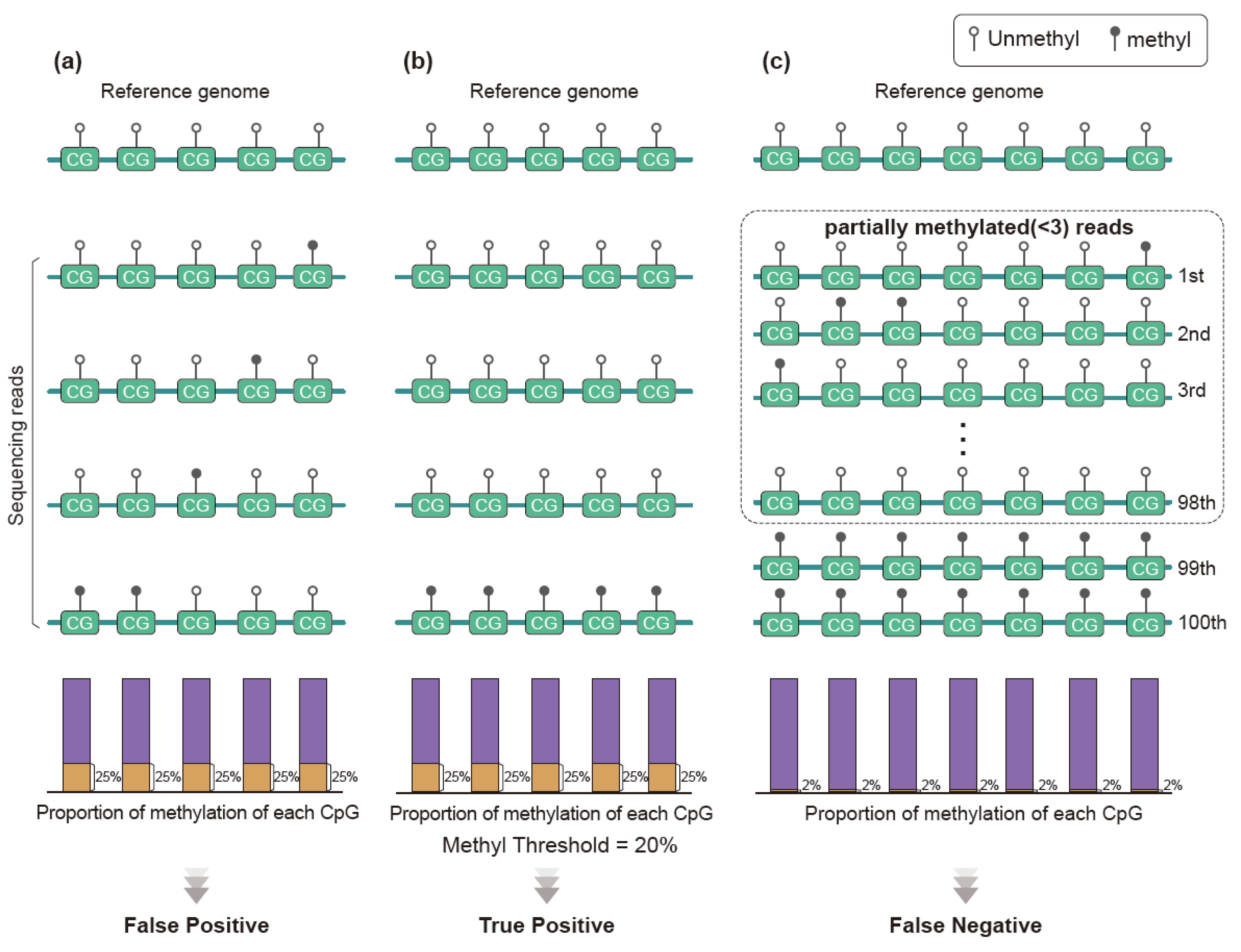
3.3. Methylation Analysis Using Methylation Level
3.4. Methylation Analysis Using Methylation Pattern of Sequence Reads
4. Application
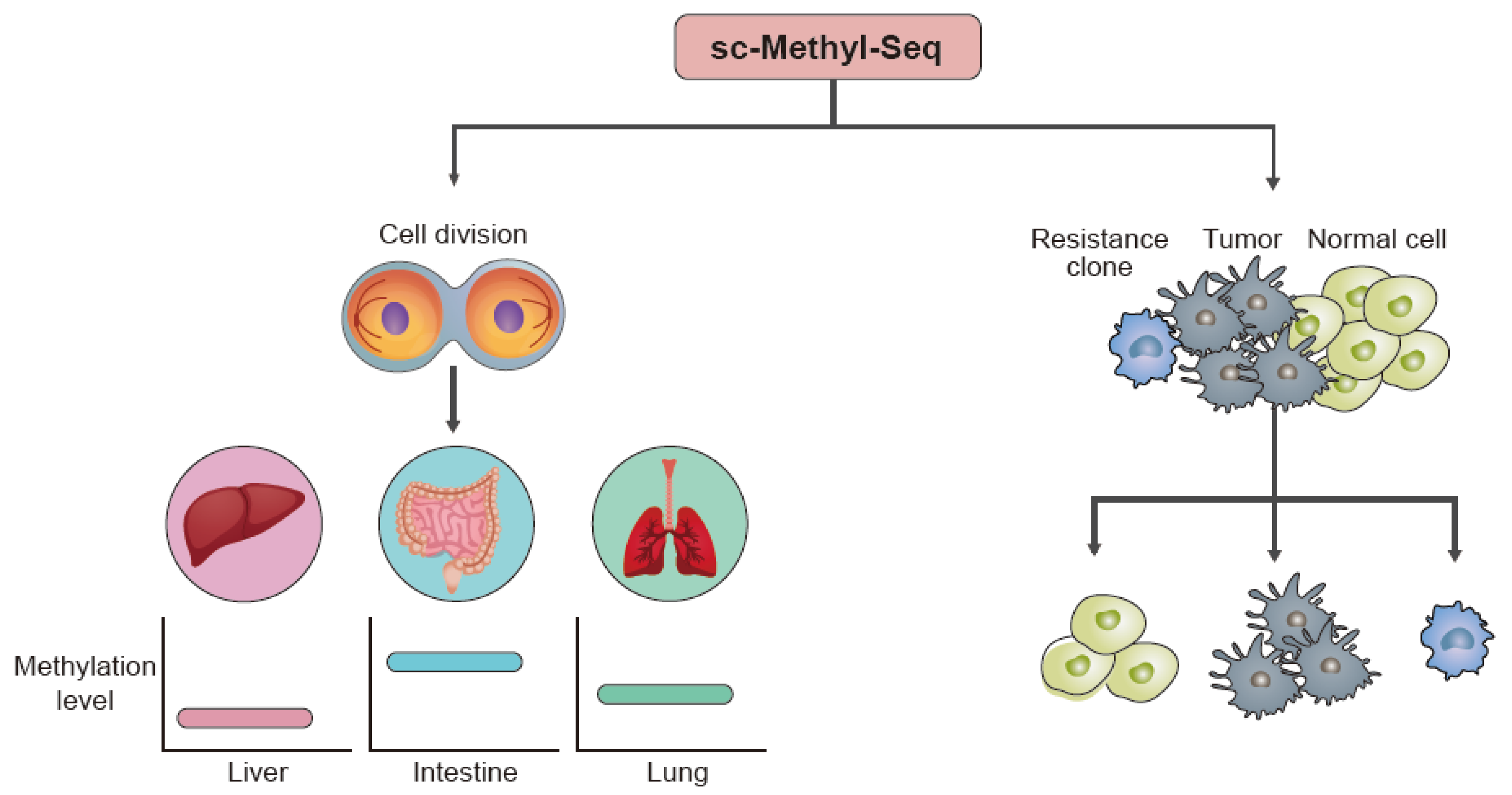
4.1. Cell Development
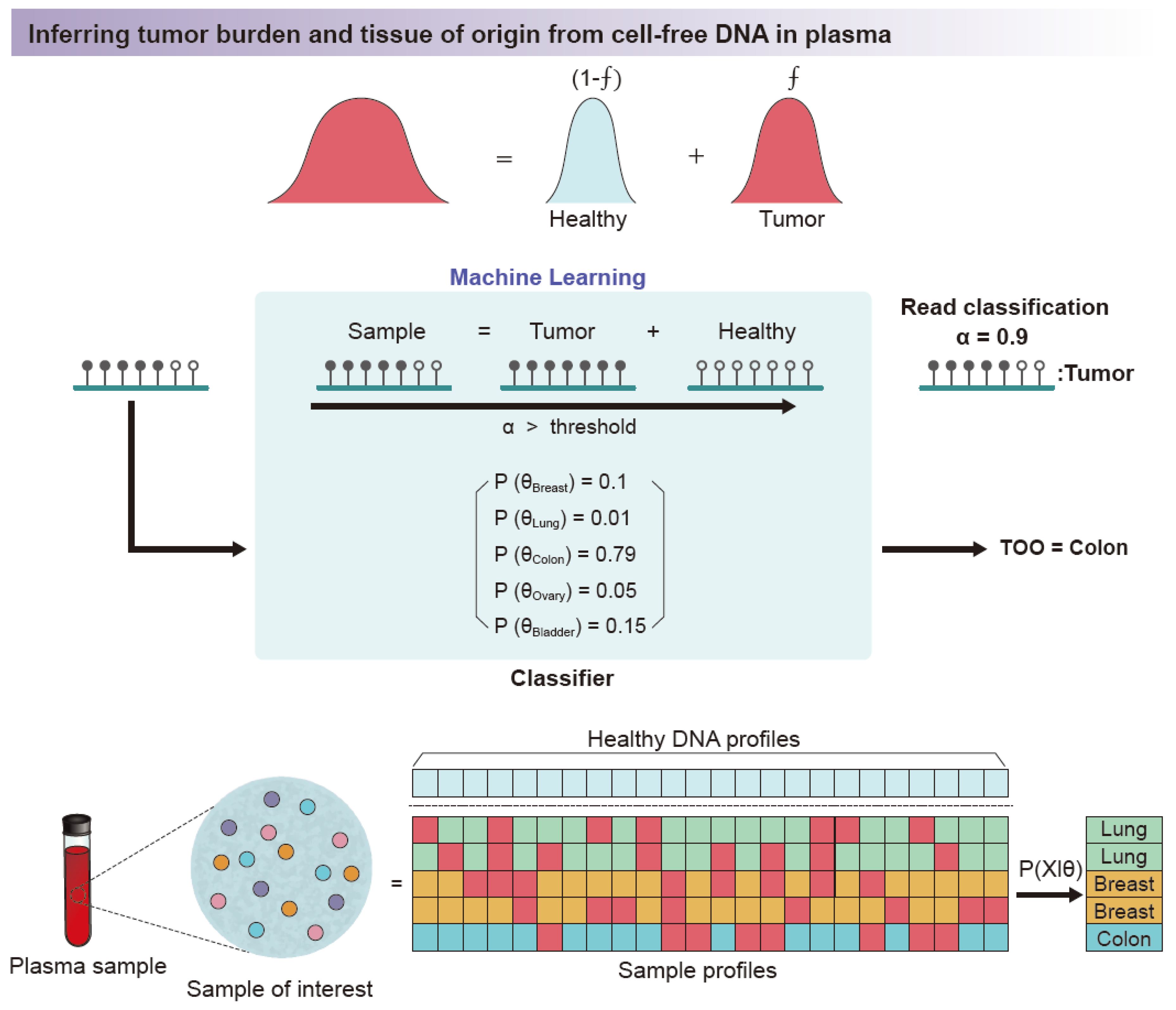
4.2. Disease-Associated Studies
5. Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341. [Google Scholar] [CrossRef] [Green Version]
- Hernando-Herraez, I.; Garcia-Perez, R.; Sharp, A.J.; Marques-Bonet, T. DNA Methylation: Insights into Human Evolution. PLoS Genet. 2015, 11, e1005661. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.N.; Trautner, T.A.; Kornberg, A. Enzymatic Synthesis of Deoxyribonucleic Acid: XI. further studies on nearest neighbor base sequences in deoxyribonucleic acids. J. Biol. Chem. 1962, 237, 1961–1967. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG Islands in Vertebrate Genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Bock, C.; Walter, J.; Paulsen, M.; Lengauer, T. CpG Island Mapping by Epigenome Prediction. PLoS Comput. Biol. 2007, 3, e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; He, F.; Hu, S.; Yu, J. On the Nature of Human Housekeeping Genes. Trends Genet. 2008, 24, 481–484. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A Genome-Wide Analysis of CpG Dinucleotides in the Human Genome Distinguishes Two Distinct Classes of Promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaton, A.M.; Bird, A. CpG Islands and the Regulation of Transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of Sequencing-Based Methods to Profile DNA Methylation and Identification of Monoallelic Epigenetic Modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA Methylation: The Nuts and Bolts of Repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA Methylation in Genomic Imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef]
- Heard, E.; Clerc, P.; Avner, P. X-Chromosome Inactivation in Mammals. Annu. Rev. Genet. 1997, 31, 571–610. [Google Scholar] [CrossRef]
- Gaudet, F.; Rideout, W.M.; Meissner, A.; Dausman, J.; Leonhardt, H.; Jaenisch, R. Dnmt1 Expression in Pre- and Postimplantation Embryogenesis and the Maintenance of IAP Silencing. Mol. Cell. Biol. 2004, 24, 1640–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; LeProust, E.M.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and Genome-Scale Strategies Reveal Gene-Body Methylation Signatures in Human Cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced Representation Bisulfite Sequencing for Comparative High-Resolution DNA Methylation Analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M. DNA Methylation in Cancer: Too Much, but Also Too Little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a Dynamic DNA Methylation Landscape of the Human Genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.-H.; Wang, H.-T.; Kong, Q.