
Chalcone Derivatives: Role in Anticancer Therapy


Abstract
:1. Introduction
2. Strategies Employed to Produce Anticancer Chalcones
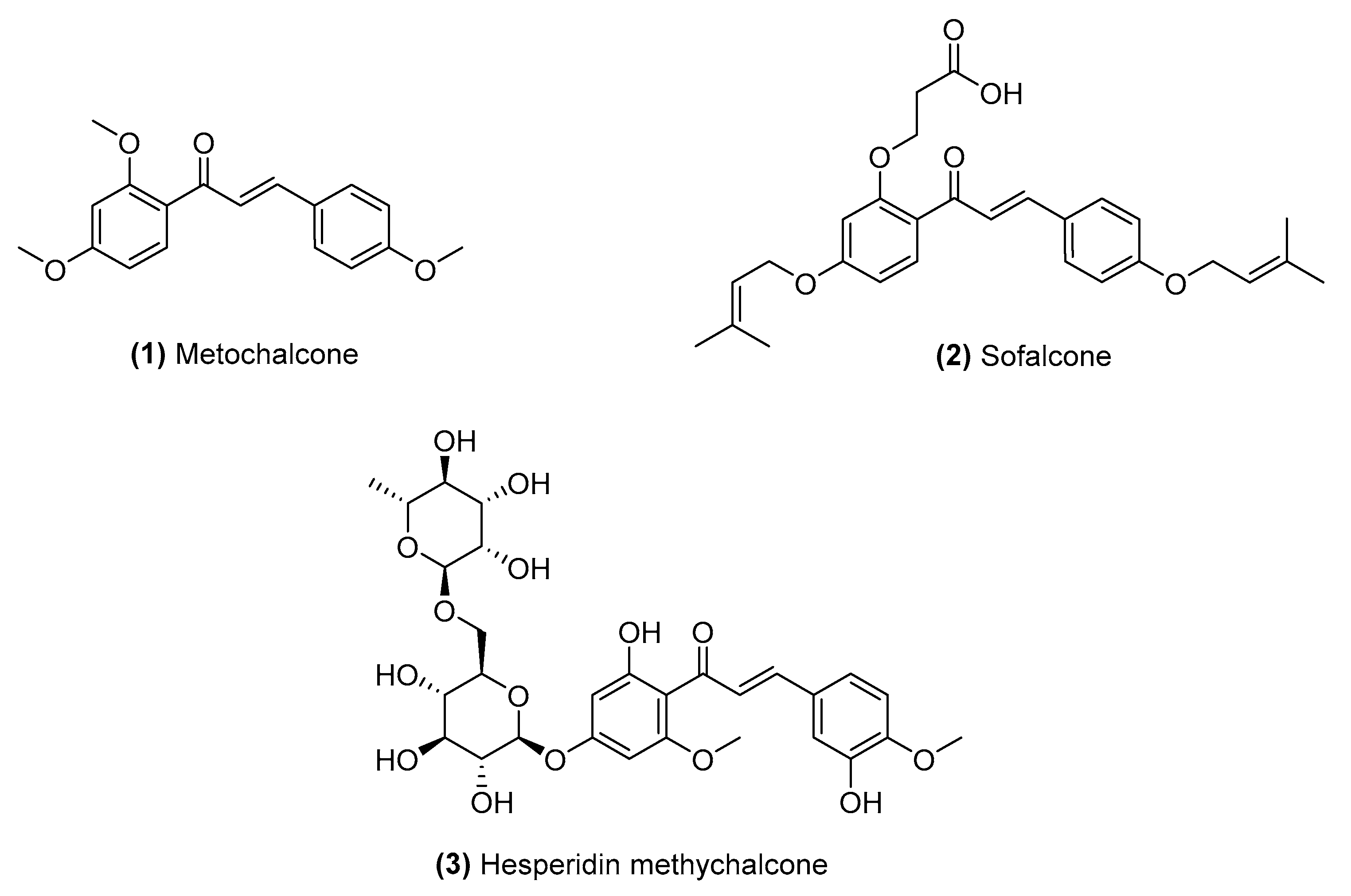
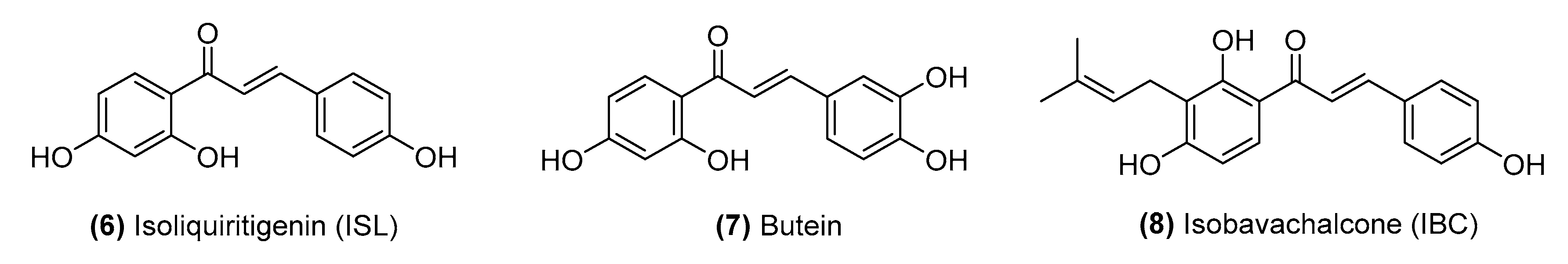
2.1. Naturally Occurring Chalcones
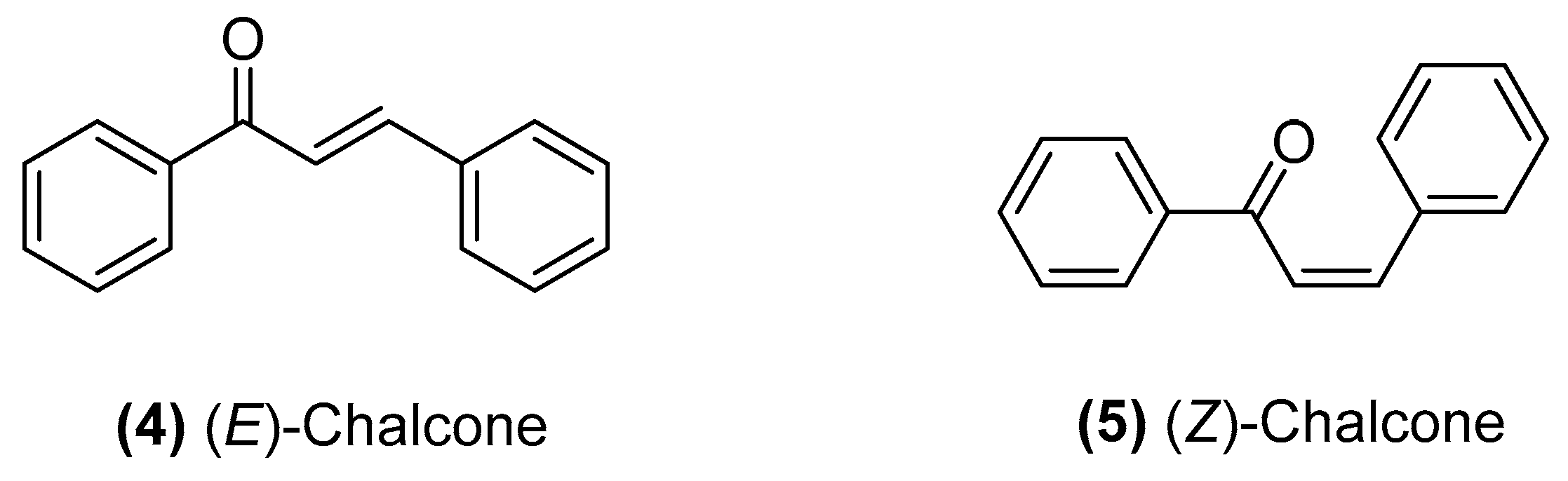
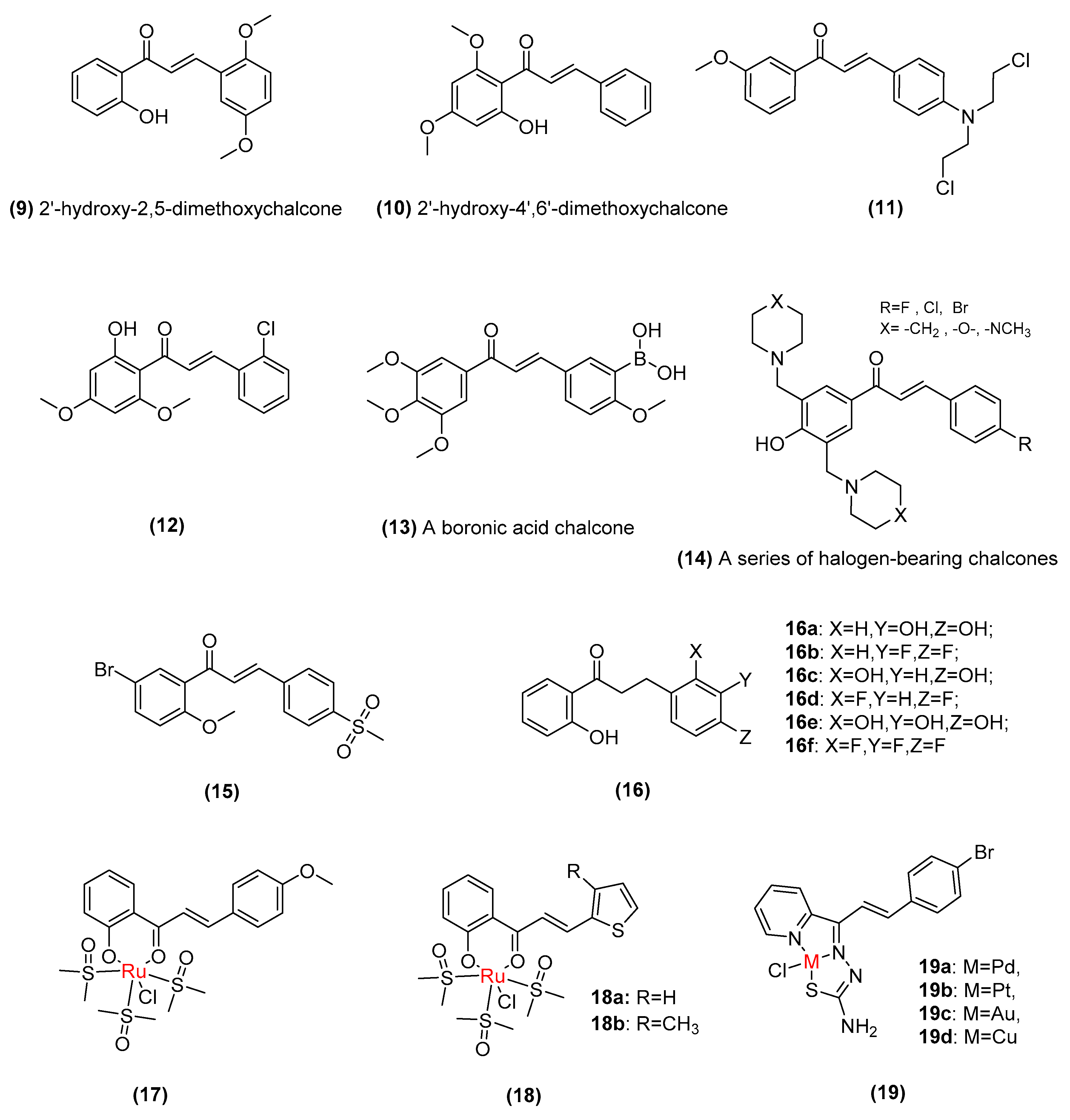
2.2. Synthetic Chalcone Derivatives
2.3. Chalcone Hybrids
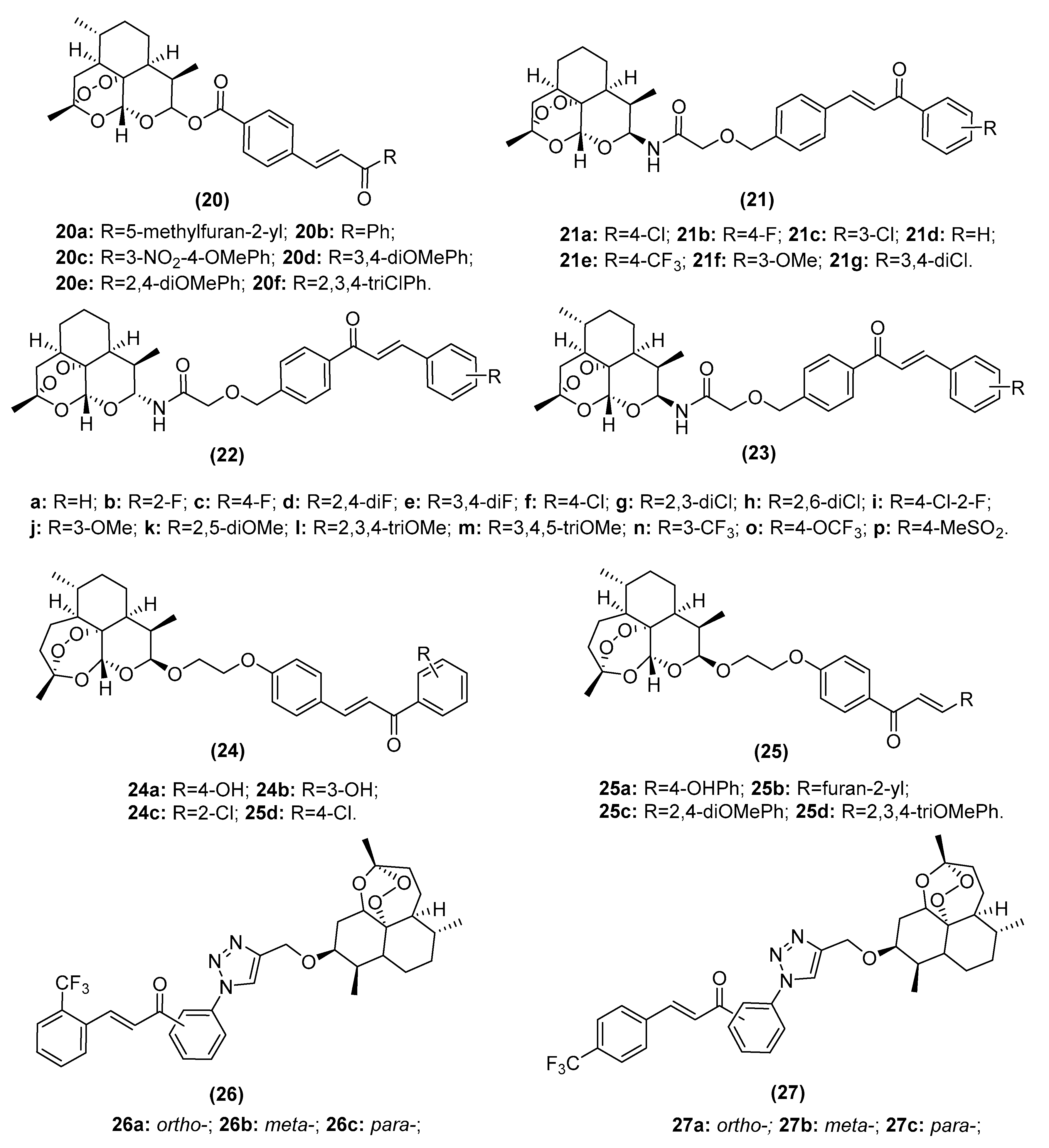
2.3.1. Artemisinin–Chalcone Hybrids
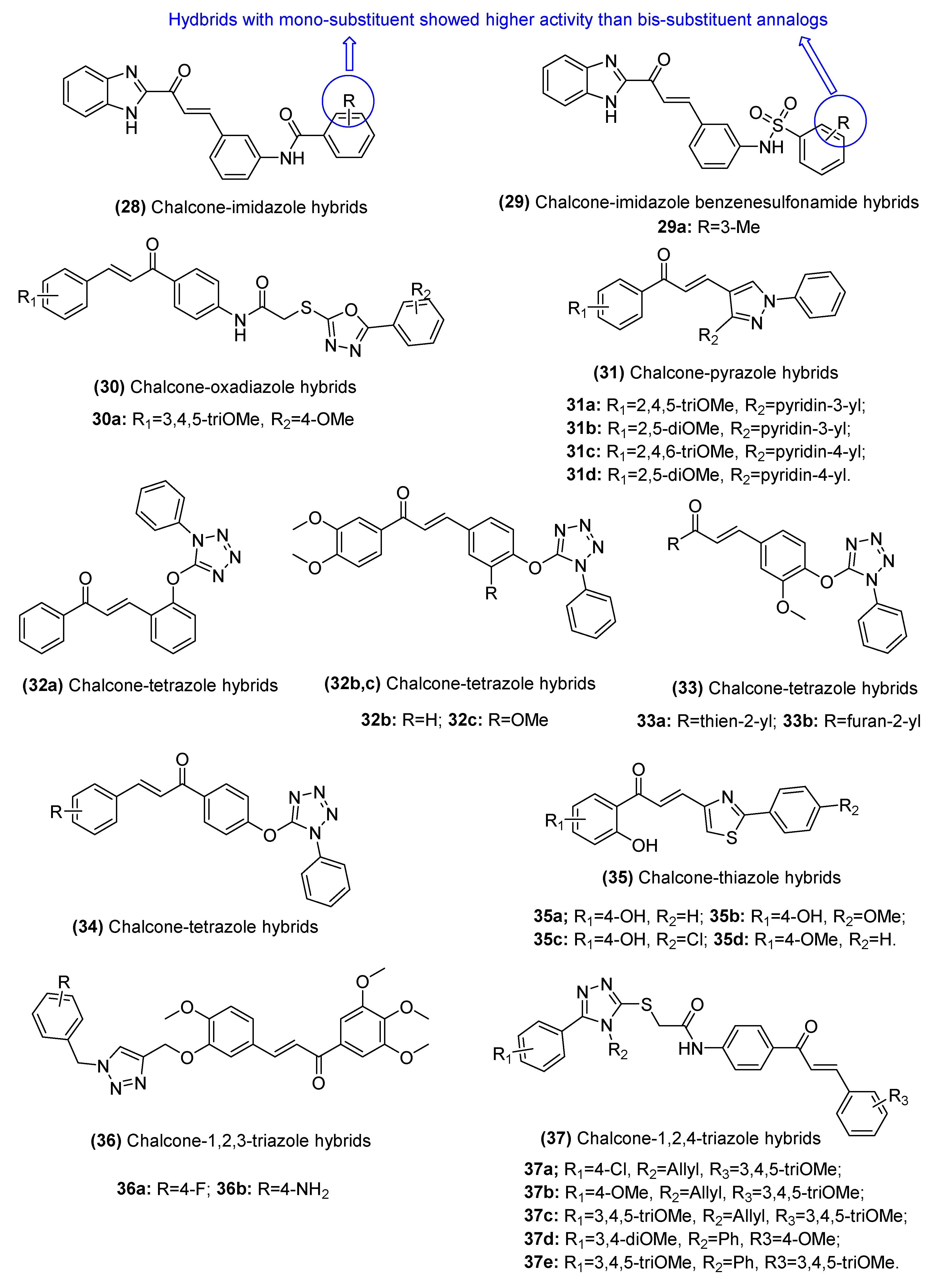
2.3.2. Chalcone–Azole Hybrids
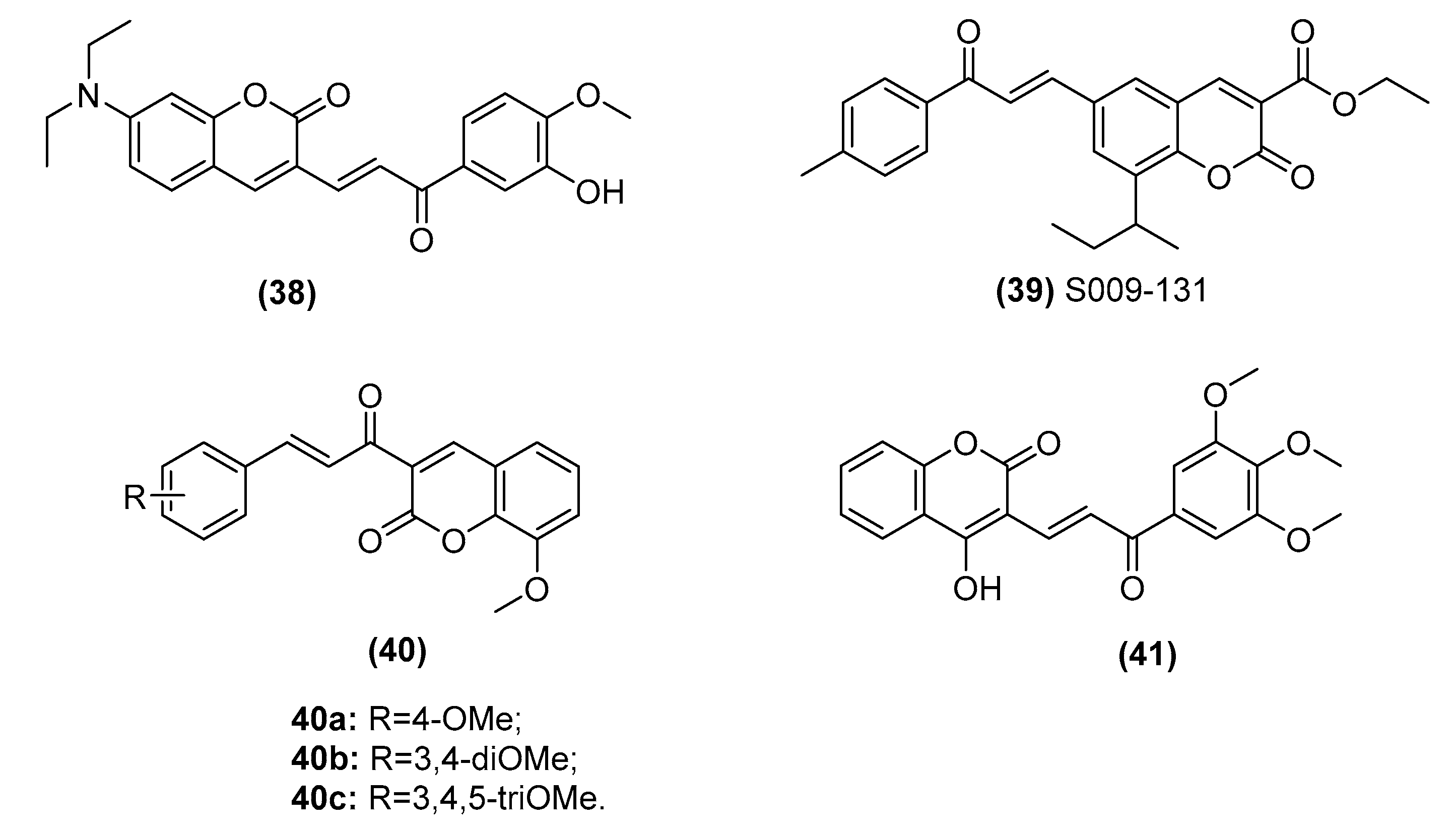
2.3.3. Chalcone–Coumarin Hybrids
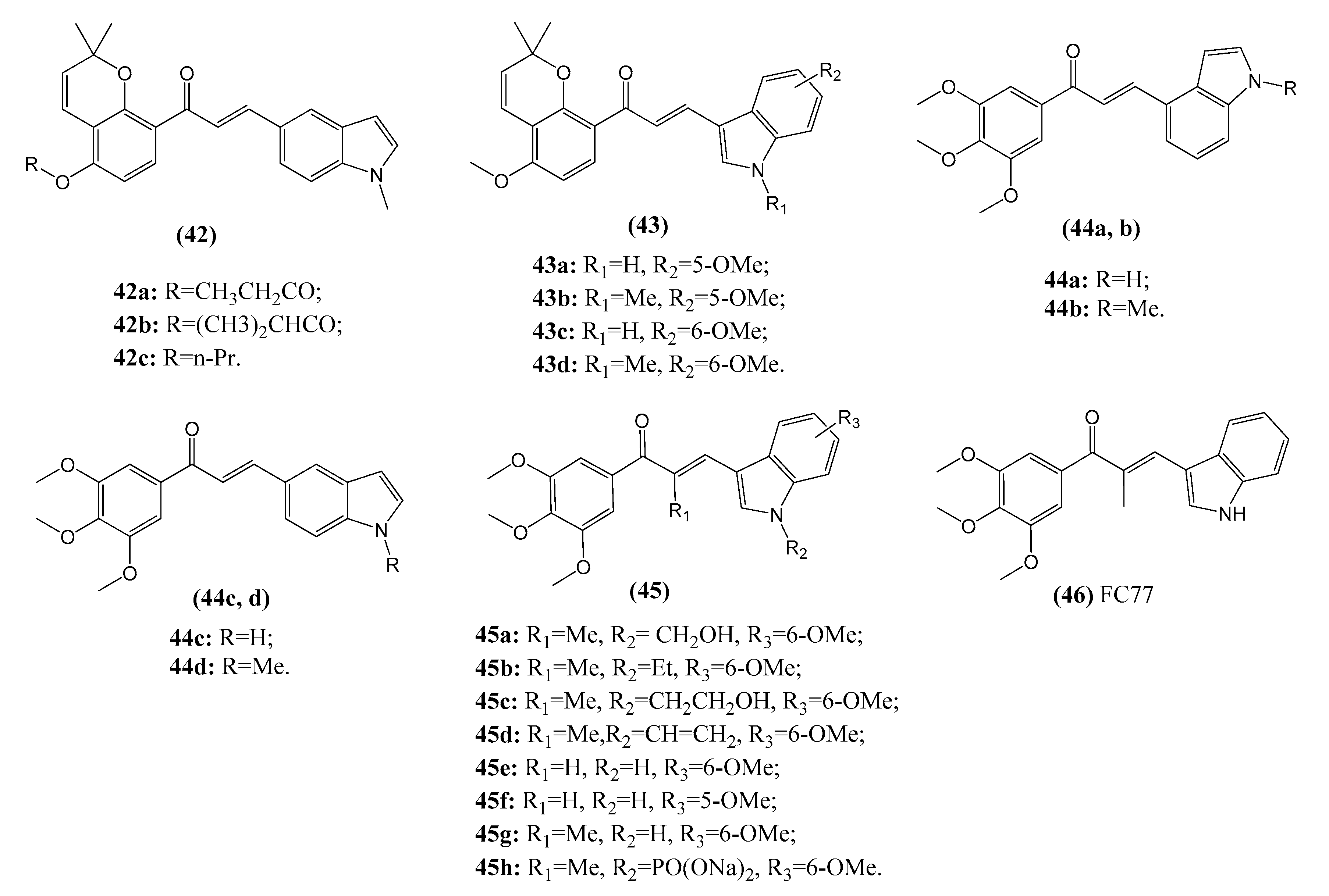
2.3.4. Chalcone–Indole Hybrids
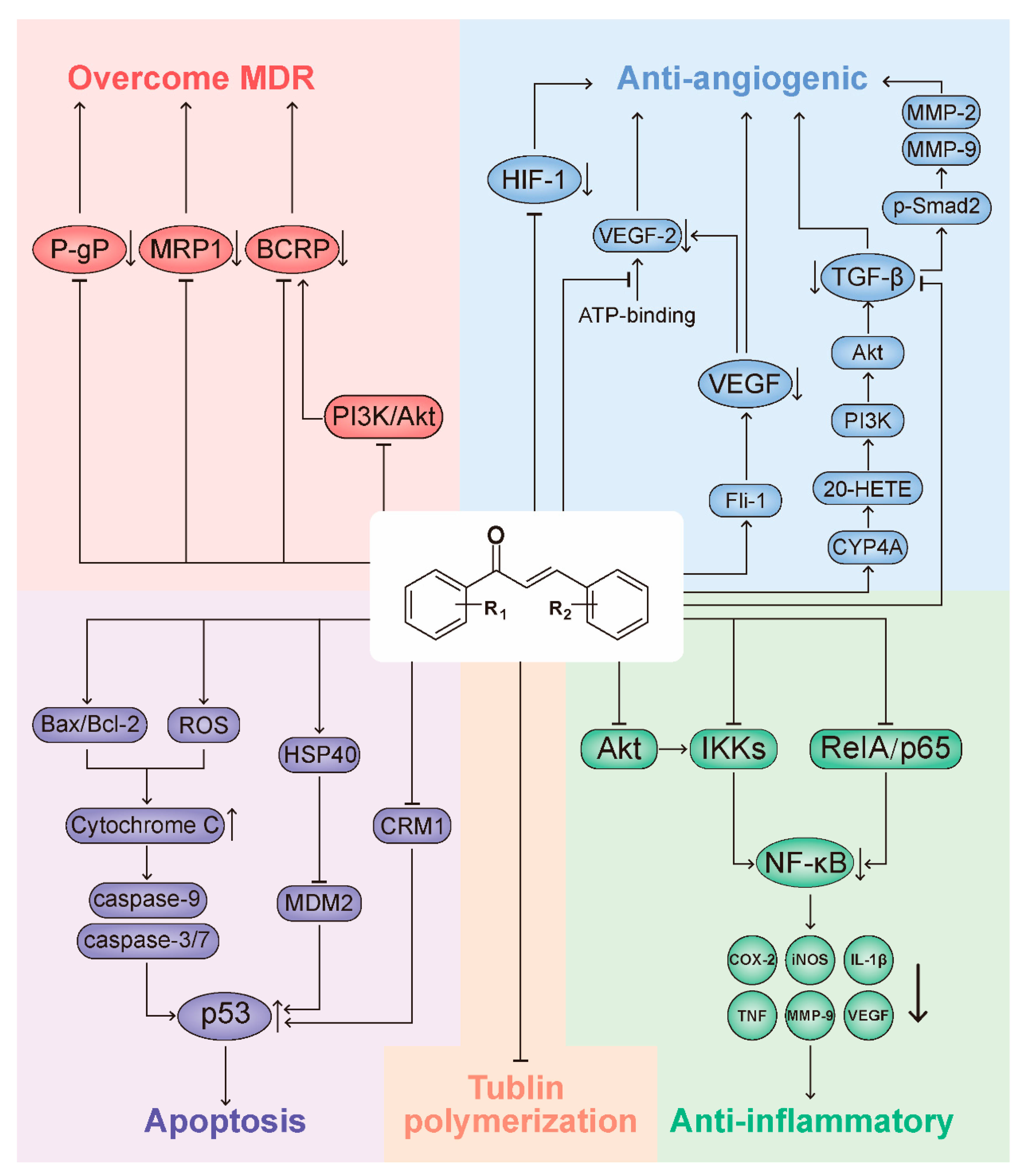
3. Representative Mechanisms of Anticancer Action of Chalcones
3.1. Chalcones Target the p53 Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lead Compounds | Mechanisms of Action | Reference |
|---|---|---|
| Trans-chalcone (1) | Enhances the expression of HSP40 and inhibits CRM1, thereby blocking MDM2-mediated ubiquitination of p53 and enhancing p53 accumulation in the nucleus. | Silva et al. [128,130] |
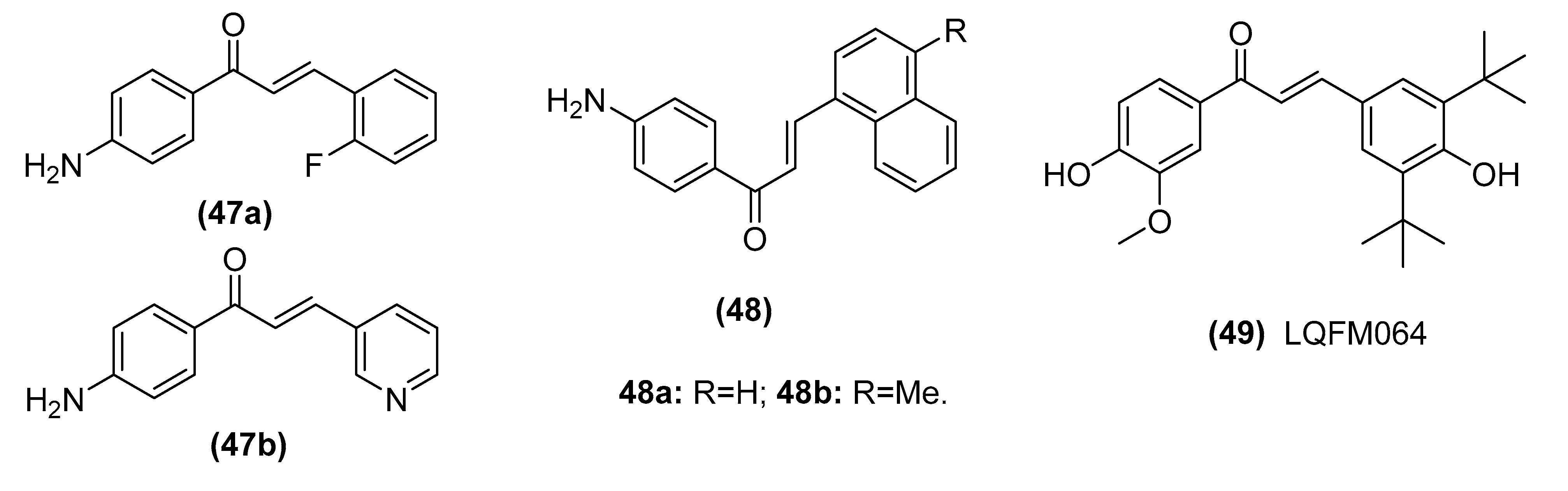
| 2-fluoro-4’-aminochalcone (47a) and 3-pyridyl-4’-aminochalcone (47b) | Induces apoptosis by up-regulating p53 expression and without Sp1 expression alteration in MCF-7 cells | Dos Santos et al. [129] |
| Trans-chalcone (1) | Induces death by autophagy mediated by p53 up-regulation and Wnt/β-catenin down-regulation on human hepatocellular carcinoma HuH7.5 cell line | Siqueira et al. [131] |
| Chalcone derivative 4′-aminochalcone (48) | Suppresses migration and invasion of osteosarcoma cells mediated by p53 regulating EMT-related genes | Seba et al. [132] |
| Coumarlcone hybrid (39, S009-131) | Instigates DNA damage, disrupts p53-MDM2 interaction and stabilizes p53 through post-translational modifications in both vitro and vivo of HeLa cells | Sashidhara et al. [81,82] |
| (E)-3-(3, 5-di-ter-butyl-4-hydroxyphenyl)-1-(4-hydroxy-3-methoxyphenyl) prop-2-en-1-one (49, LQFM064) | Induces cell cycle arrest at the G0/G1 phase with up-regulation of p53 and p21 | Cabral et al. [133] |
3.2. Chalcones Target Tubulin Polymerization
3.3. Chalcones and the NF-κB Pathway
3.4. Chalcones as Inhibitors of Angiogenesis
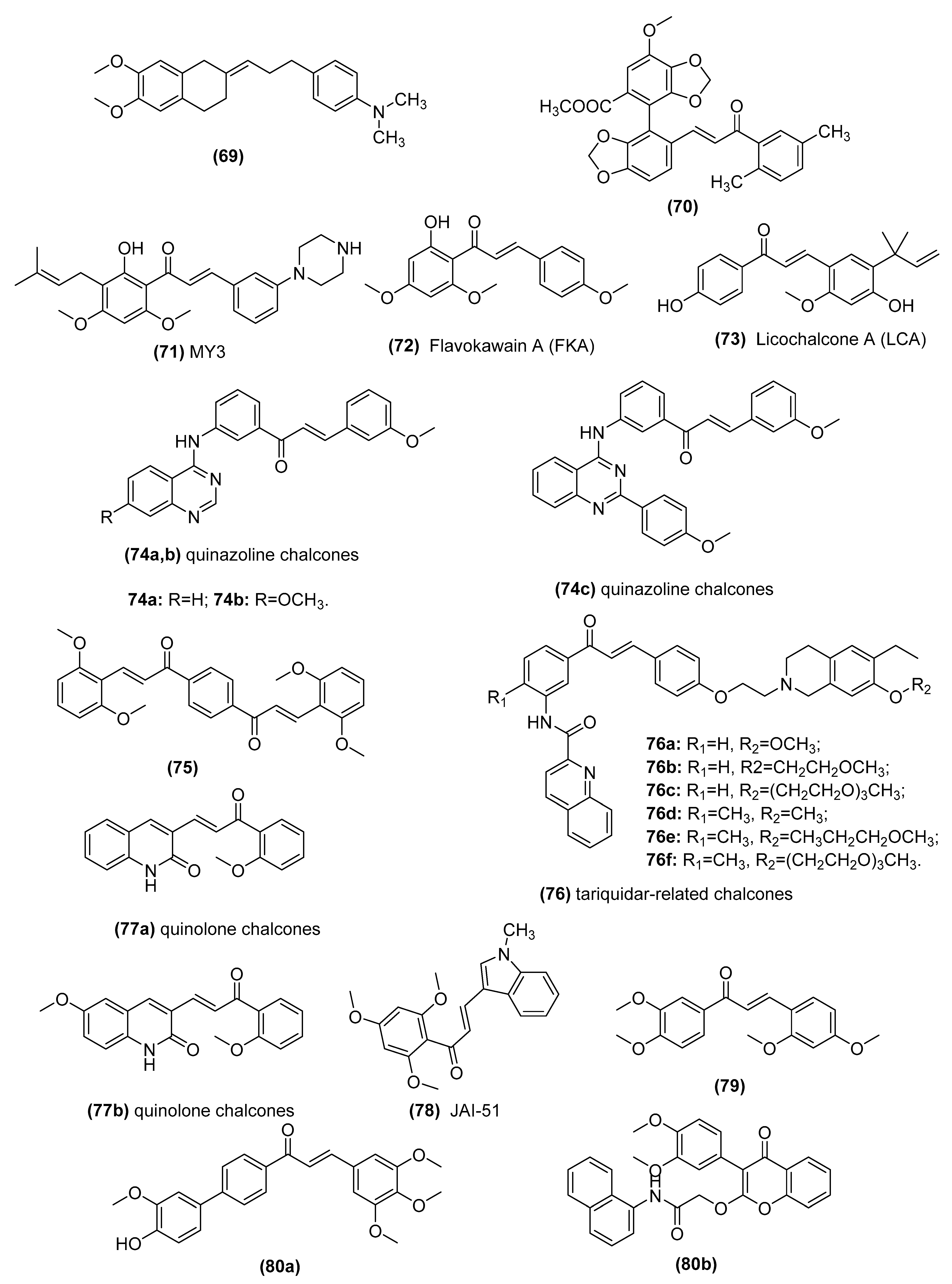
3.5. Chalcones as Inhibitors of MDR Channels
| Lead Compounds | Mechanisms of Action | Reference |
|---|---|---|
| 2-[3-(4-Dimethylaminophenyl)-prop-2-en-yliden]-5,6- dimethoxyindan-1-one (69) | Inhibitors of human P-glycoprotein | Parveen et al. [176] and Ngo et al. [177] |
| Bifendate chalcone hybrids (70) | P-gp inhibitors in K562/A02 cells whichoverexpress P-gp (induced by adriamycin) | Gu et al. [178] |
| MY3 (71) | Inhibits expression of P-gp and enhances the efficacy of DOX against the tumor xenografts bearing MCF-7/DOX cells | Yin et al. [179] |
| Flavokawain A (FKA) (72) | Inhibits P-gp protein expression by blocking the PI3K/Akt pathway in PTX-resistant lung cancer A549 cells | Li et al. [180,181] |
| Licochalcone A (LCA) (73) | Binds ABCG2 in the transmembrane substrate-binding pocket and reverses ABCG2-mediated multidrug resistance in human multidrug-resistant cancer cell lines | Wu et al. [182] |
| Quinazoline chalcones (74) | Modulators of breast cancer resistance protein (BCRP/ABCG2) in P-gp overexpressing MDCK II cells | Kraege et al. [114] |
| Symmetric bis-chalcones (75) | Inhibits mitoxantrone efflux from ABCG2-transfected HEK293 cells by stimulating ABCG2 basal ATPase activity | Winter et al. [183] |
| Tariquidar-related chalcones (76) | ABCG2 Modulators in ABCG2-overexpressing MCF-7/Topo cells | Peña-Solórzano et al. [109] |
| Quinolone chalcones (77) | Targets colchicine-binding pocket and kills multidrug-resistant cancer cells by inhibiting tubulin activity and MRP1 function | Lindamulage et al. [111] |
| A novel chalcone derivative, JAI-51 (78) | A microtubule-depolymerizing agent and an inhibitor of P-gp and BCRP in vitro and in vivo of glioblastoma models | Boumendjel et al. [184] |
| Nonbasic chalcone (79) | A dual ABCG2/ABCB1 inhibitor in S1-M1-80 (mitoxantrone (MX)-selected ABCG2-overexpressing of human colorectal cancer cell line S1), NCI-H460/MX20 (MX-selected ABCG2-overexpressing of human lung cancer cell line NCI-H460) | Han et al. [185] and Cai et al. [186] |
| Novel chalcone and flavone derivatives (80) | Selective and dual inhibitors of the transport proteins ABCB1 and ABCG2 in ABCG2-overexpressing MDCK II BCRP cells | Silbermann et al. [99] |
3.6. Other Molecular Cancer Targets Modulated by Chalcones and Target Identification
4. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hussain, S.; Singh, A.; Nazir, S.U.; Tulsyan, S.; Khan, A.; Kumar, R.; Bashir, N.; Tanwar, P.; Mehrotra, R. Cancer drug resistance: A fleet to conquer. J. Cell. Biochem. 2019, 120, 14213–14225. [Google Scholar] [CrossRef]
- Jandial, D.D.; Blair, C.A.; Zhang, S.; Krill, L.S.; Zhang, Y.-B.; Zi, X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, P.; Sharghi, M.; Mansourkhani, S.M.; Ardekan, A.P.; Jouybari, L.; Daraei, N.; Peiro, K.; Mohamadian, S.; Rezaei, M.; Heidari, M.; et al. Medicinal Plants in the Prevention and Treatment of Colon Cancer. Oxid. Med. Cell. Longev. 2019, 2019, 2075614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imperatore, C.; Della Sala, G.; Casertano, M.; Luciano, P.; Aiello, A.; Laurenzana, I.; Piccoli, C.; Menna, M. In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products. Mar. Drugs 2019, 17, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzo, E. Synthesis of Marine Natural Products and Molecules Inspired by Marine Substances. Mar. Drugs 2021, 19, 208. [Google Scholar] [CrossRef]
- Karthikeyan, C.; Moorthy, N.S.H.N.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anticancer Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Zhou, B. Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. 2015, 5, 388–404. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Gao, F.; Huang, G.; Xiao, J. Chalcone hybrids as potential anticancer agents: Current development, mechanism of action, and structure-activity relationship. Med. Res. Rev. 2020, 40, 2049–2084. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone Derivatives: Anti-inflammatory Potential and Molecular Targets Perspectives. Curr. Top. Med. Chem. 2017, 17, 3146–3169. [Google Scholar] [CrossRef]
- Rocha, S.; Ribeiro, D.; Fernandes, E.; Freitas, M. A Systematic Review on Anti-diabetic Properties of Chalcones. Curr. Med. Chem. 2020, 27, 2257–2321. [Google Scholar] [CrossRef]
- Xu, S.; Chen, M.; Chen, W.; Hui, J.; Ji, J.; Hu, S.; Zhou, J.; Wang, Y.; Liang, G. Chemopreventive effect of chalcone derivative, L2H17, in colon cancer development. BMC Cancer 2015, 15, 870. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhang, M.; Lu, Q.; Xie, J.; Wu, J.; Chen, C. A novel chalcone derivative exerts anti-inflammatory and anti-oxidant effects after acute lung injury. Aging 2019, 11, 7805–7816. [Google Scholar] [CrossRef]
- Henry, E.J.; Bird, S.J.; Gowland, P.; Collins, M.; Cassella, J.P. Ferrocenyl chalcone derivatives as possible antimicrobial agents. J. Antibiot. 2020, 73, 299–308. [Google Scholar] [CrossRef]
- de Mello, M.V.P.; Abrahim-Vieira, B.A.; Domingos, T.F.S.; de Jesus, J.B.; de Sousa, A.C.C.; Rodrigues, C.R.; Souza, A.M.T. A comprehensive review of chalcone derivatives as antileishmanial agents. Eur. J. Med. Chem. 2018, 150, 920–929. [Google Scholar] [CrossRef]
