
Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus


1
Immunology Laboratory, Virology Section, Evandro Chagas Institute, Ananindeua, Pará 67030-000, Brazil
2
Postgraduate Program in Virology (PPGV), Evandro Chagas Institute, Ananindeua, Pará 67030-000, Brazil
3
Virology Laboratory, Institute of Biological Sciences, Federal University of Pará, Belém 66075-110, Brazil
*
Author to whom correspondence should be addressed.
Biomolecules 2021, 11(12), 1792; https://doi.org/10.3390/biom11121792
Submission received: 15 October 2021
/
Revised: 21 November 2021
/
Accepted: 23 November 2021
/
Published: 30 November 2021
(This article belongs to the Collection Feature Papers in Enzymology)
Abstract
:Epstein–Barr virus (EBV) is an oncovirus ubiquitously distributed and associated with different types of cancer. The reason why only a group of infected people develop cancer is still unknown. EBV-associated cancers represent about 1.8% of all cancer deaths worldwide, with more than 150,000 new cases of cancer being reported annually. Since EBV-associated cancers are described as more aggressive and more resistant to the usual treatment compared to EBV-negative ones, the recent introduction of monoclonal antibodies (mAbs) targeting immune checkpoints (ICs) in the treatment of cancer patients represents a possible therapy for EBV-associated diseases. However, the current mAb therapies available still need improvement, since a group of patients do not respond well to treatment. Therefore, the main objective of this review is to summarize the progress made regarding the contribution of EBV infection to the expression of the IC indoleamine 2,3-dioxygenase (IDO) thus far. This IC has the potential to be used as a target in new immune therapies, such as mAbs. We hope that this work helps the development of future immunotherapies, improving the prognosis of EBV-associated cancer patients.
1. Introduction
Epstein–Barr (EBV) is the first human oncovirus ever described and known to transform primary B cells in vitro [1,2]. Although it infects approximately 95% of the population worldwide [3], for reasons that are not completely understood, not all of the infected people develop cancer [4]. EBV has a significant health impact since EBV-associated cancers represent about 1.8% of all cancer deaths worldwide, with more than 150,000 new cases of cancer being reported annually [5,6]. Moreover, EBV-associated cancers are usually described as more aggressive and more resistant to the usual treatments [7,8,9,10,11,12]. It is clear that new approaches are necessary in order to improve the prognosis of these patients.
The recent introduction of monoclonal antibodies (mAbs) targeting immune checkpoints (ICs) as a treatment for cancer patients represents a possible therapy for EBV-associated diseases. However, this mAb therapy still needs improvement, since a group of patients showed serious adverse effects and/or did not respond to the actual mAb therapy [13,14,15,16,17,18]. Therefore, our review aims to summarize the progress made regarding the contribution of EBV infection to the expression of the IC indoleamine 2,3-dioxygenase (IDO) so far. In order for a complete understanding of how immunotherapy is a promising treatment for EBV-associated cancers, we also summarize (a) EBV infection, (b) IDO expression in EBV associated-diseases, (c) EBV regulation of IDO, and (d) concomitantly, of an important immune therapy axis, programmed cell death 1 (PD-1)/ programmed cell death 1 ligand 1 (PD-L1).
2. EBV Infection and Its Associated Diseases
2.1. EBV Virus
EBV is a double-stranded DNA Gammaherpesvirus composed of a nucleoid, capsid, membrane, and tegument [4,19], with a genome of 172 kb that encodes about 85 genes [19,20]. The virus has been described to infect B lymphocytes, epithelial, and NK cells. The initial steps during infection are not well described in the last two cell types. In B lymphocytes, upon infection, EBV gp350 interacts with complement C3d receptor 2 (CR2), located on the surface of B cells, followed by EBV gp42 binding with major histocompatibility complex (MHC) class II molecules, culminating in the fusion of the virus and cell membrane [9,21,22,23,24]. In CR2-negative epithelial cells, EBV may utilize viral proteins BMRF-2 or gH/gL to attach integrins to the cell surface. Ephrin receptor 2 has been identified as the entry receptor that interacts with viral gH/gL. Interestingly, EBV gp350 and gp42 are non-essential for epithelial cell infection [22,24,25,26].
2.2. EBV Infection and EBV-Associated Tumors
Infection by this oncovirus has been reported to be associated with several types of cancers, such as lymphomas and carcinomas [20,28,29]. Most EBV-associated cancers are lymphomas of B cell origin and carcinomas derived from epithelial cells. Moreover, a group of T/NK cell origin lymphomas is also considered EBV-associated cancer [29]. The closest association between EBV and cancers is seen in undifferentiated nasopharyngeal carcinoma (NPC) and T/NK lymphomas that are 100% EBV positive [29]. It is interesting that each one of the EBV-associated diseases presents a particular profile of viral latency, in which the expression of a set of viral proteins and RNAs is expressed, and other sets are silenced [20,28], demonstrating that EBV expression is elegantly controlled (Table 1).
Some of the EBV-associated cancers are aggressive types, showing resistance to the usual treatments [8,10,11,12]. In fact, EBV-associated cancers are associated with the patient’s immune status [30,31]. For instance, certain EBV-associated malignancies, such as PTLDs, tend to develop in immunocompromised patients [20,28]. In this case, EBV-infected cells show latency III status, which is characterized by the expression of the full spectrum of EBV proteins, which is opposite of what is observed in other EBV-associated cancers that present more restricted forms of latency (latencies II, III, and 0) and are often developed in the absence of such immunosuppression [29]. These factors indicate that the immune response is crucial to EBV-associated cancers.
Multiple strategies of immune escape by this virus have been reported [32]. Most recently, growing evidence shows that EBV proteins have the potential to upregulate ICs [33,34,35], which are important regulators of immune homeostasis and, therefore, can prevent pathologies [36]. Through these mechanisms, but not solely by them, EBV infection could influence the setting of an immune suppressive profile, contributing to cancer development and aggressiveness.
3. Targeting ICs
ICs are essential in controlling the immune response. They prevent ligand–receptor engagement, avoiding diseases such as autoimmune ones. Its applicability in the treatment of malignancies such as cancer has greatly improved the survival of patients and has been revolutionizing the field of cancer treatment [14].
