
Natriuretic Peptides Regulate Prostate Cells Inflammatory Behavior: Potential Novel Anticancer Agents for Prostate Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Drug Treatments
2.3. Extracellular Vesicle Isolation
2.4. Western Blot Analysis
2.5. Measurements of Secreted IL-1β
2.6. Quantitative Real Time PCR Analysis
2.7. Statistical Analysis
3. Results
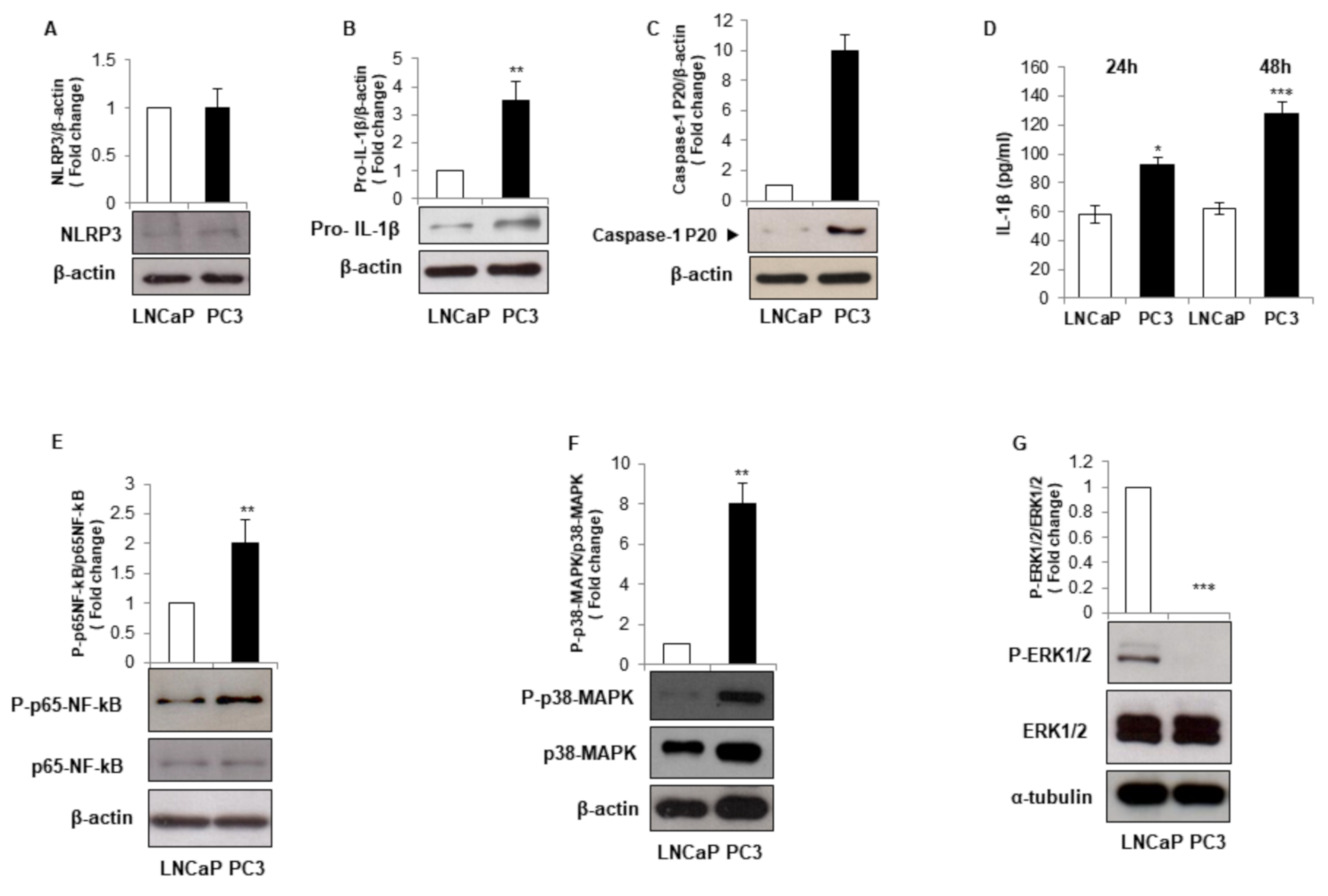
3.1. LNCaP and PC3 Cells Display Different Inflammatory Phenotype
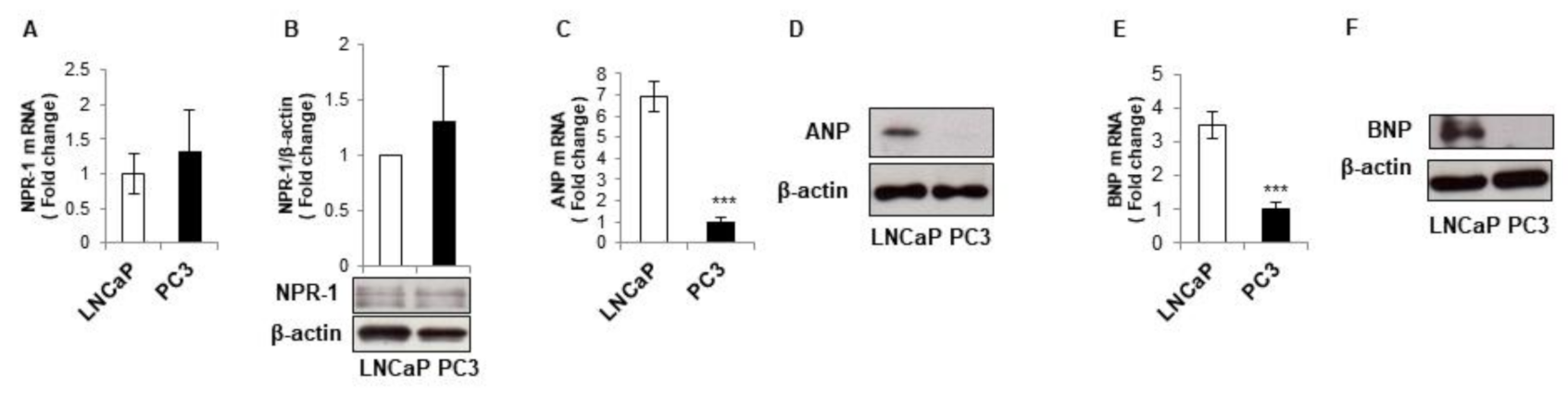
3.2. LNCaP and PC3 Cells Display Different Natriuretic Peptides Expression Levels
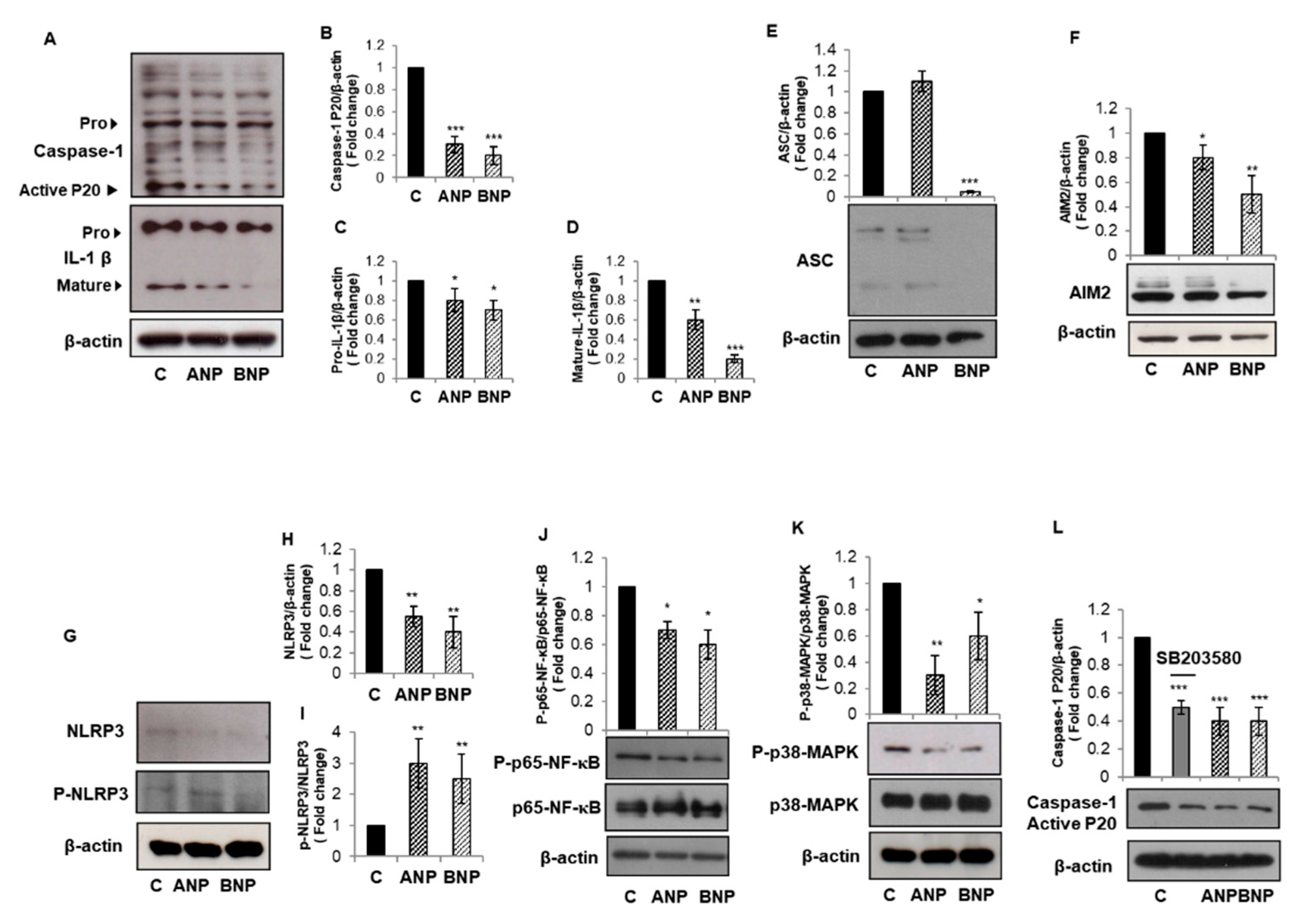
3.3. ANP and BNP Counteract Constitutive Inflammasome Activation in PC3 Cells
3.4. ANP and BNP Counteract PC3-Derived EVs-Induced Inflammasome Activation in PNT2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Strope, J.D.; Price, D.K.; Figg, W.D. Building a Hit List for the Fight against Metastatic Castration Resistant Prostate Cancer. Cancer Biol. Ther. 2016, 17, 231–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I.; Scarpelli, P.; Pizzo, S.V.; Grottelli, S.; Costanzi, E.; Minelli, A. ROS-Independent Nrf2 Activation in Prostate Cancer. Oncotarget 2017, 8, 67506–67518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karan, D.; Dubey, S. From Inflammation to Prostate Cancer: The Role of Inflammasomes. Adv. Urol. 2016, 2016, 3140372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, J.; Beyaert, R. Inflammation and NF-ΚB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells 2018, 7, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaeminck-Guillem, V. Extracellular Vesicles in Prostate Cancer Carcinogenesis, Diagnosis, and Management. Front. Oncol. 2018, 8, 222. [Google Scholar] [CrossRef]
- Sahlén, G.; Ahlander, A.; Frost, A.; Ronquist, G.; Norlén, B.J.; Nilsson, B.O. Prostasomes Are Secreted from Poorly Differentiated Cells of Prostate Cancer Metastases: Prostasomes Are Secreted from Prostate Cancer Metastases. Prostate 2004, 61, 291–297. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Costanzi, E.; Scarpelli, P.; Talesa, V.N.; Bellezza, I. Extracellular Vesicles from Human Advanced-Stage Prostate Cancer Cells Modify the Inflammatory Response of Microenvironment-Residing Cells. Cancers 2019, 11, 1276. [Google Scholar] [CrossRef] [Green Version]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.N.; Voronov, E. Immunotherapeutic Approaches of IL-1 Neutralization in the Tumor Microenvironment. J. Leukoc. Biol. 2017, 102, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 Inflammasome in Fibroblasts Links Tissue Damage with Inflammation in Breast Cancer Progression and Metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Geng, Y.; Zhao, L.; Li, R.; Zhang, Z.; Li, K.; Liang, R.; Shao, X.; Huang, M.; Zuo, D.; et al. NLRP3 Inflammasomes in Macrophages Drive Colorectal Cancer Metastasis to the Liver. Cancer Lett. 2019, 442, 21–30. [Google Scholar] [CrossRef]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively Active Inflammasome in Human Melanoma Cells Mediating Autoinflammation via Caspase-1 Processing and Secretion of Interleukin-1β. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [Green Version]
- Eiró, N.; Bermudez-Fernandez, S.; Fernandez-Garcia, B.; Atienza, S.; Beridze, N.; Escaf, S.; Vizoso, F.J. Analysis of the Expression of Interleukins, Interferon β, and Nuclear Factor-κ B in Prostate Cancer and Their Relationship with Biochemical Recurrence. J. Immunother. 2014, 37, 366–373. [Google Scholar] [CrossRef]
- Liu, Q.; Russell, M.R.; Shahriari, K.; Jernigan, D.L.; Lioni, M.I.; Garcia, F.U.; Fatatis, A. Interleukin-1β Promotes Skeletal Colonization and Progression of Metastatic Prostate Cancer Cells with Neuroendocrine Features. Cancer Res. 2013, 73, 3297–3305. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Anti-Inflammatory Agents: Present and Future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Tschopp, J. Inflammatory Caspases and Inflammasomes: Master Switches of Inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef]
- Veeranki, S. Role of Inflammasomes and Their Regulators in Prostate Cancer Initiation, Progression and Metastasis. Cell Mol. Biol. Lett. 2013, 18, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-Y.; Tsai, M.-C.; Tu, W.; Yeh, H.-C.; Wang, S.-C.; Huang, S.-P.; Li, C.-Y. Role of the NLRP3 Inflammasome: Insights into Cancer Hallmarks. Front. Immunol. 2020, 11, 610492. [Google Scholar] [CrossRef]