- Cheng, P.; Yang, L.; Huang, X.; Wang, X.; Gong, M. Chalcone hybrids and their antimalarial activity. Arch. Pharm. 2020, 353, e1900350. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.A.; Jasamai, M.; Jantan, I. Synthesis and biological evaluation of chalcone derivatives (mini review). Mini Rev. Med. Chem. 2012, 12, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.H.; Wang, R.F.; Guo, S.Z.; Liu, B. An update on antitumor activity of naturally occurring chalcones. Evid. Based Complement. Alternat. Med. 2013, 2013, 815621. [Google Scholar] [CrossRef] [PubMed]
- Rozmer, Z.; Perjési, P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2014, 15, 87–120. [Google Scholar] [CrossRef]
- Wang, K.L.; Yu, Y.C.; Hsia, S.M. Perspectives on the Role of Isoliquiritigenin in Cancer. Cancers 2021, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Tang, H.; Liu, P.; Shen, J.; Guan, X.; Xie, X.; Gao, J.; Xiong, L.; Jia, L.; Chen, J.; et al. Isoliquiritigenin modulates miR-374a/PTEN/Akt axis to suppress breast cancer tumorigenesis and metastasis. Sci. Rep. 2017, 7, 9022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.-H.; Chiang, Y.-F.; Shieh, T.-M.; Chen, H.-Y.; Shih, C.-K.; Wang, T.-H.; Wang, K.-L.; Huang, T.-C.; Hong, Y.-H.; Li, S.-C.; et al. Dietary Compound Isoliquiritigenin, an Antioxidant from Licorice, Suppresses Triple-Negative Breast Tumor Growth via Apoptotic Death Program Activation in Cell and Xenograft Animal Models. Antioxidants 2020, 9, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.-Y.; Huang, T.-C.; Shieh, T.-M.; Wu, C.-H.; Lin, L.-C.; Hsia, S.-M. Isoliquiritigenin Induces Autophagy and Inhibits Ovarian Cancer Cell Growth. Int. J. Mol. Sci. 2017, 18, 2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Lai, Y.; Li, Y.; Shu, N.; Wang, Z.; Wang, Y.; Li, Y.; Chen, Z. Antineoplastic activity of isoliquiritigenin, a chalcone compound, in androgen-independent human prostate cancer cells linked to G2/M cell cycle arrest and cell apoptosis. Eur. J. Pharmacol. 2018, 821, 57–67. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Wang, Y.; Liu, X.; Liu, Y.; Li, Y.; Chen, H.; Fan, C.; Wu, D.; Yang, J. Inhibition of COX-2, mPGES-1 and CYP4A by isoliquiritigenin blocks the angiogenic Akt signaling in glioma through ceRNA effect of miR-194-5p and lncRNA NEAT1. J. Exp. Clin. Cancer Res. CR 2019, 38, 371. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.; Zeng, H.; Xia, F.; Ji, Q.; Xue, J.; Ren, R.; Que, F.; Zhou, B. The dietary flavonoid isoliquiritigenin induced apoptosis and suppressed metastasis in melanoma cells: An in vitro and in vivo study. Life Sci. 2021, 264, 118598. [Google Scholar] [CrossRef]
- Wang, N.; Wang, Z.; Peng, C.; You, J.; Shen, J.; Han, S.; Chen, J. Dietary compound isoliquiritigenin targets GRP78 to chemosensitize breast cancer stem cells via β-catenin/ABCG2 signaling. Carcinogenesis 2014, 35, 2544–2554. [Google Scholar] [CrossRef] [Green Version]
- Jayasooriya, R.G.P.T.; Molagoda, I.M.N.; Park, C.; Jeong, J.-W.; Choi, Y.H.; Moon, D.-O.; Kim, M.-O.; Kim, G.-Y. Molecular chemotherapeutic potential of butein: A concise review. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 112, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, G.; Rathnakaram, S.R.; Monisha, J.; Bordoloi, D.; Roy, N.K.; Kunnumakkara, A.B. Potential of butein, a tetrahydroxychalcone to obliterate cancer. Phytomedicine 2015, 22, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Di, S.; Fan, C.; Ma, Z.; Li, M.; Guo, K.; Han, D.; Li, X.; Mu, D.; Yan, X. PERK/eIF-2α/CHOP Pathway Dependent ROS Generation Mediates Butein-induced Non-small-cell Lung Cancer Apoptosis and G2/M Phase Arrest. Int. J. Biol. Sci. 2019, 15, 1637–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Li, M.; Yu, X.; Liu, T.; Li, T.; Zhou, L.; Liu, W.; Li, W.; Gao, F. Butein suppresses hepatocellular carcinoma growth via modulating Aurora B kinase activity. Int. J. Biol. Sci. 2018, 14, 1521–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, D.-O.; Kim, M.-O.; Choi, Y.H.; Hyun, J.W.; Chang, W.Y.; Kim, G.-Y. Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett. 2010, 288, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.-O.; Choi, Y.H.; Moon, S.-K.; Kim, W.-J.; Kim, G.-Y. Butein suppresses the expression of nuclear factor-kappa B-mediated matrix metalloproteinase-9 and vascular endothelial growth factor in prostate cancer cells. Toxicol. In Vitro 2010, 24, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Akhtar, N.; Khan, A.; Khan, K.A.; Haqqi, T.M. Butrin, isobutrin, and butein from medicinal plant Butea monosperma selectively inhibit nuclear factor-kappaB in activated human mast cells: Suppression of tumor necrosis factor-alpha, interleukin (IL)-6, and IL-8. J. Pharmacol. Exp. Ther. 2010, 333, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-S.; Jeong, G.-S. Butein provides neuroprotective and anti-neuroinflammatory effects through Nrf2/ARE-dependent haem oxygenase 1 expression by activating the PI3K/Akt pathway. Br. J. Pharmacol. 2016, 173, 2894–2909. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.; Cho, S.-G.; Liu, M.; Aggarwal, B.B. Butein, a tetrahydroxychalcone, suppresses cancer-induced osteoclastogenesis through inhibition of receptor activator of nuclear factor-kappaB ligand signaling. Int. J. Cancer 2011, 129, 2062–2072. [Google Scholar] [CrossRef] [Green Version]
- Kuete, V.; Sandjo, L.P. Isobavachalcone: An overview. Chin. J. Integr. Med. 2012, 18, 543–547. [Google Scholar] [CrossRef]
- Li, B.; Xu, N.; Wan, Z.; Ma, L.; Li, H.; Cai, W.; Chen, X.; Huang, Z.; He, Z. Isobavachalcone exerts anti-proliferative and pro-apoptotic effects on human liver cancer cells by targeting the ERKs/RSK2 signaling pathway. Oncol. Rep. 2019, 41, 3355–3366. [Google Scholar] [CrossRef]
- Li, K.; Zheng, Q.; Chen, X.; Wang, Y.; Wang, D.; Wang, J. Isobavachalcone Induces ROS-Mediated Apoptosis Targeting Thioredoxin Reductase 1 in Human Prostate Cancer PC-3 Cells. Oxid. Med. Cell. Longev. 2018, 2018, 1915828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Qin, X.; Li, P.; Zhang, H.; Lin, T.; Miao, Z.; Ma, S. Isobavachalcone isolated from inhibits cell proliferation and induces apoptosis via inhibiting the AKT/GSK-3β/β-catenin pathway in colorectal cancer cells. Drug Des. Dev. Ther. 2019, 13, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Chen, Y.; Chen, W.; Tang, C.; Zhang, H.; Chen, Y.; Yang, X.; Xu, Z.; Wei, J.; Chen, J. Isobavachalcone sensitizes cells to E2-induced paclitaxel resistance by down-regulating CD44 expression in ER+ breast cancer cells. J. Cell. Mol. Med. 2018, 22, 5220–5230. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Tokuda, H.; Hasegawa, D.; Ukiya, M.; Kimura, Y.; Enjo, F.; Suzuki, T.; Nishino, H. Chalcones and other compounds from the exudates of Angelica keiskei and their cancer chemopreventive effects. J. Nat. Prod. 2006, 69, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Tabata, K.; Arakawa, M.; Ito, Y.; Kimura, Y.; Akihisa, T.; Nagai, H.; Sakuma, A.; Kohno, H.; Suzuki, T. Isobavachalcone, a chalcone constituent of Angelica keiskei, induces apoptosis in neuroblastoma. Biol. Pharm. Bull. 2007, 30, 1878–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, A.; Henklewska, M.; Hernandez Suarez, B.; Luzny, M.; Kozlowska, E.; Obminska-Mrukowicz, B.; Janeczko, T. Chalcone Methoxy Derivatives Exhibit Antiproliferative and Proapoptotic Activity on Canine Lymphoma and Leukemia Cells. Molecules 2020, 25, 4362. [Google Scholar] [CrossRef]
- Elkhalifa, D.; Siddique, A.B.; Qusa, M.; Cyprian, F.S.; El Sayed, K.; Alali, F.; Al Moustafa, A.E.; Khalil, A. Design, synthesis, and validation of novel nitrogen-based chalcone analogs against triple negative breast cancer. Eur. J. Med. Chem. 2020, 187, 111954. [Google Scholar] [CrossRef]