The use of mAbs against, at first, Cytotoxic T-lymphocyte protein 4 (CTLA-4) was associated with several adverse effects. Later on, the outcome of patients improved with the emergence of mAbs targeting the axis of PD-1/PD-L1. Currently, there are seven different IC inhibitors that have been approved for the treatment of patients as first- and second-line therapies [13,14,37]; however, there is a subset of individuals that still do not respond to this therapy by presenting de novo and/or adaptive responses [14,15,16,17,18,38]. For instance, treatment targeting anti-PD-1/PD-L1 therapy in certain types of cancers with microsatellite stability shows poor efficiency [39]. Moreover, the recently approved first-line treatment for metastatic non-small cell lung cancer expressing PD-L1 (≥1%) presented a median overall survival of 17.1 months and overall survival rates of 40%. The median duration of response was 23.2 and 6.2 months in chemotherapy patients [40,41]. In melanoma, the combination therapy group which received anti-PD-1 and anti-CTLA-4 mAbs presented a 3-year overall survival rate of 58%. In monoclonal therapy cohorts, rates of 52% and 38% were observed in anti-PD-1 and anti-CTLA-4 groups, respectively [42]. Although these therapies are extremely important and have shaped the treatment of cancer patients in an unprecedented way, the above results indicate the need for the improvement of current mAbs therapies [13] and/or the discovery of other ICs that could be used as targets for immune therapies [36,43].
EBV-associated malignancies have the potential to be specifically treated with IC targeted immune therapy. In fact, several EBV-associated malignancies present higher expression of ICs such as IDO [44] and PD-L1 [45,46,47] when compared to EBV(−) tumor samples. Additionally, in vitro and in vivo reports show a better response to IC blockades in EBV(+) cases [48,49,50,51].
EBV(+) refractory or relapsed non-Hodgkin lymphoma (NHL) patients responded to PD-1 blockade, as opposed to EBV(−) NHL patients [49], and antitumor activity was more evident after PD-1 blockade in EBV(+) DLBCL than in EBV(−) DLBCL cells [50]. A more detailed approach of how EBV is able to regulate the IC PD-L1 is reviewed elsewhere [52].
A group of tumors has the T cell-inflamed tumor phenotype, which is characterized by a large number of tumor-infiltrating lymphocytes (TILs) and interferon (IFN)-transcriptional profiles, including the expression of PD-L1 [53,54], and a high mutation burden responding to checkpoint immunotherapy [55]. On the other hand, the so-called “cold” tumors, because they lack this immune setting, do not respond well to IC-targeting therapy [56]. This possibly indicates that other immunosuppressive mechanisms contribute to the immune-mediated tumor escape and, consequently, tumor regression besides the PD-1/PD-L1 axis. Studies have identified immunosuppressive mechanisms other than the ones directly associated with the PD-1/PD-L1 axis that are present in the T cell–inflamed tumors. These tumors are characterized, for instance, by the presence of Foxp3+Tregs and the tryptophan–kynurenine–aryl hydrocarbon (Trp–Kyn–AhR) pathway [57,58].
4. IDO
4.1. Trp–Kyn–AhR Pathway
The Trp–Kyn–AhR pathway converts the essential amino acid tryptophan into kynurenine (Kyn) and other secondary metabolites [59]. It is the primary route of tryptophan (Trp) catabolism [60,61]. T cells are extremely sensitive to the local depletion of tryptophan; as kyn binds to AhR, causing T cell differentiation into Tregs, reducing antigen presentation by APCs, and increasing IL-10 production [58,59,62,63,64,65].
IDO-1, by diminishing the availability of Trp and increasing the accumulation of metabolites, also transduces its signaling through the mammalian target of apamycin (mTORC) and general control non-depressible 2 (GNC2). This leads to cell-cycle arrest and/or apoptosis through GCN2 phosphorylation and the inhibition of eukaryotic initiation factor 2α kinase, contributing to the immunosuppressive effects [58,66,67].
IDO-1 is a rate-limiting enzyme that catalyzes tryptophan into Kyn. Later on, it was discovered that IDO-2 is an isoform of IDO-1 and is also able to catalyze Trp; however, functional differences between them have been reported [59,68,69,70]. For instance, it appears that the catalytic activity of IDO-2 is much lower than IDO-1, and IDO2 expression varies according to cancer type [68,71]. Actually, most of the literature does not differentiate between IDO1 and IDO2 [72], and studies are mostly focused on IDO1, so these enzymes will be referred to collectively as IDO unless otherwise specified.
IDO1 is not normally expressed under physiological conditions in human cells, but it is expressed in subsets of antigen-presenting cells (APCs), endothelial, and tumor cells [57,66]. IDO expression can be modulated by other ICs, such as PD-1, PD-L1, and CTLA-4 [73,74,75,76], and cytokines, such as IFN-γ and transforming growth factor-beta (TGF-β), pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and prostaglandin E2 (PGE2) [63,77,78,79,80,81].
Overall, high levels of IDO have been described as a poor prognosis in cancer patients [82,83,84,85,86]. A recent analysis of the Cancer Genome Atlas (TCGA) database indicated that T cell infiltration was more associated with IDO1 than PD-L1, and EBV-associated cancers were correlated with IDO1. Other than that, EBV(+) gastric cancer was also associated with the over-expression of IDO2 [87]. To clarify, among the EBV-associated cancers that were studied, only EBV(+) GC was associated with both IDO2 and IDO1. Overall, EBV-associated cancers usually are associated with the enhanced expression of IDO1 [87]. Either way, this ratifies the fact that EBV(+) gastric cancer is a good candidate for immune therapy treatment, as it has been described as highly immunogenetic [47,88,89]. Interestingly, EBV was not associated with tryptophan-2,3-dioxygenase (TDO) gene expression [87]. TDO encodes an enzyme that is also able to catabolize Trp [90]. Altogether, literature shows that IDO1 might play a key role in EBV-associated cancers.
4.2. IDO Expression in EBV-Associated Cancers
IDO is found to be upregulated in several cancer types [66,72,87]. Regarding EBV infection, IDO expression may be induced by EBV in a tumor microenvironment. Cells from the NPC and HL milieu have been described to present stronger IDO expression when compared to tumor cells [33,44]. Importantly, IDO expression was found to be upregulated in EBV-associated HL, particularly in the mixed cellularity subtype [44] and also in EBV(+) oral squamous cell carcinoma when compared to EBV(−) samples [91]. Interestingly, no difference was found regarding IDO expression when the plasmablastic lymphoma (PL) microenvironment from EBV(+) and EBV(−) were compared [45]. This indicates that IDO expression may be correlated with EBV-associated cancers presenting latency II and/or III, since PL is the only cancer of the above-mentioned ones that presents latency I. A summary of the detection of IDO expression in EBV-associated malignancies is in Table 2.
