
Conformational Plasticity-Rigidity Axis of the Coagulation Factor VII Zymogen Elucidated by Atomistic Simulations of the N-Terminally Truncated Factor VIIa Protease Domain


Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics (MD) Simulations
2.1.1. Temperature-Replica-Exchange Molecular Dynamics (T-REMD) Simulations
2.1.2. The In Silico Zymogenized Variants
2.2. Analyses
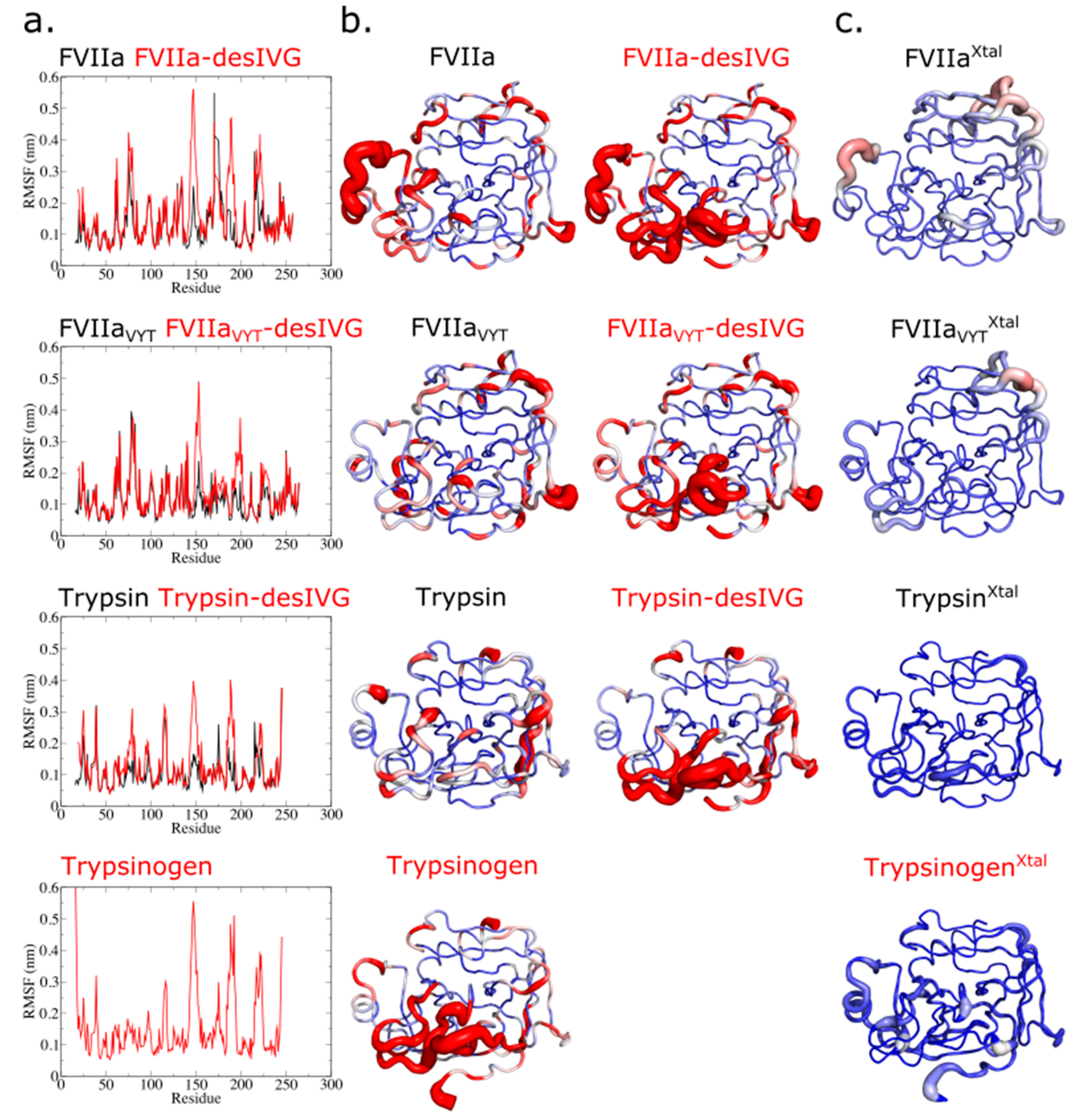
2.2.1. Root-Mean-Squared Deviation (RMSD) and Root-Mean-Squared Fluctuation (RMSF)
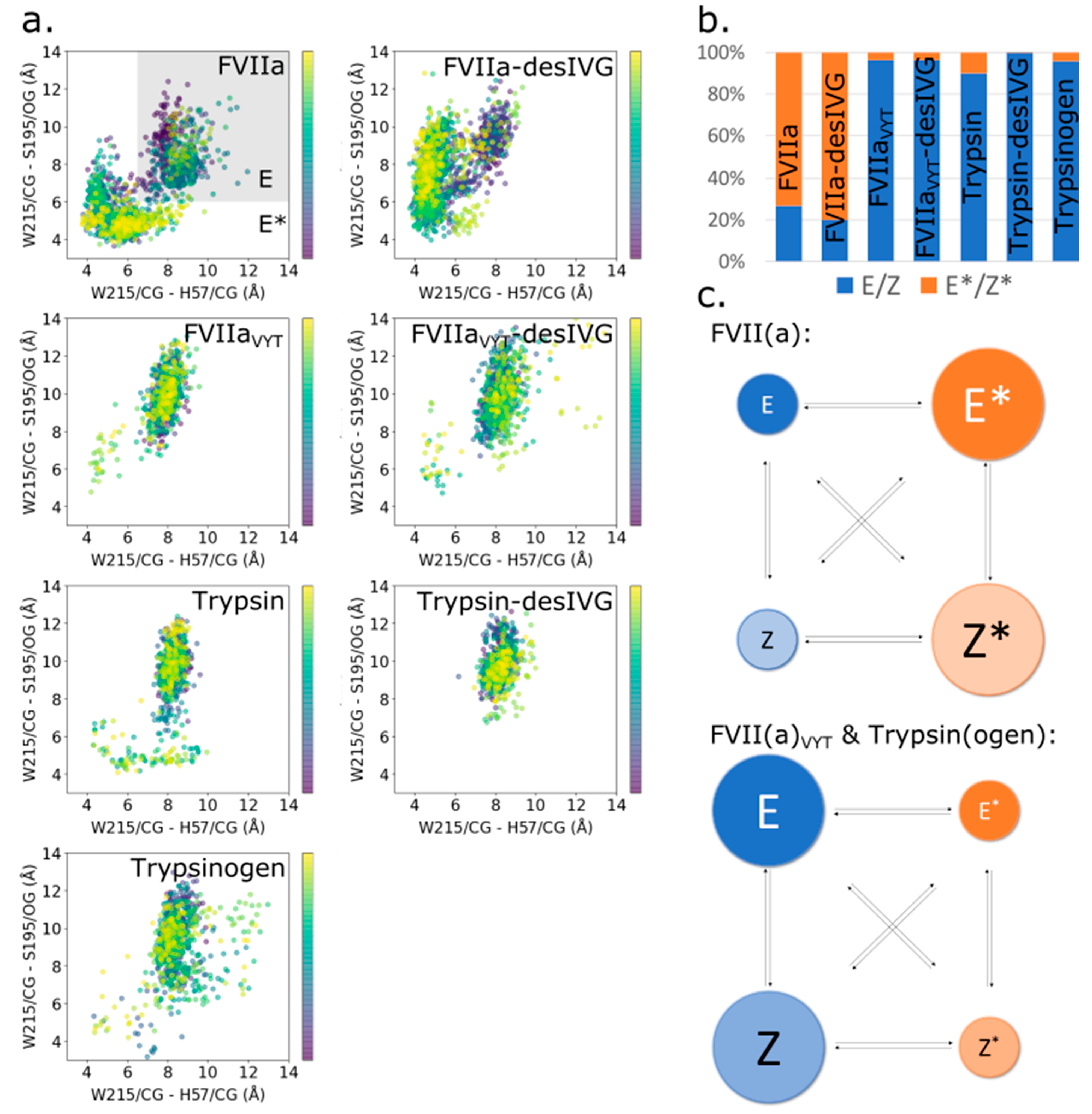
2.2.2. E/Z-E*/Z* Distributions
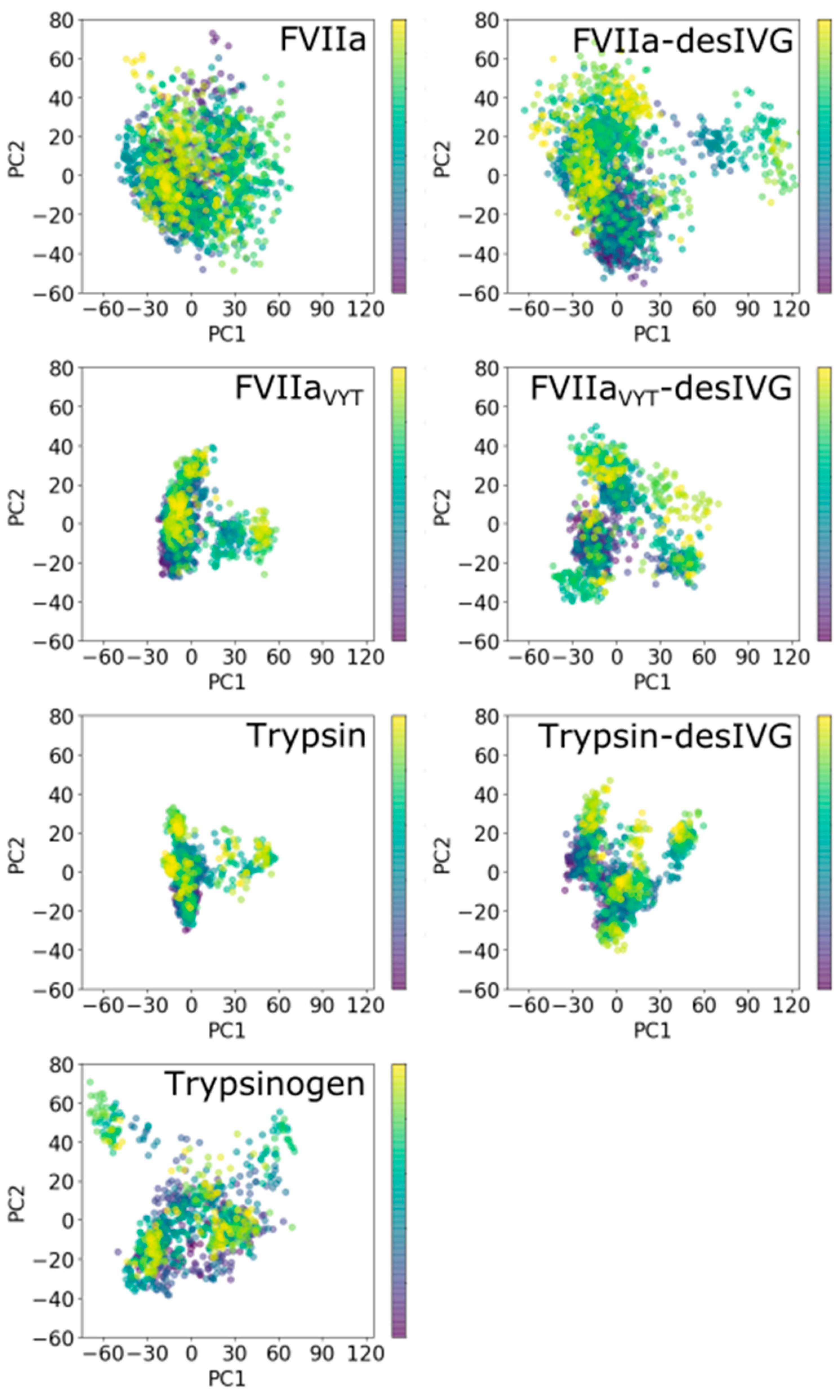
2.2.3. Principal Components Analysis (PCA)
2.2.4. Hierarchical Clustering Analysis
2.2.5. Dynamic Cross-Correlations
2.2.6. Plotting and Visualization
3. Results and Discussion
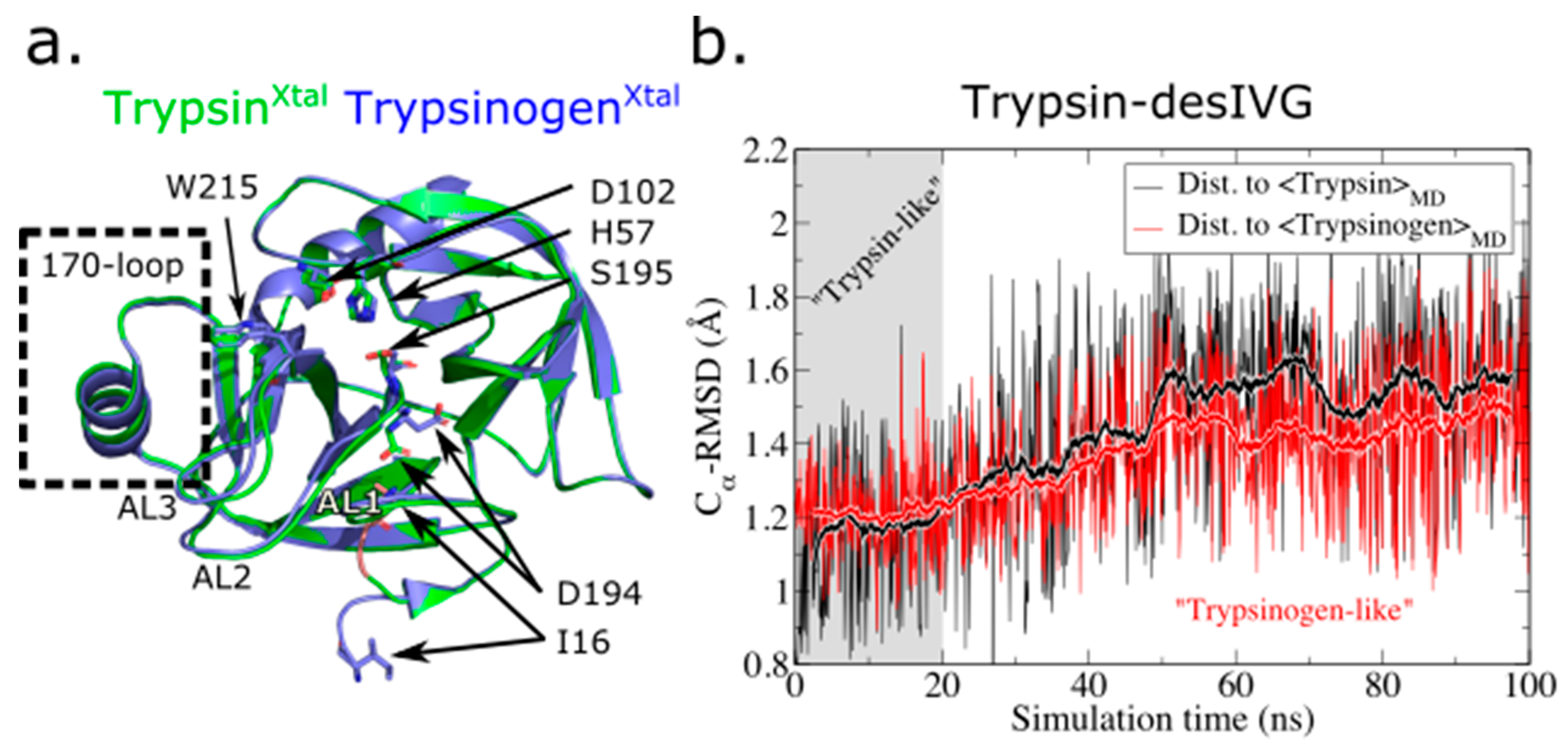
3.1. The N-Terminally Truncated Trypsin Gradually Adopts Structural and Dynamic Features Characteristic to Trypsinogen
3.2. Conformational Flexibility of FVIIa, the TF-Independent Variant FVIIaVYT, and Trypsin
3.3. The Allosteric E-E* and Z-Z* Equilibria Are Influenced by the 170-Loop, but Independent of N-Terminus Insertion
3.4. Conformational Plasticity-Rigidity Axis Intrinsically Governed by the 170-Loop with the N-Terminus Insertion as Extrinsic Modulator
3.5. A Glimpse of the Elusive FVII Zymogen Exposed at Last?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- MacFarlane, R. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature 1964, 202, 498–499. [Google Scholar] [CrossRef]
- Davie, E.W.; Ratnoff, O.D. Waterfall Sequence for Intrinsic Blood Clotting. Science 1964, 145, 1310–1312. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C. The molecular basis of blood coagulation. Cell 1988, 53, 505–518. [Google Scholar] [CrossRef]
- Davie, E.W.; Fujikawa, K.; Kisiel, W. The coagulation cascade: Initiation, maintenance, and regulation. Biochemistry 1991, 30, 10363–10370. [Google Scholar] [CrossRef] [PubMed]
- Neuenschwander, P.F.; Morrissey, J.H. Roles of the membrane-interactive regions of factor VIIa and tissue factor. The factor VIIa Gla domain is dispensable for binding to tissue factor but important for activation of factor X. J. Biol. Chem. 1994, 269, 8007–8013. [Google Scholar] [CrossRef]
- Leonard, B.J.N.; Clarke, B.J.; Sridhara, S.; Kelley, R.; Ofosu, F.A.; Blajchman, M.A. Activation and active site occupation alter conformation in the region of the first epidermal growth factor-like domain of human factor VII. J. Biol. Chem. 2000, 275, 34894–34900. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Sen, P. Structural modulation of factor VIIa by full-length tissue factor (TF1-263): Implication of novel interactions between EGF2 domain and TF. J. Biomol. Struct. Dyn. 2018, 36, 621–633. [Google Scholar] [CrossRef]