-P. Dynamic DNA Methylation During Aging: A “Prophet” of Age-Related Outcomes. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, S.; Fernald, R.D.; Storey, K.B.; Szyf, M. The Dynamic Nature of DNA Methylation: A Role in Response to Social and Seasonal Variation. Integr. Comp. Biol. 2014, 54, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lax, E.; Szyf, M. The Role of DNA Methylation in Drug Addiction: Implications for Diagnostic and Therapeutics. In Progress in Molecular Biology and Translational Science; Epigenetics and Psychiatric Disease; Grayson, D.R., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 157, pp. 93–104. [Google Scholar]
- Voisin, S.; Eynon, N.; Yan, X.; Bishop, D.J. Exercise Training and DNA Methylation in Humans. Acta Physiol. 2015, 213, 39–59. [Google Scholar] [CrossRef]
- Lim, U.; Song, M.-A. Dietary and Lifestyle Factors of DNA Methylation. In Cancer Epigenetics: Methods and Protocols; Methods in Molecular Biology; Dumitrescu, R.G., Verma, M., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 359–376. ISBN 978-1-61779-612-8. [Google Scholar]
- Chuang, Y.-H.; Quach, A.; Absher, D.; Assimes, T.; Horvath, S.; Ritz, B. Coffee Consumption Is Associated with DNA Methylation Levels of Human Blood. Eur. J. Hum. Genet. 2017, 25, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D. DNA Methylation and Human Disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-Cell Genome-Wide Bisulfite Sequencing for Assessing Epigenetic Heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J.; et al. Dynamic Changes in the Human Methylome during Differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the Methylome: Erasing Memory and Creating Diversity. Cell Stem Cell 2014, 14, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A Genomic Sequencing Protocol That Yields a Positive Display of 5-Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Siejka-Zielińska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C.-X. Bisulfite-Free Direct Detection of 5-Methylcytosine and 5-Hydroxymethylcytosine at Base Resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. EM-Seq: Detection of DNA Methylation at Single Base Resolution from Picograms of DNA. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tollefsbol, T.O. DNA Methylation Detection: Bisulfite Genomic Sequencing Analysis. In Epigenetics Protocols; Methods in Molecular Biology; Tollefsbol, T.O., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 11–21. ISBN 978-1-61779-316-5. [Google Scholar]