- Kolb, R.; Liu, G.-H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in Cancer: A Double-Edged Sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 Inflammasome in Cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzasoma, L.; Talesa, V.N.; Romani, R.; Bellezza, I. ANP and BNP Exert Anti-Inflammatory Action via NPR-1/CGMP Axis by Interfering with Canonical, Non-Canonical, and Alternative Routes of Inflammasome Activation in Human THP1 Cells. Int. J. Mol. Sci. 2020, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic Peptides: Their Structures, Receptors, Physiologic Functions and Therapeutic Applications. Handb. Exp. Pharmacol. 2009, 191, 341–366. [Google Scholar] [CrossRef] [Green Version]
- Potter, L.R. Regulation and Therapeutic Targeting of Peptide-Activated Receptor Guanylyl Cyclases. Pharmacol. Ther. 2011, 130, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojiri, T.; Hosoda, H.; Tokudome, T.; Miura, K.; Ishikane, S.; Otani, K.; Kishimoto, I.; Shintani, Y.; Inoue, M.; Kimura, T.; et al. Atrial Natriuretic Peptide Prevents Cancer Metastasis through Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4086–4091. [Google Scholar] [CrossRef] [Green Version]
- Serafino, A.; Moroni, N.; Psaila, R.; Zonfrillo, M.; Andreola, F.; Wannenes, F.; Mercuri, L.; Rasi, G.; Pierimarchi, P. Anti-Proliferative Effect of Atrial Natriuretic Peptide on Colorectal Cancer Cells: Evidence for an Akt-Mediated Cross-Talk between NHE-1 Activity and Wnt/β-Catenin Signaling. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Bando, S.; Soeki, T.; Matsuura, T.; Tobiume, T.; Ise, T.; Kusunose, K.; Yamaguchi, K.; Yagi, S.; Fukuda, D.; Iwase, T.; et al. Plasma Brain Natriuretic Peptide Levels Are Elevated in Patients with Cancer. PLoS ONE 2017, 12, e0178607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzasoma, L.; Peirce, M.J.; Minelli, A.; Bellezza, I. Natriuretic Peptides: The Case of Prostate Cancer. Molecules 2017, 22, 1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, P.; Vincent Rajkumar, S.; Merlini, G.; Kumar, S.; Gertz, M.A.; Palladini, G.