4.3. Regulation of IDO Expression by EBV
Reports have shown that EBV infection induces IDO expression. In fact, in vitro infection of primary B cells induced IDO expression [35]. In monocyte-derived macrophages (MDMs), activation of the retinoic acid-inducible gene I (RIG-I) pathway by viral EBER1 delivered from exosomes derived from EBV-infected cells culminated in IDO upregulation through IL-6 and TNF-α (Figure 1). These IDO+ cells suppressed the proliferation and cytolytic activity of T cells [91]. Interestingly, EBV infection, through p38 MAPK and NF-kB pathways, increased the production of these cytokines from MDMs, which also resulted in IDO expression [33]. IDO induction did not require IFN-γ, previously known as the main IDO inducer, although it required a synergism between two inflammatory cytokines [33]. This implies that the pathway RIG-I/IL-6/TNF-α/IDO may cross-talk with the MAPK and NF-kB pathways at some point during EBV infection, contributing to IDO expression.
EBER1 is a non-polyadenylated, untranslated RNA, 167 bp in length and is one of the viral RNAs that has been reported to interact with RIG-I. EBERs are known to induce type I interferon production through NF-kB and Interferon regulatory factor 3 (IRF3) [98]. It is interesting to note that the increased expression of RIG-I is observed in EBV(+) cells of cHL of the elderly; however, no differences in the level of IRF3 and IFN-β, two of RIG-I known targets, were detected when compared to EBV(−) samples [99]. This indicates that the recognition of EBER by RIG-I/IL-6/TNF-a is important for IDO expression and, therefore, contributes to an immunosuppressive setting; at the same time, the usual downstream pathway of RIG-I, particularly the IFN production, which is known as a potent antiviral, is affected by several other viral proteins and RNAs at some point during infection. In fact, several reports show the IFN response being hampered directly by EBV [98,100,101].
RIG-I is a cytosolic pathogen-recognition receptor (PRR) that initiates a type-1 interferon (IFN1) response [102,103]. It recognizes short viral double-stranded RNA (dsRNA) and other non-coding RNAs, such as viral RNA and replication transcripts, although it remains difficult to predict exactly which type of motifs and sequences trigger the RIG-I response. Overall, RIG-I has been linked to a response to West Nile virus, Sendai virus, and coronaviruses [102].
EBV, by inducing IDO expression in the infected cell, is able to regulate a series of events in the microenvironment. For instance, EBV-infected MDMexpressingIDO has been shown to impair T cell proliferation and CD8+T cytotoxic activity [33]; further, incubation with L-kynurenine, an IDO metabolite, suppresses the JNK pathway in NK cells, reducing NKG2-D type II integral membrane protein and (NKG2D) type II integral membrane protein expression [35]. This activating receptor has been reported to eliminate target cells through granule-dependent cytotoxicity [104]. It also has been implicated in modulating immune responses in important events, such as cancer, viral infections, and chronic inflammatory diseases [105,106]. Interestingly, the EBV-genome-encoded miR-BART7 has been implicated in inhibiting the expression of MHC class I polypeptide-related sequence A, a ligand of NKG2D, partially through the TGF-β/c-myc pathway in NPC cells [106]. Therefore, by upregulating IDO and, consequently, impairing T- and NK-mediated killing, an EBV infection is able to induce a suppressive microenvironment by affecting both innate and adaptive immune responses.
4.4. IDO and PD-L1 Regulation
Several reports have pointed to the activation of PD-1/PD-L1 and IDO pathways in cancer samples. In EBV(+) large B-cell lymphoma (LBCL), PD-L1-positive tumors express high levels of IDO [94]. In undifferentiated NPC, significantly increased gene expression of IDO and PD-L1 was observed when compared to normal tissues [96]. Interestingly, it was shown that IFN-γ regulates the expression of both PD-L1, IDO1, and IDO2 through AhR in murine oral cancer cells [107]. In addition, IFN-γ produced by CD8+T cells induces the production of Kyn by tumor cells, which induces and activates AhR and upregulates PD-1 expression in CD8+T cells [75]. PD-1 is the known receptor of PD-L1 [108].
It seems that IL-27 induces the expression of both IDO and PD-L1 in human cancer cells [109]. Specifically, in cancer cells, IDO1 upregulation is mostly dependent on STAT1, and IL-27 regulation of PD-L1 occurs through signal transducer and activator of transcription 3 (STAT3) [109]. IL-27 is a heterodimer consisting of p28 (IL-27A) and EBV-induced gene 3 (EBI3) chains. EBI3 was found to be upregulated in several B-cell lymphomas and can be regulated by viral LMP1 through NF-κB [110,111]. Indeed, EBI3 has been associated with tumor metastasis and cancer progression [35,112]. In lymphoma cells, EBI3 is expressed without detectable p28 [113,114]. Surrounding macrophages are the probable source of IL-27p28, resulting in the possible upregulation of PD-L1 and PD-L2. It is interesting that the IL-27 heterodimer was able to induce PD-L1/2 expression more efficiently than EBI3 alone [113], meaning that PD-L1/2 expression may be further increased by interaction of EBI3 and macrophage-derived p28.
5. Targeting IDO in EBV-Associated Malignancies
Targeting ICs has revolutionized cancer treatment. Although the current available IC-targeting therapies have greatly improved the prognosis of patients [13,115], a significant group of recipients still face serious adverse reactions and/or do not respond to the therapy [116,117,118,119,120,121]; additionally, even the patients who initially respond to mAb therapies may eventually develop a progressive disease with clinical complications [122]. Even though the initial results of preclinical and phase I/II clinical trials with the inhibition of IDO showed promising results [123,124,125,126,127,128,129,130], IDO inhibition during phase III clinical trials has not shown a similar outcome [131]. This may have occurred because of the drug of choice, among other factors that might have influenced the results [132,133]; regardless, new effective treatments with less toxicity are still strongly needed. In this scenario, the IDO and PD-1/PD-L1 cross-talk represents an opportunity for the development of new therapies. Indeed, delivery of siRNAs targeting IDO-1 in mouse DCs has improved the immune response by reducing the number of Treg cells [132]. Other than that, the recent report of a phase I/II clinical trial targeting both of these pathways in melanoma patients is on course through an innovative drug that modulates both PD-L1/PD-1 and IDO pathways and has shown exciting results [134].
In the future, it would also be important to determine during which latency and/or lytic replication programs these phenomena occur, facilitating the development of new target antibodies that could be associated with other current, or soon to be developed, immunotherapies, such as EBV-targeted vaccines. Describing the molecular pathways induced by viral proteins would also be beneficial since they could guide and/or help in the development of new treatments.
In summary, this review showed that the regulation of IDO expression is one of the many mechanisms of immune escape driven by the virus that has the potential of significantly impacting the prognosis of EBV-associated cancer patients. We hope that this review can help with the development and improvement of therapies.