- Leontiou, C.A.; Hadjidaniel, M.D.; Mina, P.; Antoniou, P.; Ioannides, M.; Patsalis, P.C. Bisulfite Conversion of DNA: Performance Comparison of Different Kits and Methylation Quantitation of Epigenetic Biomarkers That Have the Potential to Be Used in Non-Invasive Prenatal Testing. PLoS ONE 2015, 10, e0135058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogino, S.; Kawasaki, T.; Brahmandam, M.; Cantor, M.; Kirkner, G.J.; Spiegelman, D.; Makrigiorgos, G.M.; Weisenberger, D.J.; Laird, P.W.; Loda, M.; et al. Precision and Performance Characteristics of Bisulfite Conversion and Real-Time PCR (MethyLight) for Quantitative DNA Methylation Analysis. J. Mol. Diagn. 2006, 8, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kint, S.; Spiegelaere, W.D.; Kesel, J.D.; Vandekerckhove, L.; Criekinge, W.V. Evaluation of Bisulfite Kits for DNA Methylation Profiling in Terms of DNA Fragmentation and DNA Recovery Using Digital PCR. PLoS ONE 2018, 13, e0199091. [Google Scholar] [CrossRef]
- Miura, F.; Enomoto, Y.; Dairiki, R.; Ito, T. Amplification-Free Whole-Genome Bisulfite Sequencing by Post-Bisulfite Adaptor Tagging. Nucleic Acids Res. 2012, 40, e136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; de la Fuente Revenga, M.; Sun, Z.; Sun, C.; Murphy, T.W.; Xie, H.; González-Maeso, J.; Lu, C. Cell-Type-Specific Brain Methylomes Profiled via Ultralow-Input Microfluidics. Nat. Biomed. Eng. 2018, 2, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Herranz, D.E.; Ribeiro, A.J.M.; Krueger, F.; Thornton, J.M.; Reik, W.; Stubbs, T.M. CuRRBS: Simple and Robust Evaluation of Enzyme Combinations for Reduced Representation Approaches. Nucleic Acids Res. 2017, 45, 11559–11569. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-R.; Ryu, D.-S.; Park, S.-J.; Choe, S.-H.; Cho, H.-M.; Lee, S.-R.; Kim, S.-U.; Kim, Y.-H.; Huh, J.-W. Successful Application of Human-Based Methyl Capture Sequencing for Methylome Analysis in Non-Human Primate Models. BMC Genom. 2018, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhu, P.; Guo, F.; Li, X.; Wu, X.; Fan, X.; Wen, L.; Tang, F. Profiling DNA Methylome Landscapes of Mammalian Cells with Single-Cell Reduced-Representation Bisulfite Sequencing. Nat. Protoc. 2015, 10, 645–659. [Google Scholar] [CrossRef]
- Wang, K.; Li, X.; Dong, S.; Liang, J.; Mao, F.; Zeng, C.; Wu, H.; Wu, J.; Cai, W.; Sun, Z.S. Q-RRBS: A Quantitative Reduced Representation Bisulfite Sequencing Method for Single-Cell Methylome Analyses. Epigenetics 2015, 10, 775–783. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-Cell Triple Omics Sequencing Reveals Genetic, Epigenetic, and Transcriptomic Heterogeneity in Hepatocellular Carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, K.; An, Q.; Du, G.; Hu, G.; Xue, J.; Zhu, X.; Wang, C.-Y.; Xue, Z.; Fan, G. Simultaneous Profiling of Transcriptome and DNA Methylome from a Single Cell. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Smallwood, S.A.; Lee, H.J.; Krueger, F.; Reik, W.; Kelsey, G. Genome-Wide Base-Resolution Mapping of DNA Methylation in Single Cells Using Single-Cell Bisulfite Sequencing (ScBS-Seq). Nat. Protoc. 