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Leung, N.; et al. N-terminal Fragment of the Type-B Natriuretic Peptide (NT-proBNP) Contributes to a Simple New Frailty Score in Patients with Newly Diagnosed Multiple Myeloma. Am. J. Hematol. 2016, 91, 1129–1134. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Aisa, M.C.; Palazzo, R.; Costanzi, E.; Mearini, E.; Minelli, A. Extracellular Matrix Degrading Enzymes at the Prostasome Surface. Prostate Cancer Prostatic Dis. 2005, 8, 344–348. [Google Scholar] [CrossRef] [Green Version]
- Mezzasoma, L.; Antognelli, C.; Talesa, V.N. Atrial Natriuretic Peptide Down-Regulates LPS/ATP-Mediated IL-1β Release by Inhibiting NF-KB, NLRP3 Inflammasome and Caspase-1 Activation in THP-1 Cells. Immunol. Res. 2016, 64, 303–312. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Antognelli, C.; Talesa, V.N. A Novel Role for Brain Natriuretic Peptide: Inhibition of IL-1β Secretion via Downregulation of NF-KB/Erk 1/2 and NALP3/ASC/Caspase-1 Activation in Human THP-1 Monocyte. Mediat. Inflamm. 2017, 2017, 5858315. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ren, W.; Jiang, Z.; Zhu, L. Regulation of the NLRP3 Inflammasome and Macrophage Pyroptosis by the P38 MAPK Signaling Pathway in a Mouse Model of Acute Lung Injury. Mol. Med. Rep. 2018, 18, 4399–4409. [Google Scholar] [CrossRef] [Green Version]
- Tartey, S.; Kanneganti, T.-D. Differential Role of the NLRP3 Inflammasome in Infection and Tumorigenesis. Immunology 2019, 156, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the Use of Therapeutic Peptides for Cancer Treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef] [Green Version]
- Fish-Trotter, H.; Ferguson, J.F.; Patel, N.; Arora, P.; Allen, N.B.; Bachmann, K.N.; Daniels, L.B.; Reilly, M.P.; Lima, J.A.C.; Wang, T.J.; et al. Inflammation and Circulating Natriuretic Peptide Levels. Circ. Heart Fail. 2020, 13, e006570. [Google Scholar] [CrossRef]
- Zhang, J.; Li, M.; Yang, Y.; Yan, Y.; Li, J.; Qu, J.; Wang, J. NPR-A: A Therapeutic Target in Inflammation and Cancer. Crit. Rev. Eukaryot Gene Expr. 2015, 25, 41–46. [Google Scholar] [CrossRef]