- Kachadourian, R.; Day, B.J.; Pugazhenti, S.; Franklin, C.C.; Genoux-Bastide, E.; Mahaffey, G.; Gauthier, C.; Di Pietro, A.; Boumendjel, A. A synthetic chalcone as a potent inducer of glutathione biosynthesis. J. Med. Chem. 2012, 55, 1382–1388. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Wang, K.; Edler, M.C.; Hamel, E.; Mooberry, S.L.; Paige, M.A.; Brown, M.L. A boronic acid chalcone analog of combretastatin A-4 as a potent anti-proliferation agent. Bioorg. Med. Chem. 2010, 18, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Schobert, R.; Biersack, B.; Dietrich, A.; Knauer, S.; Zoldakova, M.; Fruehauf, A.; Mueller, T. Pt(II) complexes of a combretastatin A-4 analogous chalcone: Effects of conjugation on cytotoxicity, tumor specificity, and long-term tumor growth suppression. J. Med. Chem. 2009, 52, 241–246. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Sakagami, H.; Supuran, C.T. Synthesis and bioactivities of halogen bearing phenolic chalcones and their corresponding bis Mannich bases. J. Enzym. Inhib. Med. Chem. 2016, 31, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, T.; Zhang, Y.; Xu, H.; Li, Y.; Zi, X.; Yu, H.; Li, J.; Jin, C.Y.; Liu, H.M. A new brominated chalcone derivative suppresses the growth of gastric cancer cells in vitro and in vivo involving ROS mediated up-regulation of DR5 and 4 expression and apoptosis. Toxicol. Appl. Pharmacol. 2016, 309, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padhye, S.; Ahmad, A.; Oswal, N.; Dandawate, P.; Rub, R.A.; Deshpande, J.; Swamy, K.V.; Sarkar, F.H. Fluorinated 2′-hydroxychalcones as garcinol analogs with enhanced antioxidant and anticancer activities. Bioorg. Med. Chem. Lett. 2010, 20, 5818–5821. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V.; Singh, S.K. Perspectives of medicinally privileged chalcone based metal coordination compounds for biomedical applications. Eur. J. Med. Chem. 2019, 174, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Saxena, G.; Dixit, S.; Hamidullah; Singh, S.K.; Singh, S.K.; Arshad, M.; Konwar, R. Synthesis, characterization and biological activities of some Ru(II) complexes with substituted chalcones and their applications as chemotherapeutics against breast cancer. J. Mol. Struct. 2016, 1111, 90–99. [Google Scholar] [CrossRef]
- Jovanovic, K.K.; Gligorijevic, N.; Gaur, R.; Mishra, L.; Radulovic, S. Anticancer activity of two ruthenium(II)-DMSO-chalcone complexes: Comparison of cytotoxic, pro-apoptotic and antimetastatic potential. J. BUON 2016, 21, 482–490. [Google Scholar]
- Vutey, V.; Castelli, S.; D’Annessa, I.; Samia, L.B.; Souza-Fagundes, E.M.; Beraldo, H.; Desideri, A. Human topoisomerase IB is a target of a thiosemicarbazone copper(II) complex. Arch. Biochem. Biophys. 2016, 606, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Samia, L.B.; Parrilha, G.L.; Da Silva, J.G.; Ramos, J.P.; Souza-Fagundes, E.M.; Castelli, S.; Vutey, V.; Desideri, A.; Beraldo, H. Metal complexes of 3-(4-bromophenyl)-1-pyridin-2-ylprop-2-en-1-one thiosemicarbazone: Cytotoxic activity and investigation on the mode of action of the gold(III) complex. Biometals 2016, 29, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Shaveta; Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.-S.; Xu, Z.; Chang, L.; Li, C.; Yan, X.-F.; Gao, C.; Ding, C.; Zhao, F.; Shi, F.; Wu, X. Hybrid molecules with potential in vitro antiplasmodial and in vivo antimalarial activity against drug-resistant Plasmodium falciparum. Med. Res. Rev. 2020, 40, 931–971. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, X.; Chen, W.; Chen, Y.; Zhang, Q.; Mo, S.; Lu, J. Dihydroartemisinin: A Potential Natural Anticancer Drug. Int. J. Biol. Sci. 2021, 17, 603–622. [Google Scholar] [CrossRef]
- Wong, Y.K.; Xu, C.; Kalesh, K.A.; He, Y.; Lin, Q.; Wong, W.S.F.; Shen, H.-M.; Wang, J. Artemisinin as an anticancer drug: Recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017, 37, 1492–1517. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Çapcı Karagöz, A.; Reiter, C.; Tsogoeva, S.B. Artemisinin-Derived Dimers: Potent Antimalarial and Anticancer Agents. J. Med. Chem. 2016, 59, 7360–7388. [Google Scholar] [CrossRef] [PubMed]
- Smit, F.J.; van Biljon, R.A.; Birkholtz, L.M.; N’Da, D.D. Synthesis and in vitro biological evaluation of dihydroartemisinyl-chalcone esters. Eur. J. Med. Chem. 2015, 90, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhai, X.; Ren, L.; Meng, H.; Liu, C.; Zhu, W.; Zhao, Y. Design, synthesis and antitumor activity of novel artemisinin derivatives using hybrid approach. Chem. Pharm. Bull. 2011, 59, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zhai, X.; Liu, C.; Li, P.; Li, Y.; Guo, G.; Gong, P. Anti-tumor activity of new artemisinin-chalcone hybrids. Arch. Pharm. 2011, 344, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Gaur, R.; Pathania, A.S.; Malik, F.A.; Bhakuni, R.S.; Verma, R.K. Synthesis of a series of novel dihydroartemisinin monomers and dimers containing chalcone as a linker and their anticancer activity. Eur. J. Med. Chem. 2016, 122, 232–246. [Google Scholar] [CrossRef]
- Kapkoti, D.S.; Singh, S.; Luqman, S.; Bhakuni, R.S. Synthesis of novel 1,2,3-triazole based artemisinin derivatives and their antiproliferative activity. New J. Chem. 2018, 42, 5978–5995. [Google Scholar] [CrossRef]
- Ahmad, K.; Khan, M.K.A.; Baig, M.H.; Imran, M.; Gupta, G.K. Role of Azoles in Cancer Prevention and Treatment: Present and Future Perspectives. Anticancer Agents Med. Chem. 2018, 18, 46–56. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, S.; Li, R.; Zheng, Z.; Yi, H.; Li, Z. Synthesis and biological evaluation of novel synthetic chalcone derivatives as anti-tumor agents targeting Cat L and Cat K. Bioorg. Med. Chem. 2018, 26, 8–16. [Google Scholar] [CrossRef]
- Fathi, M.A.A.; Abd El-Hafeez, A.A.; Abdelhamid, D.; Abbas, S.H.; Montano, M.M.; Abdel-Aziz, M. 1,3,4-oxadiazole/chalcone hybrids: Design, synthesis, and inhibition of leukemia cell growth and EGFR, Src, IL-6 and STAT3 activities. Bioorg. Chem. 2019, 84, 150–163. [Google Scholar] [CrossRef]
- Hawash, M.M.A.; Kahraman, D.C.; Eren, F.; Cetin Atalay, R.; Baytas, S.N. Synthesis and biological evaluation of novel pyrazolic chalcone derivatives as novel hepatocellular carcinoma therapeutics. Eur. J. Med. Chem. 2017, 129, 12–26. [Google Scholar] [CrossRef]
- ElMonaem, H.S.A.; Abdel-Aziz, N.I.; Morsy, M.A.; Badria, F.A.; ElSenduny, F.; El-Ashmawy, M.B.; Moustafa, M.A. Synthesis, In Vitro Antiproliferative Evaluation and Molecular Docking of New tetrazole-chalcone and tetrazole-pyrazoline Hybrids. J. Appl. Pharm. Sci. 2018, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Coman, F.-M.; Mbaveng, A.T.; Leonte, D.; Bencze, L.C.; Vlase, L.; Imre, S.; Kuete, V.; Efferth, T.; Zaharia, V. Heterocycles 44. Synthesis, characterization and anticancer activity of new thiazole ortho-hydroxychalcones. Med. Chem. Res. 2018, 27, 1396–1407. [Google Scholar] [CrossRef]
- Hussaini, S.M.; Yedla, P.; Babu, K.S.; Shaik, T.B.; Chityal, G.K.; Kamal, A. Synthesis and Biological Evaluation of 1,2,3-triazole tethered Pyrazoline and Chalcone Derivatives. Chem. Biol. Drug. Des. 2016, 88, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.F.; Abd El-Hafeez, A.A.; Abbas, S.H.; Abdelhamid, D.; Abdel-Aziz, M. New 1,2,4-triazole-Chalcone hybrids induce Caspase-3 dependent apoptosis in A549 human lung adenocarcinoma cells. Eur. J. Med. Chem. 2018, 151, 705–722. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Dong, J.; Gao, J. Design, synthesis and bioactivity evaluation of coumarin-chalcone hybrids as potential anticancer agents. Bioorg. Chem. 2020, 95. [Google Scholar] [CrossRef]
- Singh, N.; Sarkar, J.; Sashidhara, K.V.; Ali, S.; Sinha, S. Anti-tumour activity of a novel coumarin-chalcone hybrid is mediated through intrinsic apoptotic pathway by inducing PUMA and altering Bax/Bcl-2 ratio. Apoptosis 2014, 19, 1017–1028. [Google Scholar] [CrossRef]