2017, 12, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Koike, T.; Sakashita, A.; Tanaka, K.; Kumamoto, S.; Kono, T. Repetitive DNA Methylome Analysis by Small-Scale and Single-Cell Shotgun Bisulfite Sequencing. Genes Cells 2016, 21, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Farlik, M.; Sheffield, N.C.; Nuzzo, A.; Datlinger, P.; Schönegger, A.; Klughammer, J.; Bock, C. Single-Cell DNA Methylome Sequencing and Bioinformatic Inference of Epigenomic Cell-State Dynamics. Cell Rep. 2015, 10, 1386–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Rivkin, A.; Zhou, J.; Sandoval, J.P.; Kurihara, L.; Lucero, J.; Castanon, R.; Nery, J.R.; Pinto-Duarte, A.; Bui, B.; et al. Robust Single-Cell DNA Methylome Profiling with SnmC-Seq2. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mulqueen, R.M.; Pokholok, D.; Norberg, S.J.; Torkenczy, K.A.; Fields, A.J.; Sun, D.; Sinnamon, J.R.; Shendure, J.; Trapnell, C.; O’Roak, B.J.; et al. Highly Scalable Generation of DNA Methylation Profiles in Single Cells. Nat. Biotechnol. 2018, 36, 428–431. [Google Scholar] [CrossRef]
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-Cell Multiomics Sequencing and Analyses of Human Colorectal Cancer. Science 2018, 362, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.A.; Ponting, C.P.; Voet, T.; et al. Parallel Single-Cell Sequencing Links Transcriptional and Epigenetic Heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Argelaguet, R.; Kapourani, C.-A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. ScNMT-Seq Enables Joint Profiling of Chromatin Accessibility DNA Methylation and Transcription in Single Cells. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Liu, S.; Wu, Q.; Zhang, L.; Guo, F. Integrative Single-Cell Analysis of Transcriptome, DNA Methylome and Chromatin Accessibility in Mouse Oocytes. Cell Res. 2019, 29, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Pott, S. Simultaneous Measurement of Chromatin Accessibility, DNA Methylation, and Nucleosome Phasing in Single Cells. eLife 2017, 6, e23203. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wu, H.-J.; Zhu, H.; Kim, K.-Y.; Marjani, S.L.; Riester, M.; Euskirchen, G.; Zi, X.; Yang, J.; Han, J.; et al. Bisulfite-Independent Analysis of CpG Island Methylation Enables Genome-Scale Stratification of Single Cells. Nucleic Acids Res. 2017, 45, e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karemaker, I.D.; Vermeulen, M. Single-Cell DNA Methylation Profiling: Technologies and Biological Applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Nordman, E.; Li, B.; Xu, N.; Bashkirov, V.I.; Lao, K.; Surani, M.A. RNA-Seq Analysis to Capture the Transcriptome Landscape of a Single Cell. Nat. Protoc. 2010, 5, 516–535. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length RNA-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Doherty, R.; Couldrey, C. Exploring Genome Wide Bisulfite Sequencing for DNA Methylation Analysis in Livestock: A Technical Assessment. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Paun, O.; Verhoeven, K.J.F.; Richards, C.L. Opportunities and Limitations of Reduced Representation Bisulfite Sequencing in Plant Ecological Epigenomics. New Phytol. 2019, 221, 738–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Keown, C.L.; Kurihara, L.; Zhou, J.; He, Y.; Li, J.; Castanon, R.; Lucero, J.; Nery, J.R.; Sandoval, J.P.; et al. Single-Cell Methylomes Identify Neuronal Subtypes and Regulatory Elements in Mammalian Cortex. Science 2017, 357, 600–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-Seq: Parallel Sequencing of Single-Cell Genomes and Transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-Wide Mapping of Nucleosome Positioning and DNA Methylation within Individual DNA Molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Li, L.; Li, J.; Wu, X.; Hu, B.; Zhu, P.; Wen, L.; Tang, F. Single-Cell Multi-Omics Sequencing of Mouse Early Embryos and Embryonic Stem Cells. Cell Res. 2017, 27, 967–988. [Google Scholar] [CrossRef]
- Brinkman, A.B.; Simmer, F.; Ma, K.; Kaan, A.; Zhu, J.; Stunnenberg, H.G. Whole-Genome DNA Methylation Profiling Using MethylCap-Seq. Methods 2010, 52, 232–236. [Google Scholar] [CrossRef]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of Methyl-DNA Immunoprecipitation (MeDIP) and Methyl-CpG Binding Domain (MBD) Protein Capture for Genome-Wide DNA Methylation Analysis Reveal CpG Sequence Coverage Bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, A.; Broche, J.; Lungu, C.; Bashtrykov, P. Biotechnological Applications of MBD Domain Proteins for DNA Methylation Analysis. J. Mol. Biol. 2020, 432, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved Role of Intragenic DNA Methylation in Regulating Alternative Promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef]
- Brunner, A.L.; Johnson, D.S.; Kim, S.W.; Valouev, A.; Reddy, T.E.; Neff, N.F.; Anton, E.; Medina, C.; Nguyen, L.; Chiao, E.; et al. Distinct DNA Methylation Patterns Characterize Differentiated Human Embryonic Stem Cells and Developing Human Fetal Liver. Genome Res. 2009, 19, 1044–1056. [Google Scholar] [CrossRef] [Green Version]
- Niemöller, C.; Wehrle, J.; Riba, J.; Claus, R.; Renz, N.; Rhein, J.; Bleul, S.; Stosch, J.M.; Duyster, J.; Plass, C.; et al. Bisulfite-Free Epigenomics and Genomics of Single Cells through Methylation-Sensitive Restriction. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 April 2021).
- Wulfridge, P.; Langmead, B.; Feinberg, A.P.; Hansen, K.D. Analyzing Whole Genome Bisulfite Sequencing Data from Highly Divergent Genotypes. Nucleic Acids Res. 2019, 47, e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhao, M.; Sun, Z.; Wu, F.; Liu, Y.; Liu, X.; He, Z.; He, Q.; He, Q. BCREval: A Computational Method to Estimate the Bisulfite Conversion Ratio in WGBS. BMC Bioinform. 2020, 21, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics—Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 3 April 2021).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C. Analysing and Interpreting DNA Methylation Data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Krueger, F.; Kreck, B.; Franke, A.; Andrews, S.R. DNA Methylome Analysis Using Short Bisulfite Sequencing Data. Nat. Methods 2012, 9, 145–151. [Google Scholar] [CrossRef]