- Gruden, G.; Landi, A.; Bruno, G. Natriuretic Peptides, Heart, and Adipose Tissue: New Findings and Future Developments for Diabetes Research. Diabetes Care 2014, 37, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Mezzasoma, L.; Cagini, L.; Antognelli, C.; Puma, F.; Pacifico, E.; Talesa, V.N. TNF-α Regulates Natriuretic Peptides and Aquaporins in Human Bronchial Epithelial Cells BEAS-2B. Mediat. Inflamm. 2013, 2013, 159349. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1beta-Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Poli, G.; Fabi, C.; Bellet, M.M.; Costantini, C.; Nunziangeli, L.; Romani, L.; Brancorsini, S. Epigenetic Mechanisms of Inflammasome Regulation. Int. J. Mol. Sci. 2020, 21, 5758. [Google Scholar] [CrossRef]
- Teng, G.; Wang, W.; Dai, Y.; Wang, S.; Chu, Y.; Li, J. Let-7b Is Involved in the Inflammation and Immune Responses Associated with Helicobacter Pylori Infection by Targeting Toll-like Receptor 4. PLoS ONE 2013, 8, e56709. [Google Scholar] [CrossRef] [Green Version]
- Costanzi, E.; Romani, R.; Scarpelli, P.; Bellezza, I. Extracellular Vesicles-Mediated Transfer of MiRNA Let-7b from PC3 Cells to Macrophages. Genes 2020, 11, 1495. [Google Scholar] [CrossRef]
- Pressly, E.D.; Pierce, R.A.; Connal, L.A.; Hawker, C.J.; Liu, Y. Nanoparticle PET/CT Imaging of Natriuretic Peptide Clearance Receptor in Prostate Cancer. Bioconjug. Chem. 2013, 24, 196–204. [Google Scholar] [CrossRef]
- Sun, Y.; Eichelbaum, E.J.; Lenz, A.; Skelton, W.P.; Wang, H.; Vesely, D.L. Atrial Natriuretic Peptide and Long-Acting Natriuretic Peptide Inhibit Ras in Human Prostate Cancer Cells. Anticancer Res. 2009, 29, 1889–1893. [Google Scholar]
- Li, X.; Peng, H.; Wu, J.; Xu, Y. Brain Natriuretic Peptide-Regulated Expression of Inflammatory Cytokines in Lipopolysaccharide (LPS)-Activated Macrophages via NF-ΚB and Mitogen Activated Protein Kinase (MAPK) Pathways. Med. Sci. Monit. 2018, 24, 3119–3126. [Google Scholar] [CrossRef]
- Abdel-Latif, G.A.; Elwahab, A.H.A.; Hasan, R.A.; ElMongy, N.F.; Ramzy, M.M.; Louka, M.L.; Schaalan, M.F. A Novel Protective Role of Sacubitril/Valsartan in Cyclophosphamide Induced Lung Injury in Rats: Impact of MiRNA-150-3p on NF-ΚB/MAPK Signaling Trajectories. Sci. Rep. 2020, 10, 13045. [Google Scholar] [CrossRef]