- Ashraf, R.; Hamidullah; Hasanain, M.; Pandey, P.; Maheshwari, M.; Singh, L.R.; Siddiqui, M.Q.; Konwar, R.; Sashidhara, K.V.; Sarkar, J. Coumarin-chalcone hybrid instigates DNA damage by minor groove binding and stabilizes p53 through post translational modifications. Sci. Rep. 2017, 7, 45287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Sarkar, J.; Sinha, S. Synthesis and in vitro evaluation of novel coumarin-chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. [Google Scholar] [CrossRef]
- Elshemy, H.A.H.; Zaki, M.A. Design and synthesis of new coumarin hybrids and insight into their mode of antiproliferative action. Bioorg. Med. Chem. 2017, 25, 1066–1075. [Google Scholar] [CrossRef]
- Patel, K.; Karthikeyan, C.; Solomon, V.R.; Moorthy, N.S.H.N.; Lee, H.; Sahu, K.; Deora, G.S.; Trivedi, P. Synthesis of Some Coumarinyl Chalcones and their Antiproliferative Activity Against Breast Cancer Cell Lines. Lett. Drug Des. Discov. 2011, 8, 308–311. [Google Scholar] [CrossRef]
- Suwito, H.; Hardiyanti, H.; Ul Haq, K.; Kristanti, A.; Khasanah, M. (E)-3-[3-(4-Morpholinophenyl)acryloyl]-2H-chromen-2-one. Molbank 2018, 2018, M1027. [Google Scholar] [CrossRef] [Green Version]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Miller, D.D. Indole molecules as inhibitors of tubulin polymerization: Potential new anticancer agents. Future Med. Chem. 2012, 4, 2085–2115. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Shokrzadeh, M.; Modanloo, M.; Ziar, A.; Riazi, G.H.; Emami, S. New indole-based chalconoids as tubulin-targeting antiproliferative agents. Bioorg. Chem. 2017, 75, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, C.; He, L.; Lei, K.; Wang, F.; Pu, Y.; Yang, Z.; Cao, D.; Ma, L.; Chen, J.; et al. Design, synthesis and biological evaluation of a series of pyrano chalcone derivatives containing indole moiety as novel anti-tubulin agents. Bioorg. Med. Chem. 2014, 22, 2060–2079. [Google Scholar] [CrossRef]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef] [PubMed]
- Cong, H.; Zhao, X.; Castle, B.T.; Pomeroy, E.J.; Zhou, B.; Lee, J.; Wang, Y.; Bian, T.; Miao, Z.; Zhang, W.; et al. An Indole-Chalcone Inhibits Multidrug-Resistant Cancer Cell Growth by Targeting Microtubules. Mol. Pharm. 2018, 15, 3892–3900. [Google Scholar] [CrossRef] [PubMed]
- Smit, F.J.; Bezuidenhout, J.J.; Bezuidenhout, C.C.; N’Da, D.D. Synthesis and in vitro biological activities of ferrocenyl–chalcone amides. Med. Chem. Res. 2016, 25, 568–584. [Google Scholar] [CrossRef]
- Pérès, B.; Nasr, R.; Zarioh, M.; Lecerf-Schmidt, F.; Di Pietro, A.; Baubichon-Cortay, H.; Boumendjel, A. Ferrocene-embedded flavonoids targeting the Achilles heel of multidrug-resistant cancer cells through collateral sensitivity. Eur. J. Med. Chem. 2017, 130, 346–353. [Google Scholar] [CrossRef]
- Farzaneh, S.; Zeinalzadeh, E.; Daraei, B.; Shahhosseini, S.; Zarghi, A. New Ferrocene Compounds as Selective Cyclooxygenase (COX-2) Inhibitors: Design, Synthesis, Cytotoxicity and Enzyme-inhibitory Activity. Anti-cancer agents Med. Chem. 2018, 18, 295–301. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Maluleka, M.M.; Parbhoo, N.; Malindisa, S.T. Synthesis, Evaluation for Cytotoxicity and Molecular Docking Studies of Benzo[c]furan-Chalcones for Potential to Inhibit Tubulin Polymerization and/or EGFR-Tyrosine Kinase Phosphorylation. Int. J. Mol. Sci. 2018, 19, 2552. [Google Scholar] [CrossRef] [Green Version]
- Silbermann, K.; Shah, C.P.; Sahu, N.U.; Juvale, K.; Stefan, S.M.; Kharkar, P.S.; Wiese, M. Novel chalcone and flavone derivatives as selective and dual inhibitors of the transport proteins ABCB1 and ABCG2. Eur. J. Med. Chem. 2019, 164, 193–213. [Google Scholar] [CrossRef]
- Jeon, K.-H.; Yu, H.-B.; Kwak, S.Y.; Kwon, Y.; Na, Y. Synthesis and topoisomerases inhibitory activity of heteroaromatic chalcones. Bioorg. Med. Chem. 2016, 24, 5921–5928. [Google Scholar] [CrossRef]
- Bandeira, P.N.; Lemos, T.L.; Santos, H.S.; de Carvalho, M.C.; Pinheiro, D.P.; de Moraes Filho, M.O.; Pessoa, C.; Barros-Nepomuceno, F.W.; Rodrigues, T.H.; Ribeiro, P.R.; et al. Synthesis, structural characterization, and cytotoxic evaluation of chalcone derivatives. Med. Chem. Res. 2019, 28, 2037–2049. [Google Scholar] [CrossRef]
- Bagul, C.; Rao, G.K.; Makani, V.K.K.; Tamboli, J.R.; Pal-Bhadra, M.; Kamal, A. Synthesis and biological evaluation of chalcone-linked pyrazolo[1,5-]pyrimidines as potential anticancer agents. Medchemcomm 2017, 8, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.Z.; Nguyen, M.A.; Zingales, S.K. Design, Synthesis, and Evaluation of (2-(Pyridinyl)methylene)-1-tetralone Chalcones for Anticancer and Antimicrobial Activity. Med. Chem. 2018, 14, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, S.; Wu, C.; Liu, X.; Tao, H.; Huang, Y.; Liu, Y.; Zheng, P.; Zhu, W. Design, synthesis and activity of novel sorafenib analogues bearing chalcone unit. Bioorg. Med. Chem. Lett. 2016, 26, 5450–5454. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, W.; Shuai, W.; Yang, L.; Bi, Y.; Ma, C.; Yao, H.; Xu, S.; Zhu, Z.; Xu, J. Design, synthesis and biological evaluation of pyridine-chalcone derivatives as novel microtubule-destabilizing agents. Eur. J. Med. Chem. 2019, 173, 1–14. [Google Scholar] [CrossRef]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.-H.; et al. Discovery of Novel Quinoline-Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem. 2019, 62, 993–1013. [Google Scholar] [CrossRef]
- Tabbi, A.; Tebbani, D.; Caporale, A.; Saturnino, C.; Nabavi, S.F.; Giuseppe, P.; Arra, C.; Canturk, Z.; Turan-Zitouni, G.; Merazig, H.; et al. New Adamantyl Chalcones: Synthesis, Antimicrobial and Anticancer Activities. Curr. Top. Med. Chem. 2017, 17, 498–506. [Google Scholar] [CrossRef]
- Peña-Solórzano, D.; Scholler, M.; Bernhardt, G.; Buschauer, A.; König, B.; Ochoa-Puentes, C. Tariquidar-Related Chalcones and Ketones as ABCG2 Modulators. ACS Med. Chem. Lett. 2018, 9, 854–859. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Tzeng, C.-C.; Hsu, C.-Y.; Cheng, C.-M.; Yang, C.-N.; Chen, Y.-L. Discovery of 3-phenylquinolinylchalcone derivatives as potent and selective anticancer agents against breast cancers. Eur. J. Med. Chem. 2015, 97, 306–319. [Google Scholar] [CrossRef]
- Lindamulage, I.K.; Vu, H.-Y.; Karthikeyan, C.; Knockleby, J.; Lee, Y.-F.; Trivedi, P.; Lee, H. Novel quinolone chalcones targeting colchicine-binding pocket kill multidrug-resistant cancer cells by inhibiting tubulin activity and MRP1 function. Sci. Rep. 2017, 7, 10298. [Google Scholar] [CrossRef] [Green Version]
- Wani, Z.A.; Guru, S.K.; Rao, A.V.S.; Sharma, S.; Mahajan, G.; Behl, A.; Kumar, A.; Sharma, P.R.; Kamal, A.; Bhushan, S.; et al. A novel quinazolinone chalcone derivative induces mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR signaling pathway in human colon cancer HCT-116 cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2016, 87, 1–11. [Google Scholar] [CrossRef]
- Han, X.; Peng, B.; Xiao, B.-B.; Sheng-Li, C.; Yang, C.-R.; Wang, W.-Z.; Wang, F.-C.; Li, H.-Y.; Yuan, X.-L.; Shi, R.; et al. Synthesis and evaluation of chalcone analogues containing a 4-oxoquinazolin-2-yl group as potential anti-tumor agents. Eur. J. Med. Chem. 2019, 162, 586–601. [Google Scholar] [CrossRef] [PubMed]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of breast cancer resistance protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Marković, V.; Debeljak, N.; Stanojković, T.; Kolundžija, B.; Sladić, D.; Vujčić, M.; Janović, B.; Tanić, N.; Perović, M.; Tešić, V.; et al. Anthraquinone-chalcone hybrids: Synthesis, preliminary antiproliferative evaluation and DNA-interaction studies. Eur. J. Med. Chem. 2015, 89, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.-Y.; Wang, F.; Wang, X.; Sun, W.-X.; Qi, J.-L.; Pang, Y.-J.; Yang, R.-W.; Lu, G.-H.; Wang, X.-M.; Yang, Y.-H. Design, Synthesis, and Biological Evaluation of Chalcone-Containing Shikonin Derivatives as Inhibitors of Tubulin Polymerization. ChemMedChem 2017, 12, 399–406. [Google Scholar] [CrossRef]