Author Contributions
Conceptualization, L.S.; writing—original draft preparation, L.S.; writing—review and editing, L.S., A.C.R.V. and I.B.-C.; supervision, I.B.-C.; funding acquisition, I.B.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Evandro Chagas Institute.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and Biological Studies on a Virus in Cultured Lymphoblasts from Burkitt’s Lymphoma. J. Exp. Med. 1965, 121, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Mentzer, A.J.; Brenner, N.; Allen, N.; Littlejohns, T.J.; Chong, A.Y.; Cortes, A.; Almond, R.; Hill, M.; Sheard, S.; McVean, G.; et al. Identification of host-pathogen-disease relationships using a scalable Multiplex Serology platform in UK Biobank. medRxiv 2019, 19004960. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent. Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourbon, E.; Maucort-Boulch, D.; Fontaine, J.; Mauduit, C.; Sesques, P.; Safar, V.; Ferrant, E.; Golfier, C.; Ghergus, D.; Karlin, L.; et al. Clinicopathological features and survival in EBV-positive diffuse large B-cell lymphoma not otherwise specified. Blood Adv. 2021, 5, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.P.; Shannon-Lowe, C.; Rowe, M. Deciphering the role of Epstein-Barr virus in the pathogenesis of T and NK cell lymphoproliferations. Herpesviridae 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Lee, J.; Ko, Y.H.; Han, A.; Jun, H.J.; Lee, S.C.; Hwang, I.G.; Park, Y.H.; Ahn, J.S.; Jung, C.W.; et al. The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood 2007, 110, 972–978. [Google Scholar] [CrossRef]
- Kim, Y.R.; Kim, S.J.; Cheong, J.W.; Chung, H.; Jang, J.E.; Kim, Y.; Yang, W.I.; Min, Y.H.; Kim, J.S. Pretreatment Epstein-Barr virus DNA in whole blood is a prognostic marker in peripheral T-cell lymphoma. Oncotarget 2017, 8, 92312–92323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, J.; Emile, J.F.; Mounier, N.; Gisselbrecht, C.; Martin-Garcia, N.; Petrella, T.; Bouabdallah, R.; Berger, F.; Delmer, A.; Coiffier, B.; et al. Prognostic significance of Epstein-Barr virus in nodal peripheral T-cell lymphoma, unspecified: A Groupe d’Etude des Lymphomes de l’Adulte (GELA) study. Blood 2006, 108, 4163–4169. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.J.; Ng, J.C.K.; Huang, L.; Robinson, M.; O’Hara, J.; Wilson, J.A.; Mellor, A.L. The immunotherapeutic role of indoleamine 2,3-dioxygenase in head and neck squamous cell carcinoma: A systematic review. Clin. Otolaryngol. 2021, 46, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Garcia, E.; Lee, E.K.; Lindemann, N.I.; Liu, J.F.; Matulonis, U.A.; Park, P.J.; Konstantinopoulos, P.A. Genomic Determinants of De Novo Resistance to Immune Checkpoint Blockade in Mismatch Repair-Deficient Endometrial Cancer. JCO Precis. Oncol. 2020, 4, 492–497. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Nguyen, A.; Bakhshinyan, D.; Wei, J.; Hare, D.N.; MacNeill, K.L.; Wan, Y.; Oberst, A.; Bramson, J.L.; Nasir, J.A.; et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun. Biol. 2020, 3, 645. [Google Scholar] [CrossRef]
- Renaud, S.; Lefebvre, A.; Mordon, S.; Morales, O.; Delhem, N. Novel Therapies Boosting T Cell Immunity in Epstein Barr Virus-Associated Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2020, 21, 4292. [Google Scholar] [CrossRef]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Matsuura, H.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. USA 2010, 107, 22641–22646. [Google Scholar] [CrossRef] [Green Version]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Borza, C.M.; Morgan, A.J.; Turk, S.M.; Hutt-Fletcher, L.M. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 2004, 78, 5007–5014. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. EBV Persistence--Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Myburgh, R.; Caduff, N.; Spohn, M.; Kok, Y.L.; Keller, C.W.; Murer, A.; Chatterjee, B.; Ruhl, J.; Engelmann, C.; et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci. Alliance 2020, 3, e202000640. [Google Scholar] [CrossRef]
- Papadopoulos, E.B.; Ladanyi, M.; Emanuel, D.; Mackinnon, S.; Boulad, F.; Carabasi, M.H.; Castro-Malaspina, H.; Childs, B.H.; Gillio, A.P.; Small, T.N.; et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N. Engl. J. Med. 1994, 330, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Munz, C.; Moormann, A. Immune escape by Epstein-Barr virus associated malignancies. Semin. Cancer Biol. 2008, 18, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.L.; Lin, Y.H.; Xiao, H.; Xing, S.; Chen, H.; Chi, P.D.; Zhang, G. Epstein-Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-kappaB pathways: Impairment in T cell functions. J. Virol. 2014, 88, 6660–6671. [Google Scholar] [CrossRef] [Green Version]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Park, H.; Kim, J.; Park, G.; Kim, Y.S.; Kim, S.M.; Kim, D.; Seo, S.K.; Lee, H.K.; Cho, D.; et al. IDO metabolite produced by EBV-transformed B cells inhibits surface expression of NKG2D in NK cells via the c-Jun N-terminal kinase (JNK) pathway. Immunol. Lett. 2011, 136, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F.; et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Reck, M.; Ciuleanu, T.E.; Lee, J.S.; Schenker, M.; Audigier-Valette, C.; Zurawski, B.; Linardou, H.; Otterson, G.A.; Salman, P.; Nishio, M.; et al. First-Line Nivolumab Plus Ipilimumab Versus Chemotherapy in Advanced NSCLC with 1% or Greater Tumor PD-L1 Expression: Patient-Reported Outcomes from CheckMate 227 Part 1. J. Thorac. Oncol. 