- Ohkubo, Y.Z.; Madsen, J.J. Uncovering Membrane-Bound Models of Coagulation Factors by Combined Experimental and Computational Approaches. Thromb. Haemost. 2021, in press. [Google Scholar] [CrossRef]
- Eigenbrot, C.; Kirchhofer, D.; Dennis, M. The factor VII zymogen structure reveals reregistration of β strands during activation. Structure 2001, 9, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, R.A.; Olivero, A.G.; Eigenbrot, C.; Kirchhofer, D. Inhibitors of Tissue Factor—Factor VIIa for Anticoagulant Therapy. Curr. Med. Chem. 2004, 11, 2275. [Google Scholar] [CrossRef] [PubMed]
- Colina, C.M.; Venkateswarlu, D.; Duke, R.; Perera, L.; Pedersen, L.G. What causes the enhancement of activity of factor VIIa by tissue factor? J. Thromb. Haemost. 2006, 4, 2726–2729. [Google Scholar] [CrossRef]
- Ohkubo, Y.Z.; Tajkhorshid, E. Distinct structural and adhesive roles of Ca2+ in membrane binding of blood coagulation factors. Structure 2008, 16, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Ohkubo, Y.Z.; Morrissey, J.H.; Tajkhorshid, E. Dynamical view of membrane binding and complex formation of human factor VIIa and tissue factor. J. Thromb. Haemost. 2010, 8, 1044–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.J.; Chandrasekaran, V.; Wu, S.; Duke, R.E.; Pedersen, L.G. Recent Estimates of the Structure of the Factor VIIa (FVIIa)/Tissue Factor (TF) and Factor Xa (FXa) Ternary Complex. Thromb. Res. 2010, 125, S7–S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.J.; Wu, S.; Bartolotti, L.J.; Pedersen, L.G. Molecular dynamics simulations of the binary complex of human tissue factor (TF(1-242)) and factor VIIa (TF(1-242) /fVIIa) on a 4:1 POPC/POPS lipid bilayer. J. Thromb. Haemost. 2012, 10, 2402–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, E.; Madsen, J.J.; Olsen, O.H. The length of the linker between the epidermal growth factor-like domains in factor VIIa is critical for a productive interaction with tissue factor. Protein Sci. 2014, 23, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Bode, W.; Huber, R. Induction of the bovine trypsinogen—Trypsin transition by peptides sequentially similar to the N-terminus of trypsin. FEBS Lett. 1976, 68, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Bode, W.; Schwager, P.; Huber, R. The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. J. Mol. Biol. 1978, 118, 99–112. [Google Scholar] [CrossRef]