- Guimarães, D.G.; de Assis Gonsalves, A.; Rolim, L.A.; Araújo, E.C.; Dos Anjos Santos, V.L.; Silva, M.F.S.; de Cássia Evangelista de Oliveira, F.; da Costa, M.P.; Pessoa, C.; Fonseca Goulart, M.O.; et al. Naphthoquinone-based hydrazone hybrids: Synthesis and potent activity against cancer cell lines. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Ng, H.-L.; Chen, S.; Chew, E.-H.; Chui, W.-K. Applying the designed multiple ligands approach to inhibit dihydrofolate reductase and thioredoxin reductase for anti-proliferative activity. Eur. J. Med. Chem. 2016, 115, 63–74. [Google Scholar] [CrossRef]
- Ng, H.-L.; Ma, X.; Chew, E.-H.; Chui, W.-K. Design, Synthesis, and Biological Evaluation of Coupled Bioactive Scaffolds as Potential Anticancer Agents for Dual Targeting of Dihydrofolate Reductase and Thioredoxin Reductase. J. Med. Chem. 2017, 60, 1734–1745. [Google Scholar] [CrossRef]
- Fu, D.-J.; Li, J.-H.; Yang, J.-J.; Li, P.; Zhang, Y.-B.; Liu, S.; Li, Z.-R.; Zhang, S.-Y. Discovery of novel chalcone-dithiocarbamates as ROS-mediated apoptosis inducers by inhibiting catalase. Bioorg. Chem. 2019, 86, 375–385. [Google Scholar] [CrossRef]
- Fu, D.-J.; Zhang, S.-Y.; Liu, Y.-C.; Zhang, L.; Liu, J.-J.; Song, J.; Zhao, R.-H.; Li, F.; Sun, H.-H.; Liu, H.-M.; et al. Design, synthesis and antiproliferative activity studies of novel dithiocarbamate-chalcone derivates. Bioorg. Med. Chem. Lett. 2016, 26, 3918–3922. [Google Scholar] [CrossRef] [PubMed]
- Batovska, D.I.; Todorova, I.T. Trends in utilization of the pharmacological potential of chalcones. Curr Clin Pharmacol 2010, 5, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kumar, R.; Kodwani, R.; Kapoor, S.; Khare, A.; Bansal, R.; Khurana, S.; Singh, S.; Thomas, J.; Roy, B.; et al. A Review on Mechanisms of Anti Tumor Activity of Chalcones. Anticancer Agents Med. Chem. 2015, 16, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Prives, C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018, 25, 169–179. [Google Scholar] [CrossRef]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Silva, G.; Marins, M.; Fachin, A.L.; Lee, S.-H.; Baek, S.J. Anti-cancer activity of trans-chalcone in osteosarcoma: Involvement of Sp1 and p53. Mol. Carcinog. 2016, 55, 1438–1448. [Google Scholar] [CrossRef]
- Dos Santos, M.B.; Bertholin Anselmo, D.; de Oliveira, J.G.; Jardim-Perassi, B.V.; Alves Monteiro, D.; Silva, G.; Gomes, E.; Lucia Fachin, A.; Marins, M.; de Campos Zuccari, D.A.P.; et al. Antiproliferative activity and p53 upregulation effects of chalcones on human breast cancer cells. J. Enzym. Inhib. Med. Chem. 2019, 34, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.; Marins, M.; Chaichanasak, N.; Yoon, Y.; Fachin, A.L.; Pinhanelli, V.C.; Regasini, L.O.; Dos Santos, M.B.; Ayusso, G.M.; Marques, B.C.; et al. Trans-chalcone increases p53 activity via DNAJB1/HSP40 induction and CRM1 inhibition. PLoS ONE 2018, 13, e0202263. [Google Scholar] [CrossRef]
- Siqueira, E.D.S.; Concato, V.M.; Tomiotto-Pellissier, F.; Silva, T.F.; Bortoleti, B.; Goncalves, M.D.; Costa, I.N.; Junior, W.A.V.; Pavanelli, W.R.; Panis, C.; et al. Trans-chalcone induces death by autophagy mediated by p53 up-regulation and beta-catenin down-regulation on human hepatocellular carcinoma HuH7.5 cell line. Phytomedicine 2021, 80, 153373. [Google Scholar] [CrossRef]
- Seba, V.; Silva, G.; Santos, M.B.D.; Baek, S.J.; Franca, S.C.; Fachin, A.L.; Regasini, L.O.; Marins, M. Chalcone Derivatives 4’-Amino-1-Naphthyl-Chalcone (D14) and 4’-Amino-4-Methyl-1-Naphthyl-Chalcone (D15) Suppress Migration and Invasion of Osteosarcoma Cells Mediated by p53 Regulating EMT-Related Genes. Int. J. Mol. Sci. 2018, 19, 2838. [Google Scholar] [CrossRef] [Green Version]
- Cabral, B.L.S.; da Silva, A.C.G.; de Avila, R.I.; Cortez, A.P.; Luzin, R.M.; Liao, L.M.; de Souza Gil, E.; Sanz, G.; Vaz, B.G.; Sabino, J.R.; et al. A novel chalcone derivative, LQFM064, induces breast cancer cells death via p53, p21, KIT and PDGFRA. Eur. J. Pharm. Sci. 2017, 107, 1–15. [Google Scholar] [CrossRef]
- Peyrot, V.; Leynadier, D.; Sarrazin, M.; Briand, C.; Rodriquez, A.; Nieto, J.M.; Andreu, J.M. Interaction of tubulin and cellular microtubules with the new antitumor drug MDL 27048. A powerful and reversible microtubule inhibitor. J. Biol. Chem. 1989, 264, 21296–21301. [Google Scholar] [CrossRef]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.; McGown, A.T.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Mackenzie, G.; Greedy, B.; Armitage, S.; Chabert, J.F.; Bennett, E.; Nettles, J.; Snyder, J.P.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerisation. Part 2: Structure-based discovery of alpha-aryl chalcones. Bioorg. Med. Chem. 2009, 17, 7711–7722. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Kumar, G.B.; Vishnuvardhan, M.V.; Shaik, A.B.; Reddy, V.S.; Mahesh, R.; Sayeeda, I.B.; Kapure, J.S. Synthesis of phenstatin/isocombretastatin-chalcone conjugates as potent tubulin polymerization inhibitors and mitochondrial apoptotic inducers. Org. Biomol. Chem. 2015, 13, 3963–3981. [Google Scholar] [CrossRef]
- Kode, J.; Kovvuri, J.; Nagaraju, B.; Jadhav, S.; Barkume, M.; Sen, S.; Kasinathan, N.K.; Chaudhari, P.; Mohanty, B.S.; Gour, J.; et al. Synthesis, biological evaluation, and molecular docking analysis of phenstatin based indole linked chalcones as anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 105, 104447. [Google Scholar] [CrossRef]
- Yan, W.; Xiangyu, C.; Ya, L.; Yu, W.; Feng, X. An orally antitumor chalcone hybrid inhibited HepG2 cells growth and migration as the tubulin binding agent. Invest. New Drugs 2019, 37, 784–790. [Google Scholar] [CrossRef]
- Canela, M.-D.; Noppen, S.; Bueno, O.; Prota, A.E.; Bargsten, K.; Sáez-Calvo, G.; Jimeno, M.-L.; Benkheil, M.; Ribatti, D.; Velázquez, S.; et al. Antivascular and antitumor properties of the tubulin-binding chalcone TUB091. Oncotarget 2017, 8, 14325–14342. [Google Scholar] [CrossRef] [Green Version]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa B Kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-κB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.R.; Prasad, S.; Sung, B.; Aggarwal, B.B. The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer. Int. Immunopharmacol. 2011, 11, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.K.; Sandur, S.K.; Sung, B.; Sethi, G.; Kunnumakkara, A.B.; Aggarwal, B.B. Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. J. Biol. Chem. 2007, 282, 17340–17350. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Jeon, S.H.; Kim, S.H.; Kim, C.; Lee, S.J.; Koh, D.; Lim, Y.; Ha, K.; Shin, S.Y. A new synthetic chalcone derivative, 2-hydroxy-3’,5,5’-trimethoxychalcone (DK-139), suppresses the Toll-like receptor 4-mediated inflammatory response through inhibition of the Akt/NF-kappaB pathway in BV2 microglial cells. Exp. Mol. Med. 2012, 44, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-Y.; Park, S.J.; Yun, K.-J.; Cho, Y.-W.; Park, H.-J.; Lee, K.-T. Isoliquiritigenin isolated from the roots of Glycyrrhiza uralensis inhibits LPS-induced iNOS and COX-2 expression via the attenuation of NF-kappaB in RAW 264.7 macrophages. Eur. J. Pharmacol. 2008, 584, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Chen, Y.; Ding, R.; Feng, L.; Fu, Z.; Yang, S.; Deng, X.; Xie, Z.; Zheng, S. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 2017, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Tan, R.-Z.; Li, J.-C.; Liu, T.-T.; Zhong, X.; Yan, Y.; Yang, J.-K.; Lin, X.; Fan, J.-M.; Wang, L. Isoliquiritigenin Attenuates UUO-Induced Renal Inflammation and Fibrosis by Inhibiting Mincle/Syk/NF-Kappa B Signaling Pathway. Drug Des. Dev. Ther. 2020, 14, 1455–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folmer, F.; Blasius, R.; Morceau, F.; Tabudravu, J.; Dicato, M.; Jaspars, M.; Diederich, M. Inhibition of TNFalpha-induced activation of nuclear factor kappaB by kava (Piper methysticum) derivatives. Biochem. Pharmacol. 2006, 71, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Yang, H.; Wang, Z.; Feng, H.; Deng, X.; Cheng, G.; Ci, X. Nrf2 signaling and autophagy are complementary in protecting lipopolysaccharide/d-galactosamine-induced acute liver injury by licochalcone A. Cell Death Dis. 2019, 10, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruibin, J.; Bo, J.; Danying, W.; Jianguo, F.; Linhui, G. Cardamonin induces G2/M phase arrest and apoptosis through inhibition of NF-κB and mTOR pathways in ovarian cancer. Aging 2020, 12, 25730–25743. [Google Scholar] [CrossRef] [PubMed]
- Badroon, N.A.; Abdul Majid, N.; Alshawsh, M.A. Antiproliferative and Apoptotic Effects of Cardamonin against Hepatocellular Carcinoma HepG2 Cells. Nutrients 2020, 12, 1757. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Yuan, Q.; Yu, N.; Liu, B.; Huang, G.; Yuan, X. Cardamonin attenuates chronic inflammation and tumorigenesis in colon. Cell Cycle 2019, 18, 3275–3287. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ahn, K.S.; Anand, P.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood 2009, 113, 2003–2013. [Google Scholar] [CrossRef] [Green Version]
- Venkateswararao, E.; Sharma, V.K.; Yun, J.; Kim, Y.; Jung, S.H. Anti-proliferative effect of chalcone derivatives through inactivation of NF-kappaB in human cancer cells. Bioorg. Med. Chem. 2014, 22, 3386–3392. [Google Scholar] [CrossRef]
- Gan, F.F.; Zhang, R.; Ng, H.L.; Karuppasamy, M.; Seah, W.; Yeap, W.H.; Ong, S.M.; Hadadi, E.; Wong, S.C.; Chui, W.K.; et al. Novel dual-targeting anti-proliferative dihydrotriazine-chalcone derivatives display suppression of cancer cell invasion and inflammation by inhibiting the NF-kappaB signaling pathway. Food Chem. Toxicol. 2018, 116, 238–248. [Google Scholar] [CrossRef]
- Mirossay, L.; Varinska, L.; Mojzis, J. Antiangiogenic Effect of Flavonoids and Chalcones: An Update. Int. J. Mol. Sci. 2017, 19, 27. [Google Scholar] [CrossRef] [Green Version]
- Cerezo, A.B.; Winterbone, M.S.; Moyle, C.W.A.; Needs, P.W.; Kroon, P.A. Molecular structure-function relationship of dietary polyphenols for inhibiting VEGF-induced VEGFR-2 activity. Mol. Nutr. Food Res. 2015, 59, 2119–2131. [Google Scholar] [CrossRef] [Green Version]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential Role of Natural Compounds as Anti-Angiogenic Agents in Cancer. Curr. Vasc. Pharmacol. 2017, 15, 503–519. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, B.; Yu, J.; Huang, L.; Zeng, X.; Shen, X.; Ren, C.; Ben-David, Y.; Luo, H. Fli-1 Activation through Targeted Promoter Activity Regulation Using a Novel 3′, 5′-diprenylated Chalcone Inhibits Growth and Metastasis of Prostate Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2216. [Google Scholar] [CrossRef] [Green Version]
- Iheagwam, F.N.; Ogunlana, O.O.; Ogunlana, O.E.; Isewon, I.; Oyelade, J. Potential Anti-Cancer Flavonoids Isolated From Caesalpinia bonduc Young Twigs and Leaves: Molecular Docking and In Silico Studies. Bioinform. Biol. Insights 2019, 13, 1177932218821371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, N.; Han, S.; Wang, D.; Mo, S.; Yu, L.; Huang, H.; Tsui, K.; Shen, J.; Chen, J. Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PLoS ONE 2013, 8, e68566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.J.; Park, S.Y.; Kwon, G.T.; Lee, K.W.; Kang, Y.H.; Choi, M.S.; Yun, J.W.; Jeon, J.H.; Jun, J.G.; Park, J.H. Licochalcone E present in licorice suppresses lung metastasis in the 4T1 mammary orthotopic cancer model. Cancer Prev. Res. 2013, 6, 603–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Matsuo, Y.; Imafuji, H.; Okubo, T.; Maeda, Y.; Sato, T.; Shamoto, T.; Tsuboi, K.; Morimoto, M.; Takahashi, H.; et al. Xanthohumol inhibits angiogenesis by suppressing nuclear factor-kappaB activation in pancreatic cancer. Cancer Sci. 2018, 109, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, G.; Lu, X.; Wang, S.; Han, S.; Li, Y.; Ping, G.; Jiang, X.; Li, H.; Yang, J.; et al. Novel chalcone derivatives as hypoxia-inducible factor (HIF)-1 inhibitor: Synthesis, anti-invasive and anti-angiogenic properties. Eur. J. Med. Chem. 2015, 89, 88–97. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Chen, H.; Zhang, J.; Zhang, J.; Qin, T.; Duan, C.; Chen, X.; Liu, Y.; Zhou, X.; et al. Inhibition of CYP4A by a novel flavonoid FLA-16 prolongs survival and normalizes tumor vasculature in glioma. Cancer Lett. 2017, 402, 131–141. [Google Scholar] [CrossRef]
- Jeong, J.H.; Jang, H.J.; Kwak, S.; Sung, G.J.; Park, S.H.; Song, J.H.; Kim, H.; Na, Y.; Choi, K.C. Novel TGF-beta1 inhibitor antagonizes TGF-beta1-induced epithelial-mesenchymal transition in human A549 lung cancer cells. J. Cell. Biochem. 2019, 120, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Stanojkovic, T.; Markovic, V.; Matic, I.Z.; Mladenovic, M.P.; Petrovic, N.; Krivokuca, A.; Petkovic, M.; Joksovic, M.D. Highly selective anthraquinone-chalcone hybrids as potential antileukemia agents. Bioorg. Med. Chem. Lett. 2018, 28, 2593–2598. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Bois, F.; Boumendjel, A.; Mariotte, A.M.; Conseil, G.; Di Petro, A. Synthesis and biological activity of 4-alkoxy chalcones: Potential hydrophobic modulators of P-glycoprotein-mediated multidrug resistance. Bioorg. Med. Chem. 1999, 7, 2691–2695. [Google Scholar] [CrossRef]
- Bois, F.; Beney, C.; Boumendjel, A.; Mariotte, A.M.; Conseil, G.; Di Pietro, A. Halogenated chalcones with high-affinity binding to P-glycoprotein: Potential modulators of multidrug resistance. J. Med. Chem. 1998, 41, 4161–4164. [Google Scholar] [CrossRef]
- Parveen, Z.; Brunhofer, G.; Jabeen, I.; Erker, T.; Chiba, P.; Ecker, G.F. Synthesis, biological evaluation and 3D-QSAR studies of new chalcone derivatives as inhibitors of human P-glycoprotein. Bioorg. Med. Chem. 2014, 22, 2311–2319. [Google Scholar] [CrossRef]
- Ngo, T.D.; Tran, T.D.; Le, M.T.; Thai, K.M. Computational predictive models for P-glycoprotein inhibition of in-house chalcone derivatives and drug-bank compounds. Mol. Divers. 2016, 20, 945–961. [Google Scholar] [CrossRef]
- Gu, X.; Ren, Z.; Tang, X.; Peng, H.; Ma, Y.; Lai, Y.; Peng, S.; Zhang, Y. Synthesis and biological evaluation of bifendate-chalcone hybrids as a new class of potential P-glycoprotein inhibitors. Bioorg. Med. Chem. 2012, 20, 2540–2548. [Google Scholar] [CrossRef]
- Yin, H.; Dong, J.; Cai, Y.; Shi, X.; Wang, H.; Liu, G.; Tang, Y.; Liu, J.; Ma, L. Design, synthesis and biological evaluation of chalcones as reversers of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2019, 180, 350–366. [Google Scholar] [CrossRef]
- Li, J.; Zheng, L.; Yan, M.; Wu, J.; Liu, Y.; Tian, X.; Jiang, W.; Zhang, L.; Wang, R. Activity and mechanism of flavokawain A in inhibiting P-glycoprotein expression in paclitaxel resistance of lung cancer. Oncol. Lett. 2020, 19, 379–387. [Google Scholar] [CrossRef]