2021, 16, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Choe, J.Y.; Yun, J.Y.; Jeon, Y.K.; Kim, S.H.; Park, G.; Huh, J.R.; Oh, S.; Kim, J.E. Indoleamine 2,3-dioxygenase (IDO) is frequently expressed in stromal cells of Hodgkin lymphoma and is associated with adverse clinical features: A retrospective cohort study. BMC Cancer 2014, 14, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, C.; Fabiani, B.; Do, C.; Tchernonog, E.; Cartron, G.; Gravelle, P.; Amara, N.; Malot, S.; Palisoc, M.M.; Copie-Bergman, C.; et al. Immune-checkpoint expression in Epstein-Barr virus positive and negative plasmablastic lymphoma: A clinical and pathological study in 82 patients. Haematologica 2016, 101, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.; Patel, S.P.; Kurzrock, R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol. 2017, 14, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of Efficacy of Pembrolizumab between Epstein-Barr VirusPositive and Negative Relapsed or Refractory Non-Hodgkin Lymphomas. Cancer Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef]
- Panda, A.; Mehnert, J.M.; Hirshfield, K.M.; Riedlinger, G.; Damare, S.; Saunders, T.; Kane, M.; Sokol, L.; Stein, M.N.; Poplin, E.; et al. Immune Activation and Benefit from Avelumab in EBV-Positive Gastric Cancer. J. Natl. Cancer Inst. 2018, 110, 316–320. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W. Expression of PD-L1 in EBV-associated malignancies. Int. Immunopharmacol. 2021, 95, 107553. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Padget, M.R.; Fujii, R.; Schlom, J.; Hodge, J.W. Differential combination immunotherapy requirements for inflamed (warm) tumors versus T cell excluded (cool) tumors: Engage, expand, enable, and evolve. J. Immunother. Cancer 2021, 9, e001691. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A. Tryptophan metabolism in alcoholism. Nutr. Res. Rev. 2002, 15, 123–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Indoleamine 2,3-dioxygenase depletes tryptophan, activates general control non-derepressible 2 kinase and down-regulates key enzymes involved in fatty acid synthesis in primary human CD4+ T cells. Immunology 2015, 146, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Mellor, A.L.; Chandler, P.; Baban, B.; Hansen, A.M.; Marshall, B.; Pihkala, J.; Waldmann, H.; Cobbold, S.; Adams, E.; Munn, D.H. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int. Immunol. 2004, 16, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Reznik, E.; Luna, A.; Aksoy, B.A.; Liu, E.M.; La, K.; Ostrovnaya, I.; Creighton, C.J.; Hakimi, A.A.; Sander, C. A Landscape of Metabolic Variation across Tumor Types. Cell Syst. 2018, 6, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Ball, H.J.; Sanchez-Perez, A.; Weiser, S.; Austin, C.J.; Astelbauer, F.; Miu, J.; McQuillan, J.A.; Stocker, R.; Jermiin, L.S.; Hunt, N.H. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 2007, 396, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Smith, C.; DuHadaway, J.B.; Chandler, P.; Baban, B.; Merlo, L.M.; Pigott, E.; Keough, M.P.; Rust, S.; Mellor, A.L.; et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int. Immunol. 2014, 26, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lob, S.; Konigsrainer, A.; Zieker, D.; Brucher, B.L.; Rammensee, H.G.; Opelz, G.; Terness, P. IDO1 and IDO2 are expressed in human tumors: Levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 2009, 58, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H. Blocking IDO activity to enhance anti-tumor immunity. Front. Biosci. (Elite Ed.) 2012, 4, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e487. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.P.; Ferreira, A.C.; Pinho, M.P.; de Moraes, C.J.; Bergami-Santos, P.C.; Barbuto, J.A. Tolerogenic IDO(+) Dendritic Cells Are Induced by PD-1-Expressing Mast Cells. Front. Immunol. 2016, 7, 9. [Google Scholar] [CrossRef]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef]
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; Plaen, E.; van den Eynde, B.J. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol. Res. 2017, 5, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; Sharma, M.D.; Chandler, P.R.; Duhadaway, J.B.; Everhart, M.E.; Johnson, B.A., III; Kahler, D.J.; Pihkala, J.; Soler, A.P.; Munn, D.H.; et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc. Natl. Acad. Sci. USA 2008, 105, 17073–17078. [Google Scholar] [CrossRef] [Green Version]