- Cheng, H.; Xu, Y. BitMapperBS: A Fast and Accurate Read Aligner for Whole-Genome Bisulfite Sequencing. BioRxiv 2018, 442798. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.Y.Y.; Huang, Y.-J.; Chen, P.-Y. BS-Seeker3: Ultrafast Pipeline for Bisulfite Sequencing. BMC Bioinform. 2018, 19, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Gao, F.; Xia, Y.; Tang, Y.; Zhao, W.; Jin, C.; Luo, H.; Wang, J.; Li, Q.; Wang, Y. Filtrating Colorectal Cancer Associated Genes by Integrated Analyses of Global DNA Methylation and Hydroxymethylation in Cancer and Normal Tissue. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Hong, Y.; Hong, S.-H.; Oh, Y.-M.; Shin, S.-H.; Choi, S.S.; Kim, W.J. Identification of Lung Cancer Specific Differentially Methylated Regions Using Genome-Wide DNA Methylation Study. Mol. Cell. Toxicol. 2018, 14, 315–322. [Google Scholar] [CrossRef]
- Hanley, M.P.; Hahn, M.A.; Li, A.X.; Wu, X.; Lin, J.; Wang, J.; Choi, A.H.; Ouyang, Z.; Fong, Y.; Pfeifer, G.P.; et al. Genome-Wide DNA Methylation Profiling Reveals Cancer-Associated Changes within Early Colonic Neoplasia. Oncogene 2017, 36, 5035–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Kahraman, A.; Law, C.W.; Lindsay, H.; Nowicka, M.; Weber, L.M.; Zhou, X. Statistical Methods for Detecting Differentially Methylated Loci and Regions. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.-L.; Zhang, K.; Zhang, K. Identification of Methylation Haplotype Blocks Aids in Deconvolution of Heterogeneous Tissue Samples and Tumor Tissue-of-Origin Mapping from Plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wu, X.; Zheng, J.; Dong, D. DNA Methylome Profiling of Circulating Tumor Cells in Lung Cancer at Single Base-Pair Resolution. Oncogene 2021, 40, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Zikan, M.; Wahl, B.; Lempiäinen, H.; Paprotka, T.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Eichner, J.; et al. The Potential of Circulating Tumor DNA Methylation Analysis for the Early Detection and Management of Ovarian Cancer. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Reinhardt, R.; Jeltsch, A. Accuracy of DNA Methylation Pattern Preservation by the Dnmt1 Methyltransferase. Nucleic Acids Res. 2006, 34, 1182–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzini, S.; Kurtz, D.M.; Chabon, J.J.; Moding, E.J.; Hori, S.S.; Gambhir, S.S.; Alizadeh, A.A.; Diehn, M.; Reiter, J.G. A Mathematical Model of CtDNA Shedding Predicts Tumor Detection Size. Sci. Adv. 2020, 6, eabc4308. [Google Scholar] [CrossRef]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-Invasive Cancer Diagnosis and Tissue-of-Origin Prediction Using Methylation Profiles of Cell-Free DNA. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.-C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and Non-Invasive Cancer Detection at the Resolution of Individual Reads Using Cell-Free DNA Methylation Sequencing Data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef] [Green Version]
- Huan, Q.; Zhang, Y.; Wu, S.; Qian, W. HeteroMeth: A Database of Cell-to-Cell Heterogeneity in DNA Methylation. Genom. Proteom. Bioinform. 2018, 16, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Lee, H.J.; Reik, W.; Stegle, O. DeepCpG: Accurate Prediction of Single-Cell DNA Methylation States Using Deep Learning. Genome Biol. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Guo, H.; Ren, Y.; Hou, Y.; Dong, J.; Li, R.; Lian, Y.; Fan, X.; Hu, B.; Gao, Y.; et al. Single-Cell DNA Methylome Sequencing of Human Preimplantation Embryos. Nat. Genet. 2018, 50, 12–19. [Google Scholar] [CrossRef]
- Sen, M.; Mooijman, D.; Chialastri, A.; Boisset, J.-C.; Popovic, M.; Heindryckx, B.; Chuva de Sousa Lopes, S.M.; Dey, S.S.; van Oudenaarden, A. Strand-Specific Single-Cell Methylomics Reveals Distinct Modes of DNA Demethylation Dynamics during Early Mammalian Development. Nat. Commun. 2021, 12, 1–10. [Google Scholar] [CrossRef]
- Yu, B.; Dong, X.; Gravina, S.; Kartal, Ö.; Schimmel, T.; Cohen, J.; Tortoriello, D.; Zody, R.; Hawkins, R.D.; Vijg, J. Genome-Wide, Single-Cell DNA Methylomics Reveals Increased Non-CpG Methylation during Human Oocyte Maturation. Stem Cell Rep. 2017, 9, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Richardson, B. DNA Methylation and Autoimmune Disease. Clin. Immunol. 2003, 109, 72–79. [Google Scholar] [CrossRef]
- Fuchikami, M.; Morinobu, S.; Segawa, M.; Okamoto, Y.; Yamawaki, S.; Ozaki, N.; Inoue, T.; Kusumi, I.; Koyama, T.; Tsuchiyama, K.; et al. DNA Methylation Profiles of the Brain-Derived Neurotrophic Factor (BDNF) Gene as a Potent Diagnostic Biomarker in Major Depression. PLoS ONE 2011, 6, e23881. [Google Scholar] [CrossRef]
- Uzun, Y.; Wu, H.; Tan, K. Predictive Modeling of Single-Cell DNA Methylome Data Enhances Integration with Transcriptome Data. Genome Res. 2020. [Google Scholar] [CrossRef]
- Adey, A.; Shendure, J. Ultra-Low-Input, Tagmentation-Based Whole-Genome Bisulfite Sequencing. Genome Res. 2012, 22, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET Enzymes in DNA Methylation, Development, and Cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, J.; Siejka-Zielińska, P.; Weldon, C.; Roberts, H.; Lopopolo, M.; Magri, A.; D’Arienzo, V.; Harris, J.M.; McKeating, J.A.; et al. Accurate Targeted Long-Read DNA Methylation and Hydroxymethylation Sequencing with TAPS. Genome Biol. 2020, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhou, J.; Tian, W.; Luo, C.; Bartlett, A.; Aldridge, A.; Lucero, J.; Osteen, J.K.; Nery, J.R.; Chen, H.; et al. DNA Methylation Atlas of the Mouse Brain at Single-Cell Resolution. BioRxiv 2020. [Google Scholar] [CrossRef]