- Chung, Y.-H.; Kim, D.-H.; Lee, W.-W. Monosodium Urate Crystal-Induced pro-Interleukin-1β Production Is Post-Transcriptionally Regulated via the P38 Signaling Pathway in Human Monocytes. Sci. Rep. 2016, 6, 34533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Chen, W.; Fan, H.; Chen, X.; Zhang, J.; Zhu, J.-S. Dihydroartemisinin Prevents Dextran Sodium Sulphate-Induced Colitisthrough Inhibition of the Activation of NLRP3 Inflammasome and P38 MAPK Signaling. Int. Immunopharmacol. 2020, 88, 106949. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.R.; Pucci, B.; Bruzzese, F.; Carbone, C.; Piro, G.; Costantini, S.; Capone, F.; Leone, A.; Di Gennaro, E.; Caraglia, M.; et al. Acquired Resistance to Zoledronic Acid and the Parallel Acquisition of an Aggressive Phenotype Are Mediated by P38-MAP Kinase Activation in Prostate Cancer Cells. Cell Death Dis. 2013, 4, e641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandrika, L.; Lieberman, R.; Koul, S.; Kumar, B.; Maroni, P.; Chandhoke, R.; Meacham, R.B.; Koul, H.K. Hypoxia-Associated P38 Mitogen-Activated Protein Kinase-Mediated Androgen Receptor Activation and Increased HIF-1alpha Levels Contribute to Emergence of an Aggressive Phenotype in Prostate Cancer. Oncogene 2009, 28, 1248–1260. [Google Scholar] [CrossRef] [Green Version]
- Suwa, M.; Seino, Y.; Nomachi, Y.; Matsuki, S.; Funahashi, K. Multicenter Prospective Investigation on Efficacy and Safety of Carperitide for Acute Heart Failure in the “real World” of Therapy. Circ. J. 2005, 69, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Vinnakota, S.; Chen, H.H. The Importance of Natriuretic Peptides in Cardiometabolic Diseases. J. Endocr. Soc. 2020, 4, bvaa052. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Protein (Clone) | Host | Code # | Manufacturer |
|---|---|---|---|
| mAb anti-NLRP3 (D4D8T) | Rabbit | 15101T | Cell Signalling Technology |
| mAb anti-AIM2(D5 × 7K) | Rabbit | 12948T | Cell Signalling Technology |
| mAb anti-ASC/TMS1 (E13EI) | Rabbit | 13833T | Cell Signalling Technology |
| mAb anti-Phospho-NF-κB p65 (Ser536) (93H1) | Rabbit | 3033 | Cell Signalling Technology |
| mAb anti-IL1-β (3A6) | Mouse | 12242 | Cell Signalling Technology |