- Wingender, G.; Garbi, N.; Schumak, B.; Jungerkes, F.; Endl, E.; von Bubnoff, D.; Steitz, J.; Striegler, J.; Moldenhauer, G.; Tuting, T.; et al. Systemic application of CpG-rich DNA suppresses adaptive T cell immunity via induction of IDO. Eur. J. Immunol. 2006, 36, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Nouel, A.; Pochard, P.; Simon, Q.; Segalen, I.; le Meur, Y.; Pers, J.O.; Hillion, S. B-Cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. J. Autoimmun. 2015, 59, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chamuleau, M.E.D.; van de Loosdrecht, A.A.; Hess, C.J.; Janssen, J.J.W.M.; Zevenbergen, A.; Delwel, R.; Valk, P.J.M.; Lowenberg, B.; Ossenkoppele, G.J. High INDO (Indoleamine 2,3-Dioxygenase) mRNA Level in Blasts of Acute Myeloid Leukemic Patients Predicts Poor Clinical Outcome. Blood 2007, 110, 4297. [Google Scholar] [CrossRef]
- Liu, H.; Shen, Z.; Wang, Z.; Wang, X.; Zhang, H.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Increased expression of IDO associates with poor postoperative clinical outcome of patients with gastric adenocarcinoma. Sci. Rep. 2016, 6, 21319. [Google Scholar] [CrossRef] [Green Version]
- Mitra, D.; Horick, N.K.; Brackett, D.G.; Mouw, K.W.; Hornick, J.L.; Ferrone, S.; Hong, T.S.; Mamon, H.; Clark, J.W.; Parikh, A.R.; et al. High IDO1 Expression Is Associated with Poor Outcome in Patients with Anal Cancer Treated with Definitive Chemoradiotherapy. Oncologist 2019, 24, e275–e283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, J.; Shen, H.; Wang, J. The prognostic value of IDO expression in solid tumors: A systematic review and meta-analysis. BMC Cancer 2020, 20, 471. [Google Scholar] [CrossRef]
- Panda, A.; Ganesan, S. Genomic and Immunologic Correlates of Indoleamine 2,3-Dioxygenase Pathway Expression in Cancer. Front. Genet. 2021, 12, 706435. [Google Scholar] [CrossRef]
- Dai, C.; Geng, R.; Wang, C.; Wong, A.; Qing, M.; Hu, J.; Sun, Y.; Lo, A.W.; Li, J. Concordance of immune checkpoints within tumor immune contexture and their prognostic significance in gastric cancer. Mol. Oncol. 2016, 10, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Derks, S.; Liao, X.; Chiaravalli, A.M.; Xu, X.; Camargo, M.C.; Solcia, E.; Sessa, F.; Fleitas, T.; Freeman, G.J.; Rodig, S.J.; et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget 2016, 7, 32925–32932. [Google Scholar] [CrossRef]
- Ball, H.J.; Jusof, F.F.; Bakmiwewa, S.M.; Hunt, N.H.; Yuasa, H.J. Tryptophan-catabolizing enzymes—party of three. Front. Immunol. 2014, 5, 485. [Google Scholar] [CrossRef] [Green Version]
- Burassakarn, A.; Srisathaporn, S.; Pientong, C.; Wongjampa, W.; Vatanasapt, P.; Patarapadungkit, N.; Ekalaksananan, T. Exosomes-carrying Epstein-Barr virus-encoded small RNA-1 induces indoleamine 2,3-dioxygenase expression in tumor-infiltrating macrophages of oral squamous-cell carcinomas and suppresses T-cell activity by activating RIG-I/IL-6/TNF-alpha pathway. Oral Oncol. 2021, 117, 105279. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, T.I.; Son, S.A.; Lee, H.W. Immunohistochemical Features of Indoleamine 2,3-Dioxygenase (IDO) in Various Types of Lymphoma: A Single Center Experience. Diagnostics 2020, 10, 275. [Google Scholar] [CrossRef]
- Morscio, J.; Dierickx, D.; Ferreiro, J.F.; Herreman, A.; van Loo, P.; Bittoun, E.; Verhoef, G.; Matthys, P.; Cools, J.; Wlodarska, I.; et al. Gene expression profiling reveals clear differences between EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am. J. Transplant. 2013, 13, 1305–1316. [Google Scholar] [CrossRef]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.; Raffeld, M.; Jaffe, E.S. EBV-positive large B-cell lymphomas in young patients: A nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Ben-Haj-Ayed, A.; Moussa, A.; Ghedira, R.; Gabbouj, S.; Miled, S.; Bouzid, N.; Tebra-Mrad, S.; Bouaouina, N.; Chouchane, L.; Zakhama, A.; et al. Prognostic value of indoleamine 2,3-dioxygenase activity and expression in nasopharyngeal carcinoma. Immunol. Lett. 2016, 169, 23–32. [Google Scholar] [CrossRef]
- Muraro, E.; Vaccher, E.; Furlan, C.; Fratta, E.; Fanetti, G.; Fae, D.A.; Martorelli, D.; Cangemi, M.; Polesel, J.; Navarria, F.; et al. Predictive Value of CD8 Expression and FoxP3 Methylation in Nasopharyngeal Carcinoma Patients Treated with Chemoradiotherapy in a Non-endemic Area. Pathol. Oncol. Res. 2020, 26, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhang, Y.; Jiang, W.; Chen, Y.P.; Xu, S.Y.; Liu, N.; Zhao, Y.; Li, L.; Lei, Y.; Hong, X.H.; et al. Development and validation of an immune checkpoint-based signature to predict prognosis in nasopharyngeal carcinoma using computational pathology analysis. J. Immunother. Cancer 2019, 7, 298. [Google Scholar] [CrossRef]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Wada, R.; Yajima, N.; Imaizumi, T.; Yagihashi, S. Tumor microenvironment and RIG-I signaling molecules in Epstein Barr virus-positive and -negative classical Hodgkin lymphoma of the elderly. J. Clin. Exp. Hematop. 2014, 54, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus miR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Kolakofsky, D.; Kowalinski, E.; Cusack, S. A structure-based model of RIG-I activation. RNA 2012, 18, 2118–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Magri, G.; Pende, D.; Angulo, A.; Lopez-Botet, M. Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood 2010, 115, 5170–5179. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.; Chen, S.; Zhang, M.J.; Chan, J.Y.; Gao, W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef] [Green Version]