- Li, J.; Zheng, L.; Wang, R.; Sun, D.; Liang, S.; Wu, J.; Liu, Y.; Tian, X.; Li, T.; Yang, Y.; et al. Synergistic Combination of Sodium Aescinate-Stabilized, Polymer-Free, Twin-Like Nanoparticles to Reverse Paclitaxel Resistance. Int. J. Nanomed. 2020, 15, 5839–5853. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Lusvarghi, S.; Hsiao, S.H.; Liu, T.C.; Li, Y.Q.; Huang, Y.H.; Hung, T.H.; Ambudkar, S.V. Licochalcone A Selectively Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Drugs. J. Nat. Prod. 2020, 83, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Winter, E.; Devantier Neuenfeldt, P.; Chiaradia-Delatorre, L.D.; Gauthier, C.; Yunes, R.A.; Nunes, R.J.; Creczynski-Pasa, T.B.; Di Pietro, A. Symmetric bis-chalcones as a new type of breast cancer resistance protein inhibitors with a mechanism different from that of chromones. J. Med. Chem. 2014, 57, 2930–2941. [Google Scholar] [CrossRef] [PubMed]
- Boumendjel, A.; McLeer-Florin, A.; Champelovier, P.; Allegro, D.; Muhammad, D.; Souard, F.; Derouazi, M.; Peyrot, V.; Toussaint, B.; Boutonnat, J. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer 2009, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Riwanto, M.; Go, M.L.; Ee, P.L. Modulation of breast cancer resistance protein (BCRP/ABCG2) by non-basic chalcone analogues. Eur. J. Pharm. Sci. 2008, 35, 30–41. [Google Scholar] [CrossRef]
- Cai, C.Y.; Zhang, W.; Wang, J.Q.; Lei, Z.N.; Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Tan, C.P.; Wang, B.; Chen, Z.S. Biological evaluation of non-basic chalcone CYB-2 as a dual ABCG2/ABCB1 inhibitor. Biochem. Pharmacol. 2020, 175, 113848. [Google Scholar] [CrossRef]
- Shimiz, K.; Kondo, R.; Sakai, K.; Buabarn, S.; Dilokkunanant, U. A geranylated chalcone with 5alpha-reductase inhibitory properties from Artocarpus incisus. Phytochemistry 2000, 54, 737–739. [Google Scholar] [CrossRef]
- Le Bail, J.C.; Pouget, C.; Fagnere, C.; Basly, J.P.; Chulia, A.J.; Habrioux, G. Chalcones are potent inhibitors of aromatase and 17beta-hydroxysteroid dehydrogenase activities. Life Sci. 2001, 68, 751–761. [Google Scholar] [CrossRef]
- Mourad, A.A.E.; Mourad, M.A.E.; Jones, P.G. Novel HDAC/Tubulin Dual Inhibitor: Design, Synthesis and Docking Studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity. Drug Des. Dev. Ther. 2020, 14, 3111–3130. [Google Scholar] [CrossRef]
- Mohamed, M.F.A.; Shaykoon, M.S.A.; Abdelrahman, M.H.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G.E.-D.A.A. Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors. Bioorg. Chem. 2017, 72, 32–41. [Google Scholar] [CrossRef]
- Nedungadi, D.; Binoy, A.; Pandurangan, N.; Nair, B.G.; Mishra, N. Proteasomal dysfunction and ER stress triggers 2’-hydroxy-retrochalcone-induced paraptosis in cancer cells. Cell Biol. Int. 2021, 45, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pham, V.; Tippin, M.; Fu, D.; Rendon, R.; Song, L.; Uchio, E.; Hoang, B.H.; Zi, X. Flavokawain B targets protein neddylation for enhancing the anti-prostate cancer effect of Bortezomib via Skp2 degradation. Cell Commun. Signal. 2019, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Jobst, B.; Weigl, J.; Michl, C.; Vivarelli, F.; Pinz, S.; Amslinger, S.; Rascle, A. Inhibition of interleukin-3- and interferon- α-induced JAK/STAT signaling by the synthetic α-X-2’,3,4,4’-tetramethoxychalcones α-Br-TMC and α-CF3-TMC. Biol. Chem. 2016, 397, 1187–1204. [Google Scholar] [CrossRef]
- Yuan, X.; Li, T.; Xiao, E.; Zhao, H.; Li, Y.; Fu, S.; Gan, L.; Wang, Z.; Zheng, Q.; Wang, Z. Licochalcone B inhibits growth of bladder cancer cells by arresting cell cycle progression and inducing apoptosis. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2014, 65, 242–251. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, J.; Wang, Z.; Liu, B.; Zhu, S.; Zhu, L.; Peng, B. Licorice isoliquiritigenin-encapsulated mesoporous silica nanoparticles for osteoclast inhibition and bone loss prevention. Theranostics 2019, 9, 5183–5199. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, W.; Peng, Y.; Jiang, Z.-H.; Zhang, L.; Du, Z. Design, Synthesis and Anti-Tumor Activity of Novel Benzimidazole-Chalcone Hybrids as Non-Intercalative Topoisomerase II Catalytic Inhibitors. Molecules 2020, 25, 3180. [Google Scholar] [CrossRef]
- Li, P.-H.; Jiang, H.; Zhang, W.-J.; Li, Y.-L.; Zhao, M.-C.; Zhou, W.; Zhang, L.-Y.; Tang, Y.-D.; Dong, C.-Z.; Huang, Z.-S.; et al. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur. J. Med. Chem. 2018, 145, 498–510. [Google Scholar] [CrossRef]
- Wang, T.-T.; Chen, Z.-Z.; Xie, P.; Zhang, W.-J.; Du, M.-Y.; Liu, Y.-T.; Zhu, H.-Y.; Guo, Y.-S. Isoliquiritigenin suppresses the proliferation and induced apoptosis via miR-32/LATS2/Wnt in nasopharyngeal carcinoma. Eur. J. Pharmacol. 2019, 856, 172352. [Google Scholar] [CrossRef] [PubMed]
- Predes, D.; Oliveira, L.F.S.; Ferreira, L.S.S.; Maia, L.A.; Delou, J.M.A.; Faletti, A.; Oliveira, I.; Amado, N.G.; Reis, A.H.; Fraga, C.A.M.; et al. The Chalcone Lonchocarpin Inhibits Wnt/β-Catenin Signaling and Suppresses Colorectal Cancer Proliferation. Cancers 2019, 11, 1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.H.; Li, H.H.; Li, M.; Wang, S.; Jiang, X.R.; Li, Y.; Ping, G.F.; Cao, Q.; Liu, X.; Fang, W.H.; et al. SL4, a chalcone-based compound, induces apoptosis in human cancer cells by activation of the ROS/MAPK signalling pathway. Cell Prolif. 2015, 48, 718–728. [Google Scholar] [CrossRef]
- Lee, J.A.; Kim, D.J.; Hwang, O. KMS99220 Exerts Anti-Inflammatory Effects, Activates the Nrf2 Signaling and Interferes with IKK, JNK and p38 MAPK via HO-1. Mol. Cells 2019, 42, 702–710. [Google Scholar] [CrossRef]
- Hseu, Y.-C.; Lee, M.-S.; Wu, C.-R.; Cho, H.-J.; Lin, K.-Y.; Lai, G.-H.; Wang, S.-Y.; Kuo, Y.-H.; Kumar, K.J.S.; Yang, H.-L. The chalcone flavokawain B induces G2/M cell-cycle arrest and apoptosis in human oral carcinoma HSC-3 cells through the intracellular ROS generation and downregulation of the Akt/p38 MAPK signaling pathway. J. Agric. Food Chem. 2012, 60, 2385–2397. [Google Scholar] [CrossRef]
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Qiu, S.; Wang, P.; Liang, X.; Huang, F.; Wu, H.; Zhang, B.; Zhang, W.; Tian, X.; Xu, R.; et al. Cardamonin inhibits breast cancer growth by repressing HIF-1α-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. CR 2019, 38, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Ed. Engl. 2013, 52, 2744–2792. [Google Scholar] [CrossRef] [PubMed]
- Marquina, S.; Maldonado-Santiago, M.; Sanchez-Carranza, J.N.; Antunez-Mojica, M.; Gonzalez-Maya, L.; Razo-Hernandez, R.S.; Alvarez, L. Design, synthesis and QSAR study of 2′-hydroxy-4′-alkoxy chalcone derivatives that exert cytotoxic activity by the mitochondrial apoptotic pathway. Bioorg. Med. Chem. 2019, 27, 43–54. [Google Scholar] [CrossRef]
- Song, Y.Q.; Guan, X.Q.; Weng, Z.M.; Liu, J.L.; Chen, J.; Wang, L.; Cui, L.T.; Fang, S.Q.; Hou, J.; Ge, G.B. Discovery of hCES2A inhibitors from Glycyrrhiza inflata via combination of docking-based virtual screening and fluorescence-based inhibition assays. Food Funct. 2021, 12, 162–176. [Google Scholar] [CrossRef]
- George, R.F.; Kandeel, M.; El-Ansary, D.Y.; El Kerdawy, A.M. Some 1,3,5-trisubstituted pyrazoline derivatives targeting breast cancer: Design, synthesis, cytotoxic activity, EGFR inhibition and molecular docking. Bioorg. Chem. 2020, 99, 103780. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, B.S.; Ahn, S.; Koh, D.; Lee, Y.H.; Shin, S.Y.; Lim, Y. Anticancer and structure-activity relationship evaluation of 3-(naphthalen-2-yl)-N,5-diphenyl-pyrazoline-1-carbothioamide analogs of chalcone. Bioorg. Chem. 2016, 68, 166–176. [Google Scholar] [CrossRef]
- Shin, S.Y.; Yoon, H.; Ahn, S.; Kim, D.-W.; Kim, S.H.; Koh, D.; Lee, Y.H.; Lim, Y. Chromenylchalcones showing cytotoxicity on human colon cancer cell lines and in silico docking with aurora kinases. Bioorg. Med. Chem. 2013, 21, 4250–4258. [Google Scholar] [CrossRef]