- Huber, R.; Bode, W. Structural basis of the activation and action of trypsin. Acc. Chem. Res. 1978, 266, 114–122. [Google Scholar] [CrossRef]
- Bode, W. The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding: II. The binding of the pancreatic trypsin inhibitor and of isoleucine-valine and of sequentially related peptides to trypsinogen and to p-guanidinobenzoate-trypsinoge. J. Mol. Biol. 1979, 127, 357–374. [Google Scholar] [CrossRef]
- Plattner, N.; Noé, F. Protein conformational plasticity and complex ligand-binding kinetics explored by atomistic simulations and Markov models. Nat. Commun. 2015, 6, 7653. [Google Scholar] [CrossRef] [Green Version]
- Fehlhammer, H.; Bode, W.; Huber, R. Crystal structure of bovine trypsinogen at 1·8 Å resolution: II. Crystallographic refinement, refined crystal structure and comparison with bovine trypsin. J. Mol. Biol. 1977, 111, 415–438. [Google Scholar] [CrossRef]
- Boechi, L.; Pierce, L.; Komives, E.A.; McCammon, J.A. Trypsinogen activation as observed in accelerated molecular dynamics simulations. Protein Sci. 2014, 23, 1550–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sichler, K.; Banner, D.W.; D’Arcy, A.; Hopfner, K.-P.; Huber, R.; Bode, W.; Kresse, G.-B.; Kopetzki, E.; Brandstetter, H. Crystal Structures of Uninhibited Factor VIIa Link its Cofactor and Substrate-assisted Activation to Specific Interactions. J. Mol. Biol. 2002, 322, 591–603. [Google Scholar] [CrossRef]
- Banner, D.W.; D’Arcy, A.; Chène, C.; Winkler, F.K.; Guha, A.; Konigsberg, W.H.; Nemerson, Y.; Kirchhofer, D. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature 1996, 380, 41–46. [Google Scholar] [CrossRef]
- Pike, A.C.W.; Brzozowski, A.M.; Roberts, S.M.; Olsen, O.H.; Persson, E. Structure of human factor VIIa and its implications for the triggering of blood coagulation. Proc. Natl. Acad. Sci. USA 1999, 96, 8925–8930. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.; Darden, T.A.; Pedersen, L.G. Predicted solution structure of zymogen human coagulation FVII. J. Comput. Chem. 2002, 23, 35–47. [Google Scholar] [CrossRef]
- Madsen, J.J.; Persson, E.; Olsen, O.H. Tissue factor activates allosteric networks in factor VIIa through structural and dynamic changes. J. Thromb. Haemost. 2015, 13, 262–267. [Google Scholar] [CrossRef]