- Scott, C.A.; Duryea, J.D.; MacKay, H.; Baker, M.S.; Laritsky, E.; Gunasekara, C.J.; Coarfa, C.; Waterland, R.A. Identification of Cell Type-Specific Methylation Signals in Bulk Whole Genome Bisulfite Sequencing Data. Genome Biol. 2020, 21, 1–23. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basis | PBAT | Single-Cell Method | Related Method 2 | Advanced Strategy 3 | Ref. |
|---|---|---|---|---|---|
| RRBS | No | scRRBS | None | Single-tube reaction, tRNA carrier | [46] |
| Q-RRBS | scRRBS | UMI adaptor | [47] | ||
| MID-RRBS | None | Microfluidics | [41] | ||
| scTrio-seq | scRRBS, scRNA-seq | Multi-omics (RNA, CNV) | [48] | ||
| scMT-seq | scRRBS, Smart-seq2 | Multi-omics (RNA) | [49] | ||
| WGBS | Yes 4 | scBS-seq | None | Single-tube reaction, Preamplification, SPRI bead | [50] |
| scPBAT | None | Non-preamplification (repeat-specialized) | [51] | ||
| scWGBS | None | Non-preamplification | [52] | ||
| snmC-seq2 5 | snmC-seq | Single-strand library preparation method | [53] | ||
| sci-MET | None | Transposase tagmentation, combinatorial indexing | [54] | ||
| scTrio-seq2 | scBS-seq, scTrio-seq | Multi-omics (RNA, CNV) | [55] | ||
| scM&T-seq | scBS-seq, G&T-seq | Multi-omics (RNA) | [56] | ||
| scNMT-seq | scM&T-seq, NOMe-seq | Multi-omics (RNA, chromatin accessibility) | [57] | ||
| iscCOOL-seq 5 | scCOOL-seq, NOMe-seq | Multi-omics (chromatin accessibility, CNV, ploidy) | [58] | ||
| scNOMe-seq | NOMe-seq | Multi-omics (chromatin accessibility) | [59] | ||
| MSRE | No | scCGI-seq | None | MDA | [60] |
| Aligner | Index Method | URL |
|---|---|---|
| BSMAP | Wild-card | https://code.google.com/archive/p/bsmap/ |
| RMAPBS | Wild-card | https://github.com/smithlabcode/rmap |
| Bismark | Three-letter | https://github.com/FelixKrueger/Bismark |
| BS-Seeker | Three-letter | version1: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-203 version2: https://github.com/BSSeeker/BSseeker2 version3: https://github.com/khuang28jhu/bs3/ |
| BitmapperBS | Three-letter | https://github.com/chhylp123/BitMapperBS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.; Heo, S.; Lee, J.; Bang, D. Introduction to Single-Cell DNA Methylation Profiling Methods. Biomolecules 2021, 11, 1013. https://doi.org/10.3390/biom11071013
Ahn J, Heo S, Lee J, Bang D. Introduction to Single-Cell DNA Methylation Profiling Methods. Biomolecules. 2021; 11(7):1013. https://doi.org/10.3390/biom11071013
Chicago/Turabian StyleAhn, Jongseong, Sunghoon Heo, Jihyun Lee, and Duhee Bang. 2021. "Introduction to Single-Cell DNA Methylation Profiling Methods" Biomolecules 11, no. 7: 1013. https://doi.org/10.3390/biom11071013