- Bisht, K.; Wagner, K.H.; Bulmer, A.C. Curcumin, resveratrol and flavonoids as anti-inflammatory, cyto- and DNA-protective dietary compounds. Toxicology 2010, 278, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommidi, D.R.; Pagadala, R.; Rana, S.; Singh, P.; Shintre, S.A.; Koorbanally, N.A.; Jonnalagadda, S.B.; Moodley, B. Novel carbapenem chalcone derivatives: Synthesis, cytotoxicity and molecular docking studies. Org. Biomol. Chem. 2015, 13, 4344–4350. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Syahidah, H.N.; Subarnas, A.; Yusuf, M.; Bryant, S.D.; Langer, T. Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals 2017, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Riswanto, F.D.O.; Rawa, M.S.A.; Murugaiyah, V.; Salin, N.H.; Istyastono, E.P.; Hariono, M.; Wahab, H.A. Anti-Cholinesterase Activity of Chalcone Derivatives: Synthesis, In Vitro Assay and Molecular Docking Study. Med. Chem. 2019, 17, 442–452. [Google Scholar] [CrossRef]
- Narayanapillai, S.C.; Leitzman, P.; O’Sullivan, M.G.; Xing, C. Flavokawains a and B in kava, not dihydromethysticin, potentiate acetaminophen-induced hepatotoxicity in C57BL/6 mice. Chem. Res. Toxicol. 2014, 27, 1871–1876. [Google Scholar] [CrossRef] [Green Version]
- Tugcu, G.; Kirmizibekmez, H.; Aydin, A. The integrated use of in silico methods for the hepatotoxicity potential of Piper methysticum. Food Chem. Toxicol. 2020, 145, 111663. [Google Scholar] [CrossRef]
- Qian, Y.; Yang, Y.; Wang, K.; Zhou, W.; Dang, Y.; Zhu, M.; Li, F.; Ji, G. 2’-Hydroxychalcone Induced Cytotoxicity via Oxidative Stress in the Lipid-Loaded Hepg2 Cells. Front. Pharmacol. 2019, 10, 1390. [Google Scholar] [CrossRef]
- Jeong, C.H.; Park, H.B.; Jang, W.J.; Jung, S.H.; Seo, Y.H. Discovery of hybrid Hsp90 inhibitors and their anti-neoplastic effects against gefitinib-resistant non-small cell lung cancer (NSCLC). Bioorg. Med. Chem. Lett. 2014, 24, 224–227. [Google Scholar] [CrossRef]
- Oh, Y.J.; Seo, Y.H. A novel chalcone-based molecule, BDP inhibits MDAMB231 triple-negative breast cancer cell growth by suppressing Hsp90 function. Oncol. Rep. 2017, 38, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2020, 11, 592654. [Google Scholar] [CrossRef] [PubMed]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2005, 22, 15–61. [Google Scholar] [CrossRef] [PubMed]
- De Luca, D.; Lauritano, C. In Silico Identification of Type III PKS Chalcone and Stilbene Synthase Homologs in Marine Photosynthetic Organisms. Biology 2020, 9, 110. [Google Scholar] [CrossRef]
















| Lead Compounds | Mechanisms of Action | Reference |
|---|---|---|
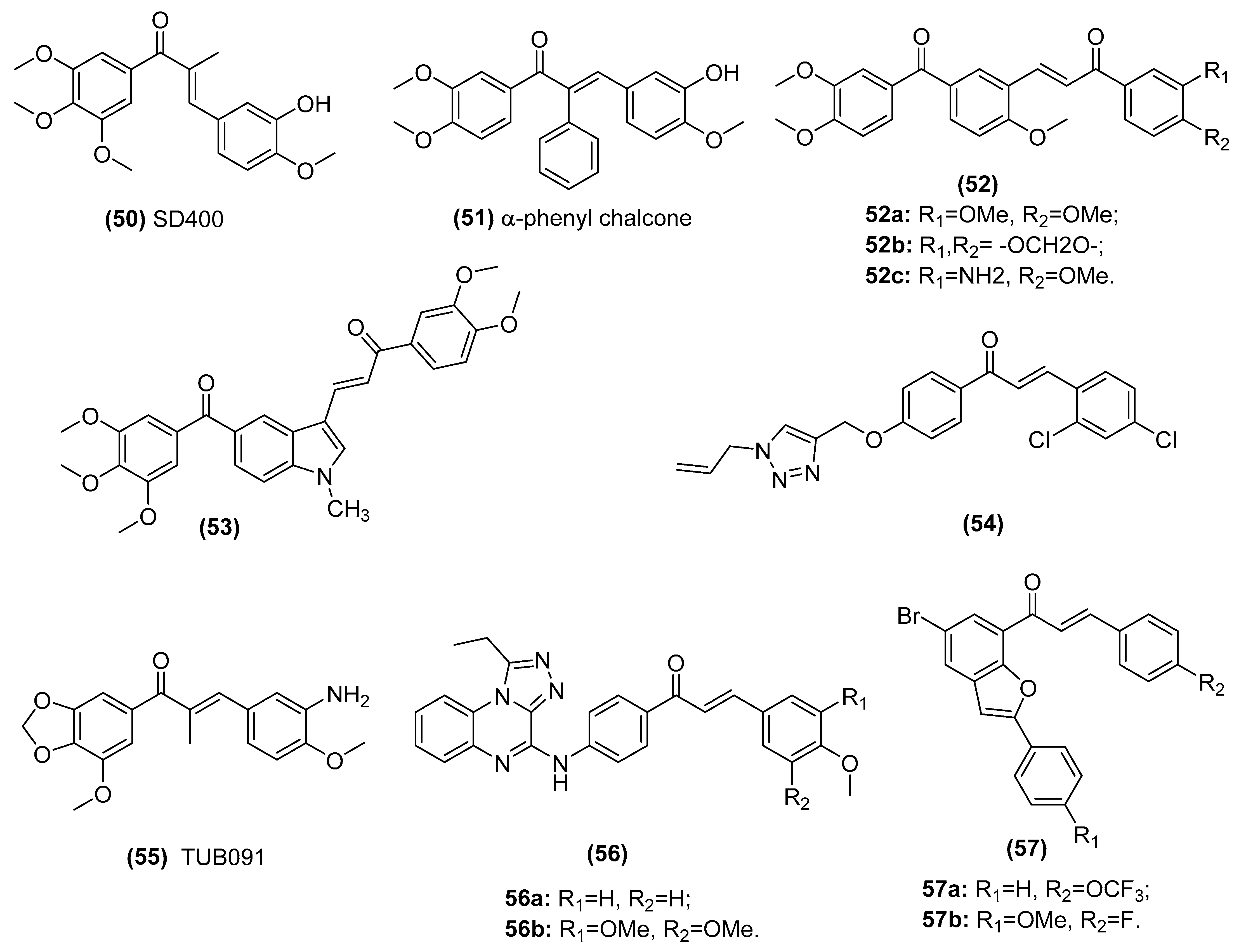
| CA-4 type chalcones SD400 (50) and α-phenyl chalcone (51) | Populate the colchicine-binding site of beta-tubulin and inhibit tubulin assembly in the K562 human chronic myelogenous leukemia cell line | Ducki et al. [135,136] |
| A series of phenstatin/isocombretastatin–chalcones (52) | Inhibit tubulin assembly, arrest in the G2/M phase and induce apoptosis in a panel of sixty human cancer cell lines | Kamal et al. [137] |
| Phenstatin based indole linked chalcone compounds (53) | Destabilizes tubulin, leading to loss of cell integrity and affecting glucose metabolism in SCC-29B human oral cancer cells, spheroids and AW13516 oral cancer xenograft model mice | Kode et al. [138] |
| Chalcone-1,2,3-triazole derivative (54) | Inhibits tubulin polymerization in liver cancer HepG2 cells | Yan et al. [139] |
| (E)-3-(3-amino-4-methoxyphenyl)-1-(5’-methoxy-3’,4’-methylendioxyphenyl)- 2-methylprop-2-en-1-one (55, TUB091) | Destabilizes microtubule, targets vascular and shows antitumor and antimetastatic activities in melanoma and breast cancer xenograft models | Canela et al. [140] |
| Triazoloquinoxaline-chalcone derivatives (56) | Displays significant antiproliferative effects against MCF-7, HCT-116 and HEPG-2 cells | Alswah et al. [141] |
| 2-arylbenzo[c]furan-chalcone hybrids (57) | Inhibits tubulin polymerization and EGFR-TK phosphorylation in the human breast cancer (MCF-7) cell line | Mphahlele et al. [98] |
| Lead Compounds | Mechanisms of Action | Reference |
|---|---|---|
| Butein (7) | Blocks the phosphorylation and degradation of IκBα and suppresses NF-κB activity in KBM-5 (human myeloid) cells | Pandey et al. [147] |
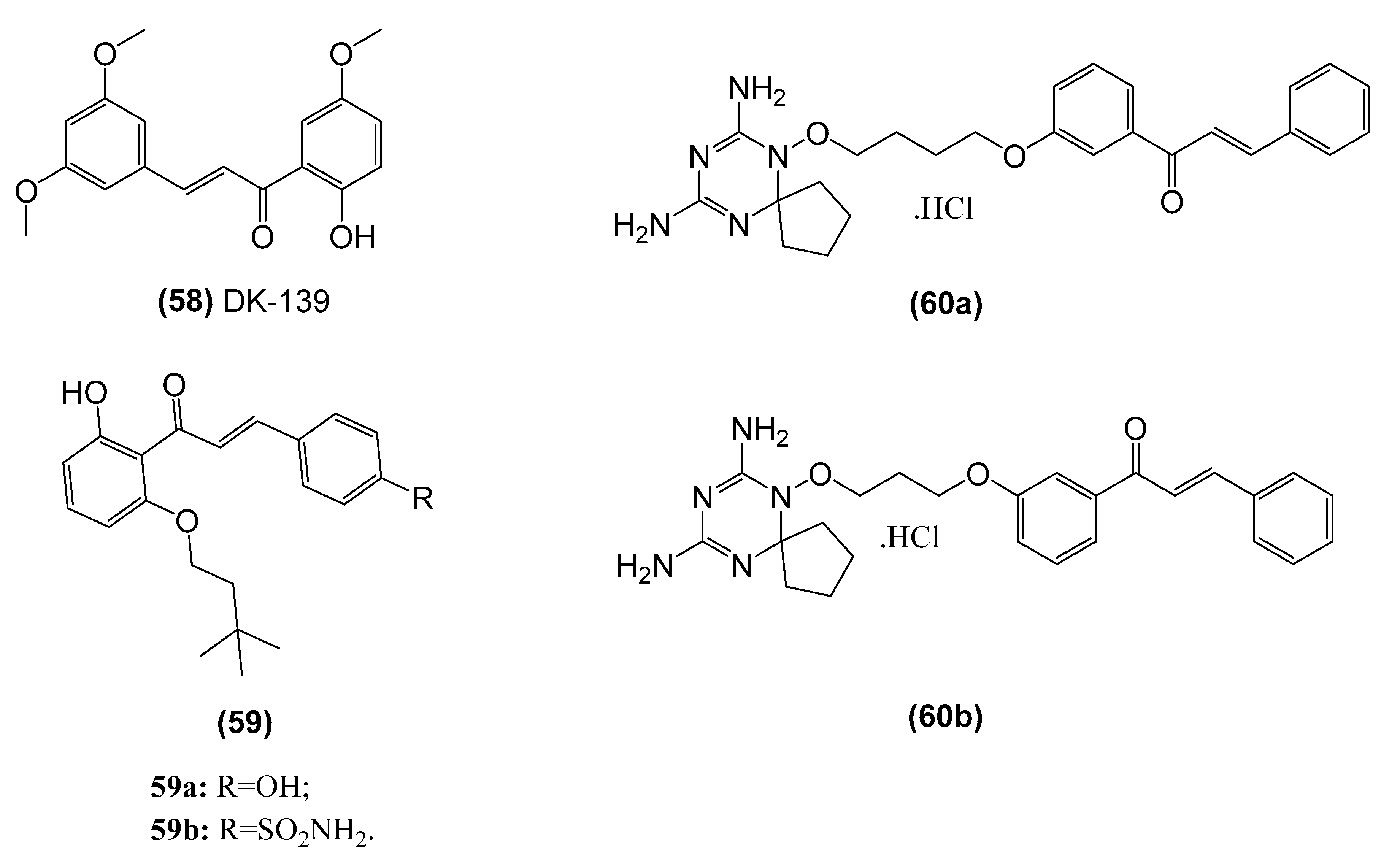
| 2-hydroxy-3’,5,5’-trimethoxychalcone (58, DK-139) | Inhibits the Akt//IKK/NF-κB signaling pathway in BV2 microglial cells | Lee et al. [148] |
| (E)-1-(2-hydroxy-6- (isopentyloxy)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (59) | Inhibits NF-κB activity in vitro and shows antiproliferative activity against various human cancer cell lines, namely ACHN (renal), NCI-H23(lung), MDA-MB-231 (breast), HCT-15 (colon), NUGC-3 (stomach) and PC-3 (prostate). | Venkateswararao et al. [158] |
| Dihydrotriazine-chalcone compounds (60) | Inhibits IKKα/β phosphorylation, leading to a reduction in phosphorylation of the p65 subunit and eventually suppression of NF-κB-dependent transcriptional activation of MMP-9 expression. in MDA-MB-231 breast carcinoma cells | Gan et al. [159] |
| Lead Compounds | Mechanisms of Action | Reference |
|---|---|---|
| 3′,5′-diprenylated chalcone (61) | An Fli-1 agonist for regulating the expression of Fli-1 target genes including VEGF-1, TGF-β2 and MMP-1 genes of prostate cancer cells | Ma et al. [163] |
| Flavonoids (62) isolated from young Caesalpinia bonduc twigs and leaves | A molecular docking analysis shows the interaction between them and cancer target proteins (TK, VEGF, and MMP) | Iheagwam et al. [164] |
| Isoliquiritigenin (6, ISL) | Inhibits the angiogenic Akt- FGF-2/TGF-β/VEGF signaling in C6 glioma cell line and the rat C6 glioma model. | Wang et al. [28] |
| Isoliquiritigenin (6, ISL) | Inhibits VEGF expression via promoting HIF-1α proteasome degradation pathway and blocks VEGFR-2 activation and the transduction of its downstream signaling in human umbilical vein endothelial cells (HUVECs), MCF-7 cells and MDA-MB-231 cells and in vivo tumor xenograft of MDA-MB-231 cells | Wang et al. [165] |
| Licochalcone E (63, LicE) | Decreases expression of VEGF-A and C, VEGF receptor 2, and HIF-1α in the mouse model of breast cancer | Kwon et al. [166] |
| Xanthohumol (64) | Inhibits angiogenesis by suppressing NF-κB activation and the subsequent production of angiogenic factors VEGF and IL-8 in vitro and in vivo of pancreatic cancer (BxPC-3) | Saito et al. [167] |
| Chalcone-based compounds with 2,2-dimethylbenzopyran (65) | Inhibits HIF-1 by downregulating the expression of HIF-1α in Hep3B and HUVEC cells and Hep3B xenograft models | Wang et al. [168] |
| 2, 3’, 4, 4’-tetrahydroxy-3, 5’-diprenylchalcone (66, FLA-16) | Inhibits CYP4A and prolongs survival and normalizes vasculature in C6 and U87 gliomas tumor models through decreasing production of TAMs and EPCs-derived VEGF and TGF-b. | Wang et al. [169] |
| Analog of chalcone (67) | Suppresses TGF β1, induces EMT markers, MMP 2 and MMP 9, and inhibits migration and invasion of A549 cells | Jeong et al. [170] |
| Anthraquinone-chalcone hybrids (68) | Decreases in the expression levels of MMP2, MMP9, and VEGF in K562 cells | Stanojkovic et al. [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. https://doi.org/10.3390/biom11060894
Ouyang Y, Li J, Chen X, Fu X, Sun S, Wu Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules. 2021; 11(6):894. https://doi.org/10.3390/biom11060894
Chicago/Turabian StyleOuyang, Yang, Juanjuan Li, Xinyue Chen, Xiaoyu Fu, Si Sun, and Qi Wu. 2021. "Chalcone Derivatives: Role in Anticancer Therapy" Biomolecules 11, no. 6: 894. https://doi.org/10.3390/biom11060894