- Peterson, F.C.; Gordon, N.C.; Gettins, P.G.W. High-level bacterial expression and 15N-alanine-labeling of bovine trypsin. Application to the study of trypsin—Inhibitor complexes and trypsinogen activation by NMR spectroscopy. Biochemistry 2001, 40, 6275–6283. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Johnson, D.J.D.; Freund, S.M.V.; Huntington, J.A. NMR resonance assignments of thrombin reveal the conformational and dynamic effects of ligation. Proc. Natl. Acad. Sci. USA 2010, 107, 14087–14092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rand, K.D.; Jørgensen, T.J.D.; Olsen, O.H.; Persson, E.; Jensen, O.N.; Stennicke, H.R.; Andersen, M.D. Allosteric activation of coagulation factor VIIa visualized by hydrogen exchange. J. Biol. Chem. 2006, 281, 23018–23024. [Google Scholar] [CrossRef] [Green Version]
- Rand, K.D.; Andersen, M.D.; Olsen, O.H.; Jørgensen, T.J.D.; Ostergaard, H.; Jensen, O.N.; Stennicke, H.R.; Persson, E. The origins of enhanced activity in factor VIIa analogs and the interplay between key allosteric sites revealed by hydrogen exchange mass spectrometry. J. Biol. Chem. 2008, 283, 13378–13387. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Olsen, O.H.; Persson, E.; Rand, K.D. Sites involved in intra- and interdomain allostery associated with the activation of factor VIIa pinpointed by hydrogen-deuterium exchange and electron transfer dissociation mass spectrometry. J. Biol. Chem. 2014, 289, 35388–35396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, O.H.; Persson, E. Cofactor-induced and mutational activity enhancement of coagulation factor VIIa. Cell. Mol. Life Sci. 2008, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.; Olsen, O.H. Allosteric activation of coagulation factor VIIa. Front. Biosci. 2011, 16, 3156–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, E.; Olsen, O.H. Activation loop 3 and the 170 loop interact in the active conformation of coagulation factor VIIa. FEBS J. 2009, 276, 3099–3109. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.; Kjalke, M.; Olsen, O.H. Rational design of coagulation factor VIIa variants with substantially increased intrinsic activity. Proc. Natl. Acad. Sci. USA 2001, 98, 13583–13588. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, A.L.; Sorensen, A.B.; Holmberg, H.L.; Gandhi, P.S.; Karlsson, J.; Buchardt, J.; Lamberth, K.; Kjelgaard-Hansen, M.; Ley, C.D.; Sørensen, B.B.; et al. Engineering of a membrane-triggered activity switch in coagulation factor VIIa. Proc. Natl. Acad. Sci. USA 2017, 114, 12454–12459. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, A.B.; Tuneew, I.; Svensson, L.A.; Persson, E.; Østergaard, H.; Overgaard, M.T.; Olsen, O.H.; Gandhi, P.S. Beating tissue factor at its own game: Design and properties of a soluble tissue factor–independent coagulation factor VIIa. J. Biol. Chem. 2020, 295, 517–528. [Google Scholar] [CrossRef]