| mAb anti-phospho-p38 MAPK (Tyr180/Tyr182) (D3F9) | Rabbit | 4511 | Cell Signalling Technology |
| mAb anti-p38 MAPK (D13E1) | Rabbit | 8690T | Cell Signalling Technology |
| pAb anti-Caspase-1 | Rabbit | 2225 | Cell Signalling Technology |
| pAb anti-Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) | Rabbit | 9101 | Cell Signalling Technology |
| anti-p44/42 MAPK (ERK1/2) | Rabbit | 9102 | Cell Signalling Technology |
| pAb anti-NF-κB p65 | Rabbit | 3034 | Cell Signalling Technology |
| pAb anti-ANP | Rabbit | PA5-29559 | Thermo Fisher Scientific |
| pAb anti-BNP | Rabbit | PA5-21321 | Thermo Fisher Scientific |
| pAb anti-NPR-1 | Rabbit | PA5-29049 | Thermo Fisher Scientific |
| pAb anti-phospho-NLRP3 (Ser 295) | Rabbit | PA5-105071 | Thermo Fisher Scientific |
| pAb anti-β-actin | Rabbit | 4967 | Cell Signalling Technology |
| mAb α-Tubulin (DM1A) | Mouse | 3873 | Cell Signalling Technology |
| Gene Name | Gene Symbol | Primer Sequences (F: Forward R: Reverse) |
|---|---|---|
| Atrial Natriuretic Peptide | ANP | F: TCAGCCCAGCCCAGAGAG R: GCTCCAATCCTGTCCATCCTG |
| Brain Natriuretic Peptide | BNP | F: GAGGGCAGGTGGGAAGCAAAC R: GCAAGAAGAGCAGGAGCAGGAG |
| Natriuretic Peptide Receptor 1 | NPR-1 | F: TAACACGCACGCACACTC R: CTATGGGAAGGAGCAGGAG |
| Glyceraldehyde-3-phosphate dehydrogenase | GAPDH | F: TGGTATCGTGGAAGGACTCATGAC R: ATGCCAGTGAGCTTCCCGTTCAGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzasoma, L.; Talesa, V.N.; Costanzi, E.; Bellezza, I. Natriuretic Peptides Regulate Prostate Cells Inflammatory Behavior: Potential Novel Anticancer Agents for Prostate Cancer. Biomolecules 2021, 11, 794. https://doi.org/10.3390/biom11060794
Mezzasoma L, Talesa VN, Costanzi E, Bellezza I. Natriuretic Peptides Regulate Prostate Cells Inflammatory Behavior: Potential Novel Anticancer Agents for Prostate Cancer. Biomolecules. 2021; 11(6):794. https://doi.org/10.3390/biom11060794
Chicago/Turabian StyleMezzasoma, Letizia, Vincenzo Nicola Talesa, Egidia Costanzi, and Ilaria Bellezza. 2021. "Natriuretic Peptides Regulate Prostate Cells Inflammatory Behavior: Potential Novel Anticancer Agents for Prostate Cancer" Biomolecules 11, no. 6: 794. https://doi.org/10.3390/biom11060794