- Kenison, J.E.; Wang, Z.; Yang, K.; Snyder, M.; Quintana, F.J.; Sherr, D.H. The aryl hydrocarbon receptor suppresses immunity to oral squamous cell carcinoma through immune checkpoint regulation. Proc. Natl. Acad. Sci. USA 2021, 118, e2012692118. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280. [Google Scholar] [CrossRef] [Green Version]
- Gonin, J.; Larousserie, F.; Bastard, C.; Picquenot, J.M.; Couturier, J.; Radford-Weiss, I.; Dietrich, C.; Brousse, N.; Vacher-Lavenu, M.C.; Devergne, O. Epstein-Barr virus-induced gene 3 (EBI3): A novel diagnosis marker in Burkitt lymphoma and diffuse large B-cell lymphoma. PLoS ONE 2011, 6, e24617. [Google Scholar] [CrossRef]
- Niedobitek, G.; Pazolt, D.; Teichmann, M.; Devergne, O. Frequent expression of the Epstein-Barr virus (EBV)-induced gene, EBI3, an IL-12 p40-related cytokine, in Hodgkin and Reed-Sternberg cells. J. Pathol. 2002, 198, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Nishino, R.; Takano, A.; Oshita, H.; Ishikawa, N.; Akiyama, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; Tsuchiya, E.; Kohno, N.; et al. Identification of Epstein-Barr virus-induced gene 3 as a novel serum and tissue biomarker and a therapeutic target for lung cancer. Clin. Cancer Res. 2011, 17, 6272–6286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horlad, H.; Ma, C.; Yano, H.; Pan, C.; Ohnishi, K.; Fujiwara, Y.; Endo, S.; Kikukawa, Y.; Okuno, Y.; Matsuoka, M.; et al. An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016, 107, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Larousserie, F.; Bardel, E.; Pflanz, S.; Arnulf, B.; Lome-Maldonado, C.; Hermine, O.; Bregeaud, L.; Perennec, M.; Brousse, N.; Kastelein, R.; et al. Analysis of interleukin-27 (EBI3/p28) expression in Epstein-Barr virus- and human T-cell leukemia virus type 1-associated lymphomas: Heterogeneous expression of EBI3 subunit by tumoral cells. Am. J. Pathol. 2005, 166, 1217–1228. [Google Scholar] [CrossRef]
- Kyi, C.; Postow, M.A. Immune checkpoint inhibitor combinations in solid tumors: Opportunities and challenges. Immunotherapy 2016, 8, 821–837. [Google Scholar] [CrossRef] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Eroglu, Z.; Kim, D.W.; Wang, X.; Camacho, L.H.; Chmielowski, B.; Seja, E.; Villanueva, A.; Ruchalski, K.; Glaspy, J.A.; Kim, K.B.; et al. Long term survival with cytotoxic T lymphocyte-associated antigen 4 blockade using tremelimumab. Eur. J. Cancer 2015, 51, 2689–2697. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Carlo-Stella, C. Are EBV-related and EBV-unrelated Hodgkin lymphomas different with regard to susceptibility to checkpoint blockade? Blood 2018, 132, 17–22. [Google Scholar] [CrossRef]
- Daud, A.; Saleh, M.N.; Hu, J.; Bleeker, J.S.; Riese, M.J.; Meier, R.; Zhou, L.; Serbest, G.; Lewis, K.D. Epacadostat plus nivolumab for advanced melanoma: Updated phase 2 results of the ECHO-204 study. J. Clin. Oncol. 2018, 36, 9511. [Google Scholar] [CrossRef]
- Gangadhar, T.C.; Schneider, B.J.; Bauer, T.M.; Wasser, J.S.; Spira, A.I.; Patel, S.P.; Balmanoukian, A.S.; Bauml, J.; Schmidt, E.V.; Zhao, Y.; et al. Efficacy and safety of epacadostat plus pembrolizumab treatment of NSCLC: Preliminary phase I/II results of ECHO-202/KEYNOTE-037. J. Clin. Oncol. 2017, 35, 9014. [Google Scholar] [CrossRef]
- Gibney, G.; Hamid, O.; Lutzky, J.; Olszanski, A.; Gangadhar, T.; Gajewski, T.; Chmielowski, B.; Hanks, B.; Boasberg, P.; Zhao, Y.; et al. 511 Updated results from a phase 1/2 study of epacadostat (INCB024360) in combination with ipilimumab in patients with metastatic melanoma. Eur. J. Cancer 2015, 51, S106–S107. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients with Advanced Solid Tumors: Phase I Results from a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Larkin, J.; Ascierto, P.A.; Long, G.V.; Hassel, J.C.; Schadendorf, D.; Hodi, F.S.; Lebbé, C.; Grob, J.J.; Grossmann, K.; et al. Characterization of complete responses (CRs) in patients with advanced melanoma (MEL) who received the combination of nivolumab (NIVO) and ipilimumab (IPI), NIVO or IPI alone. Ann. Oncol. 2017, 28, v428. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Zakharia, Y.; Rixe, O.; Ward, J.H.; Drabick, J.J.; Shaheen, M.F.; Milhem, M.M.; Munn, D.; Kennedy, E.P.; Vahanian, N.N.; Link, C.J.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 2018, 36, 9512. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Endo, R.; Nakamura, T.; Kawakami, K.; Sato, Y.; Harashima, H. The silencing of indoleamine 2,3-dioxygenase 1 (IDO1) in dendritic cells by siRNA-loaded lipid nanoparticles enhances cell-based cancer immunotherapy. Sci. Rep. 2019, 9, 11335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. A new cancer immunotherapy suffers a setback. Science 2018, 360, 588. [Google Scholar] [CrossRef] [PubMed]
- Svane, I.M.; Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Andersen, M.H. LBA48 Clinical efficacy and immunity of combination therapy with nivolumab and IDO/PD-L1 peptide vaccine in patients with metastatic melanoma: A phase I/II trial. Ann. Oncol. 2020, 31, S1176. [Google Scholar] [CrossRef]
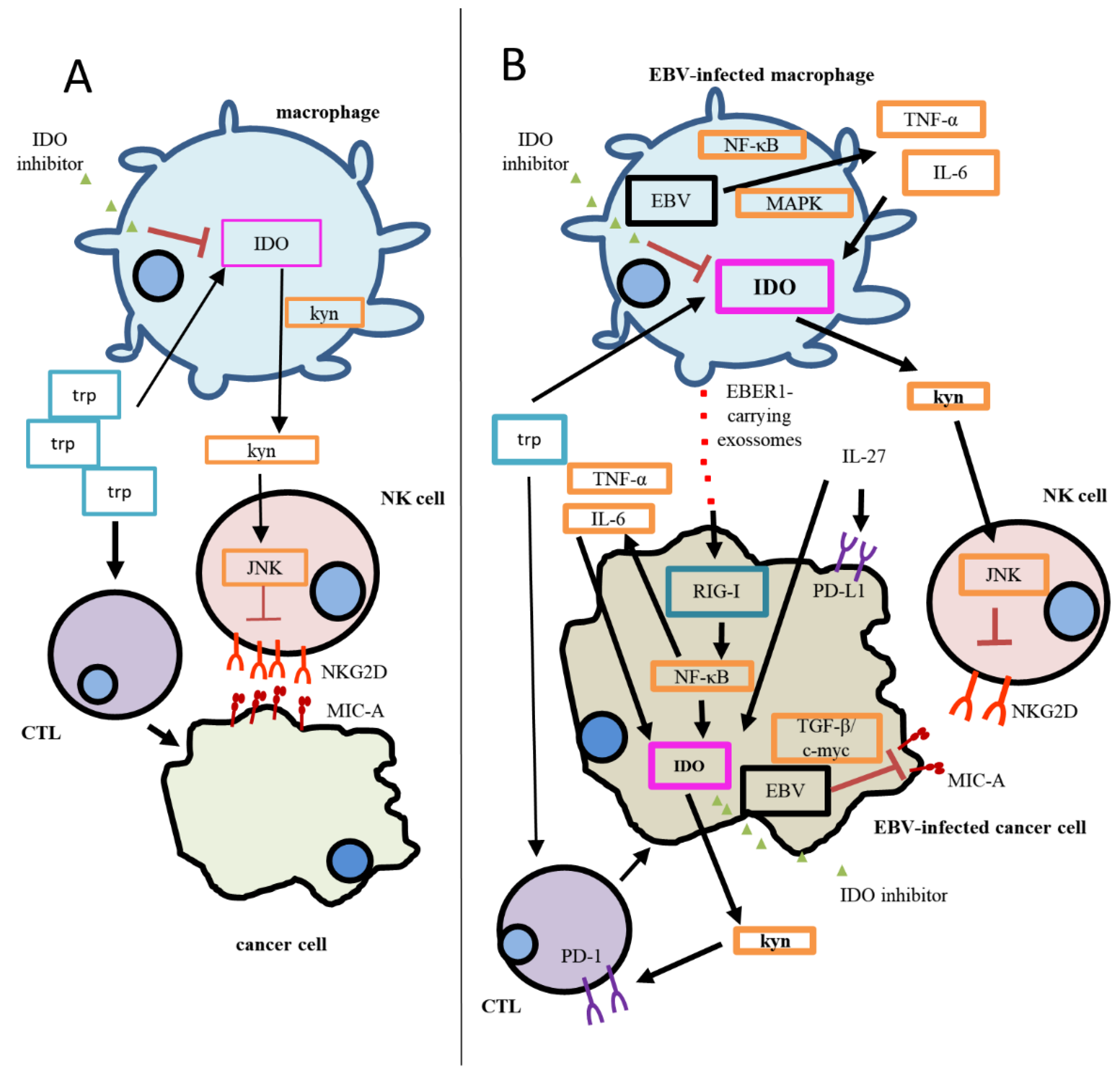
Figure 1.