- Soejima, K.; Mizuguchi, J.; Yuguchi, M.; Nakagaki, T.; Higashi, S.; Iwanaga, S. Factor VIIa modified in the 170 loop shows enhanced catalytic activity but does not change the zymogen-like property. J. Biol. Chem. 2001, 276, 17229–17235. [Google Scholar] [CrossRef] [Green Version]
- Soejima, K.; Yuguchi, M.; Mizuguchi, J.; Tomokiyo, K.; Nakashima, T.; Nakagaki, T.; Iwanaga, S. The 99 and 170 loop-modified factor VIIa mutants show enhanced catalytic activity without tissue factor. J. Biol. Chem. 2002, 277, 49027–49035. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, A.B.; Madsen, J.J.; Svensson, L.A.; Pedersen, A.A.; Østergaard, H.; Overgaard, M.T.; Olsen, O.H.; Gandhi, P.S. Molecular basis of enhanced activity in factor VIIa-trypsin variants conveys insights into tissue factor-mediated allosteric regulation of factor VIIa activity. J. Biol. Chem. 2016, 291, 4671–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; Mackerell, A.D. Charmm36M: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N*log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. [Google Scholar] [CrossRef] [Green Version]
- Nose, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Nose, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Cuesta-Seijo, J.A.; Garcia-Granda, S. Trypsin as a model for high resolution x-ray diffraction in proteins. Bol. R. Soc. Hist. Nat. Sec. Geol. 2002, 97, 123–129. [Google Scholar]
- Walter, J.; Steigemann, W.; Singh, T.P.; Bartunik, H.; Bode, W.; Huber, R. On the disordered activation domain in trypsinogen: Chemical labelling and low-temperature crystallography. Acta Crystallogr. Sect. B 1982, 1462–1472. [Google Scholar] [CrossRef]
- Yang, L.; Eyal, E.; Chennubhotla, C.; Jee, J.; Gronenborn, A.M.; Bahar, I. Insights into equilibrium dynamics of proteins from comparison of NMR and X-ray data with computational predictions. Structure 2007, 15, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquax, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Sokal, R.R.; Michener, C.D. A statistical method for evaluating systematic relationships. Univ. Kans Sci. Bull. 1958, 38, 1409–1438. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.K.; Penkler, D.L.; Amamuddy, O.S.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Bishop, Ö.T. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017, 33, 2768–2771. [Google Scholar] [CrossRef] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Gohara, D.W.; Di Cera, E. Allostery in trypsin-like proteases suggests new therapeutic strategies. Trends Biotechnol. 2011, 29, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Beeler, D.L.; Aird, W.C.; Grant, M.A. Evolutionary conservation of the allosteric activation of factor VIIa by tissue factor in lamprey. J. Thromb. Haemost. 2018, 16, 734–748. [Google Scholar] [CrossRef] [Green Version]
- Madsen, J.J.; Persson, E.; Olsen, O.H. Evolutionary conservation of the allosteric activation of factor VIIa by tissue factor in lamprey: Comment. J. Thromb. Haemost. 2018, 16, 1450–1454. [Google Scholar] [CrossRef]
- Beeler, D.L.; Aird, W.C.; Grant, M.A. Evolutionary conservation of the allosteric activation of factor VIIa by tissue factor in lamprey: Reply. J. Thromb. Haemost. 2018, 16, 1454–1456. [Google Scholar] [CrossRef] [Green Version]
- Hedstrom, L.; Lin, T.Y.; Fast, W. Hydrophobic interactions control zymogen activation in the trypsin family of serine proteases. Biochemistry 1996, 35, 4515–4523. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, A.; White, A.; Jeffery, C.J.; Medina, N.; Cahoon, M.; Ringe, D.; Hedstrom, L. The energetic cost of induced fit catalysis: Crystal structures of trypsinogen mutants with enhanced activity and inhibitor affinity. Protein Sci. 2001, 10, 1331–1342. [Google Scholar] [CrossRef] [Green Version]
- Burnley, B.T.; Afonine, P.V.; Adams, P.D.; Gros, P. Modelling dynamics in protein crystal structures by ensemble refinement. Elife 2012, 2012, 1–29. [Google Scholar] [CrossRef]
- Burnley, B.T.; Gros, P. phenix. ensemble_refinement: A test study of apo and holo BACE1. Comput. Crystallogr. Newsl. 2013, 4, 51–58. [Google Scholar]
- Sorensen, A.B.; Madsen, J.J.; Frimurer, T.M.; Overgaard, M.T.; Gandhi, P.S.; Persson, E.; Olsen, O.H. Allostery in Coagulation Factor VIIa Revealed by Ensemble Refinement of Crystallographic Structures. Biophys. J. 2019, 116, 1823–1835. [Google Scholar] [CrossRef]
- Huntington, J.A.; Esmon, C.T. The molecular basis of thrombin allostery revealed by a 1.8 Å structure of the “slow” form. Structure 2003, 11, 469–479. [Google Scholar] [CrossRef]
- Bah, A.; Garvey, L.C.; Ge, J.; Di Cera, E. Rapid kinetics of Na+ binding to thrombin. J. Biol. Chem. 2006, 281, 40049–40056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, P.S.; Chen, Z.; Mathews, F.S.; Di Cera, E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 1832–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, A.D.; Bah, A.; Di Cera, E. Evidence of the E*-E equilibrium from rapid kinetics of Na+ binding to activated protein C and factor Xa*. J. Phys. Chem. B 2010, 114, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Chen, Z.; Gandhi, P.S.; Vogt, A.D.; Pozzi, N.; Pelc, L.A.; Zapata, F.; Di Cera, E. Crystallographic and kinetic evidence of allostery in a trypsin-like protease. Biochemistry 2011, 50, 6301–6307. [Google Scholar] [CrossRef] [Green Version]