Proposed mechanism of the IDO-specific contribution to immunosuppression in EBV(−) cancer and EBV(+) cancer. (A) During homeostasis, CTLs utilize tryptophan in order to eliminate cancer cells. The degradation of tryptophan generates Kyn, which negatively contributes to NKG2D regulation of expression in NK cells. (B) EBV has been reported to increase IDO expression in infected cells; therefore, Trp degradation is enhanced, and so is Kyn accumulation. As a result, a more significant immunosuppression status is established, since CTL responses are inhibited, and stronger inhibition of NKG2D in NK cells can be observed. In addition, MIC-A, a NKG2D ligand, has been reported to be downregulated in EBV-infected cancer cells through the TGF-β/c-myc pathway. In this model, the activation of the RIG-I pathway through EBER1, delivered by exosomes, directly regulates IDO expression in cancer cells and/or also activates the NF-kB pathway in them, inducing the production of TNF-α and IL-6, culminating in the upregulation of IDO. The increased IDO expression in cancer cells would exacerbate the immunosuppression by increasing Trp utilization and Kyn accumulation. IL-27 would also play a role in this scenario, contributing to the expression of both IDO and PD-L1; the latter IC receptor expression, PD-1, increases on CD8+T cells as Kyn accumulates in the microenvironment. The use of specific IDO inhibitors would have a greater impact on EBV-associated malignancies, since IDO expression is higher when compared to EBV(−) cases. Thick lines represent stronger activation/expression than thin lines (weaker activation/expression). EBV: Epstein–Barr virus; CTL: cytotoxic T cell; JAK3/STAT3: Janus-kinase-3/signal transducer and activator of transcription-3; KYN: kynurenine; MAPK: mitogen-activated protein kinase; MIC-A: MHC class I polypeptide-related sequence A; NF-κB: nuclear factor-κB; NKG2D: NKG2-D type II integral membrane protein; PD-1: programmed cell death protein; PD-L1: programmed death-ligand 1; TGF-β/c-myc: transforming growth factor β/c-myc; TRP: tryptophan.
Figure 1.
Proposed mechanism of the IDO-specific contribution to immunosuppression in EBV(−) cancer and EBV(+) cancer. (A) During homeostasis, CTLs utilize tryptophan in order to eliminate cancer cells. The degradation of tryptophan generates Kyn, which negatively contributes to NKG2D regulation of expression in NK cells. (B) EBV has been reported to increase IDO expression in infected cells; therefore, Trp degradation is enhanced, and so is Kyn accumulation. As a result, a more significant immunosuppression status is established, since CTL responses are inhibited, and stronger inhibition of NKG2D in NK cells can be observed. In addition, MIC-A, a NKG2D ligand, has been reported to be downregulated in EBV-infected cancer cells through the TGF-β/c-myc pathway. In this model, the activation of the RIG-I pathway through EBER1, delivered by exosomes, directly regulates IDO expression in cancer cells and/or also activates the NF-kB pathway in them, inducing the production of TNF-α and IL-6, culminating in the upregulation of IDO. The increased IDO expression in cancer cells would exacerbate the immunosuppression by increasing Trp utilization and Kyn accumulation. IL-27 would also play a role in this scenario, contributing to the expression of both IDO and PD-L1; the latter IC receptor expression, PD-1, increases on CD8+T cells as Kyn accumulates in the microenvironment. The use of specific IDO inhibitors would have a greater impact on EBV-associated malignancies, since IDO expression is higher when compared to EBV(−) cases. Thick lines represent stronger activation/expression than thin lines (weaker activation/expression). EBV: Epstein–Barr virus; CTL: cytotoxic T cell; JAK3/STAT3: Janus-kinase-3/signal transducer and activator of transcription-3; KYN: kynurenine; MAPK: mitogen-activated protein kinase; MIC-A: MHC class I polypeptide-related sequence A; NF-κB: nuclear factor-κB; NKG2D: NKG2-D type II integral membrane protein; PD-1: programmed cell death protein; PD-L1: programmed death-ligand 1; TGF-β/c-myc: transforming growth factor β/c-myc; TRP: tryptophan.


{kind=link}
Table 1.
EBV-associated malignancies and viral latency programs. EBV-associated malignancies show a particular viral latency pattern.
Table 1.
EBV-associated malignancies and viral latency programs. EBV-associated malignancies show a particular viral latency pattern.
| EBV Latency Pattern | Viral Expression Pattern | EBV-Associated Malignancies |
|---|---|---|
| III | EBNA-1–6, LMP1, LMP2A-B, EBER, BART | PTLD, DLBCL |
| II | EBNA-1, LMP1, LMP2A, EBER, BART | HL, NHL, GC, ENKTCL, DLBCL, PBL and NPC |
| I | EBNA1, EBER, BART | BL, PL |
| 0 | EBER, BART | - |
Abbreviations: BART: Bam-HI A rightward transcripts; EBER: Epstein–Barrvirus-encoded RNA; EBNA: Epstein–Barr nuclear antigen; LMP: Latent membrane protein; PTLD: Post-transplant lymphoproliferative disorder; DLBCL: Diffuse large B-cell lymphoma; HL: Hodgkin lymphoma; NHL: Non-Hodgkin lymphoma; GC: Gastric cancer; ENKTCL: Extranodal NK/T-cell lymphoma; NPC: Nasopharyngeal carcinoma; BL: Burkitt lymphoma; PL: Plasmablastic lymphoma.
Table 2.
IDO expression in EBV-associated malignancies.
| EBV-Associated Malignancies | IDO Status | Reference |
|---|---|---|
| HL | Enhanced when compared to EBV(−) | [44] |
| DLBCL | Detection/enhanced when compared to EBV(−) | [92,93,94] |
| EBV(+)GC | Overexpression | [87] |
| NPC | Detection/overexpression | [33,95,96,97] |
| PL | Expression was similar to EBV(−)PL | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sawada, L.; Vallinoto, A.C.R.; Brasil-Costa, I. Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus. Biomolecules 2021, 11, 1792. https://doi.org/10.3390/biom11121792
AMA Style
Sawada L, Vallinoto ACR, Brasil-Costa I. Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus. Biomolecules. 2021; 11(12):1792. https://doi.org/10.3390/biom11121792
Chicago/Turabian StyleSawada, Leila, Antonio Carlos Rosário Vallinoto, and Igor Brasil-Costa. 2021. "Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus" Biomolecules 11, no. 12: 1792. https://doi.org/10.3390/biom11121792
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.