- Vogt, A.D.; Chakraborty, P.; Di Cera, E. Kinetic Dissection of the Pre-existing Conformational Equilibrium in the Trypsin Fold. J. Biol. Chem. 2015, 290, 22435–22445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagendra, H.G.; Sukumar, N.; Vijayan, M. Role of water in plasticity, stability, and action of proteins: The crystal structures of lysozyme at very low levels of hydration. Proteins 1998, 32, 229–240. [Google Scholar] [CrossRef]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Pochapsky, T.C.; Kazanis, S.; Dang, M. Conformational Plasticity and Structure = Function. Antioxid. Redox Signal. 2010, 13, 1273–1296. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Seeliger, M.A. Targeting conformational plasticity of protein kinases. ACS Chem. Biol. 2015, 10, 190–200. [Google Scholar] [CrossRef]
- Spoerri, P.M.; Sapra, K.T.; Zhang, C.; Mari, S.A.; Kato, H.E.; Kobilka, B.K.; Müller, D.J. Conformational Plasticity of Human Protease-Activated Receptor 1 upon Antagonist- and Agonist-Binding. Structure 2019, 27, 1517–1526. [Google Scholar] [CrossRef]
- Crean, R.M.; Gardner, J.M.; Kamerlin, S.C.L. Harnessing Conformational Plasticity to Generate Designer Enzymes. J. Am. Chem. Soc. 2020, 142, 11324–11342. [Google Scholar] [CrossRef]
- Csermely, P. Plasticity-rigidity cycles: A general adaptation mechanism. arXiv 2015, arXiv:1511.01239. [Google Scholar]
- Martín-García, F.; Papaleo, E.; Gomez-Puertas, P.; Boomsma, W.; Lindorff-Larsen, K. Comparing Molecular Dynamics Force Fields in the Essential Subspace. PLoS ONE 2015, 10, e0121114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasper, P.M.; Fuglestad, B.; Komives, E.A.; Markwick, P.R.L.; McCammon, J.A. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. USA 2012, 109, 21216–21222. [Google Scholar] [CrossRef] [Green Version]
- Fuglestad, B.; Gasper, P.M.; McCammon, J.A.; Markwick, P.R.L.; Komives, E.A. Correlated motions and residual frustration in thrombin. J. Phys. Chem. B 2013, 117, 12857–12863. [Google Scholar] [CrossRef]
- Higashi, S.; Nishimura, H.; Aita, K.; Iwanaga, S. Identification of regions of bovine factor VII essential for binding to tissue factor. J. Biol. Chem. 1994, 269, 18891–18898. [Google Scholar] [CrossRef]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef]
- Magdziarz, T.; Mitusińska, K.; Gołdowska, S.; Płuciennik, A.; Stolarczyk, M.; Lugowska, M.; Góra, A. AQUA-DUCT: A ligands tracking tool. Bioinformatics 2017, 33, 2045–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjørnelund, H.D.; Madsen, J.J.; Peters, G.H.J. Water-Intake and Water-Molecule Paths to the Active Site of Secretory Phospholipase A2 Studied Using MD Simulations and the Tracking Tool AQUA-DUCT. J. Phys. Chem. B 2020, 124, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Olsen, O.H.; Nielsen, P.F.; Persson, E. Prevention of beta strand movement into a zymogen-like position does not confer higher activity to coagulation factor VIIa. Biochemistry 2004, 43, 14096–14103. [Google Scholar] [CrossRef]
- Perera, L.; Pedersen, L.G. A reconsideration of the evidence for structural reorganization in FVII zymogen. J. Thromb. Haemost. 2005, 3, 1543–1545. [Google Scholar] [CrossRef]
- Jing, H.; Macon, K.J.; Moore, D.; DeLucas, L.J.; Volanakis, J.E.; Narayana, S.V.L. Structural basis of profactor D activation: From a highly flexible zymogen to a novel self-inhibited serine protease, complement factor D. EMBO J. 1999, 18, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Papagrigoriou, E.; McEwan, P.A.; Walsh, P.N.; Emsley, J. Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat. Struct. Mol. Biol. 2006, 13, 557–558. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | PDB ID | Simulation Protocol | Simulation Time (μs) |
|---|---|---|---|---|
| FVIIa | Human FVIIa SP domain. | 1dan_H [25] | T-REMD | 2 |
| FVIIa-desIVG | Human FVIIa SP domain. In silico zymogenized. | 1dan_H [25] | T-REMD | 2 |
| FVIIaVYT | Human FVIIaVYT SP domain, a TF-independent variant of FVIIa. | 6r2w_H [39] | T-REMD | 1 |
| FVIIaVYT-desIVG | Human FVIIaVYT SP domain, a TF-independent variant of FVIIa. In silico zymogenized. | 6r2w_H [39] | T-REMD | 1 |
| Trypsin | Bovine trypsin. | 1j8a [54] | T-REMD | 1 |
| Trypsin-desIVG | Bovine trypsin. In silico zymogenized. | 1j8a [54] | T-REMD | 1 |
| Trypsinogen | Bovine trypsinogen. | 2tgt [55] | T-REMD | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madsen, J.J.; Olsen, O.H. Conformational Plasticity-Rigidity Axis of the Coagulation Factor VII Zymogen Elucidated by Atomistic Simulations of the N-Terminally Truncated Factor VIIa Protease Domain. Biomolecules 2021, 11, 549. https://doi.org/10.3390/biom11040549
Madsen JJ, Olsen OH. Conformational Plasticity-Rigidity Axis of the Coagulation Factor VII Zymogen Elucidated by Atomistic Simulations of the N-Terminally Truncated Factor VIIa Protease Domain. Biomolecules. 2021; 11(4):549. https://doi.org/10.3390/biom11040549
Chicago/Turabian StyleMadsen, Jesper J., and Ole H. Olsen. 2021. "Conformational Plasticity-Rigidity Axis of the Coagulation Factor VII Zymogen Elucidated by Atomistic Simulations of the N-Terminally Truncated Factor VIIa Protease Domain" Biomolecules 11, no. 4: 549. https://doi.org/10.3390/biom11040549