
Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus


Abstract
:1. Introduction
2. EBV Infection and Its Associated Diseases
2.1. EBV Virus
2.2. EBV Infection and EBV-Associated Tumors
3. Targeting ICs
4. IDO
4.1. Trp–Kyn–AhR Pathway
4.2. IDO Expression in EBV-Associated Cancers
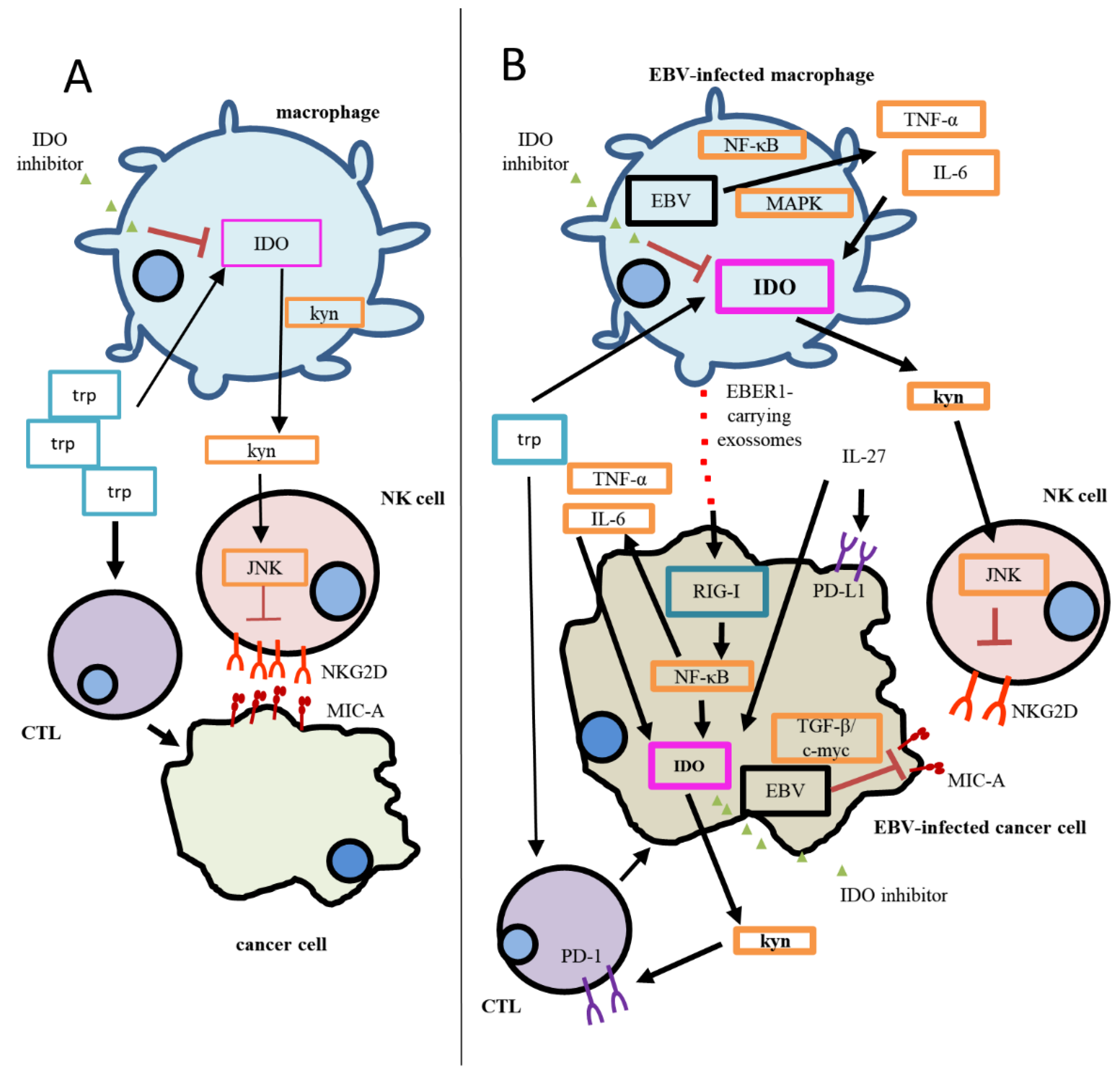
4.3. Regulation of IDO Expression by EBV
4.4. IDO and PD-L1 Regulation
5. Targeting IDO in EBV-Associated Malignancies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and Biological Studies on a Virus in Cultured Lymphoblasts from Burkitt’s Lymphoma. J. Exp. Med. 1965, 121, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Mentzer, A.J.; Brenner, N.; Allen, N.; Littlejohns, T.J.; Chong, A.Y.; Cortes, A.; Almond, R.; Hill, M.; Sheard, S.; McVean, G.; et al. Identification of host-pathogen-disease relationships using a scalable Multiplex Serology platform in UK Biobank. medRxiv 2019, 19004960. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent. Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourbon, E.; Maucort-Boulch, D.; Fontaine, J.; Mauduit, C.; Sesques, P.; Safar, V.; Ferrant, E.; Golfier, C.; Ghergus, D.; Karlin, L.; et al. Clinicopathological features and survival in EBV-positive diffuse large B-cell lymphoma not otherwise specified. Blood Adv. 2021, 5, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.P.; Shannon-Lowe, C.; Rowe, M. Deciphering the role of Epstein-Barr virus in the pathogenesis of T and NK cell lymphoproliferations. Herpesviridae 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Lee, J.; Ko, Y.H.; Han, A.; Jun, H.J.; Lee, S.C.; Hwang, I.G.; Park, Y.H.; Ahn, J.S.; Jung, C.W.; et al. The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood 2007, 110, 972–978. [Google Scholar] [CrossRef]
- Kim, Y.R.; Kim, S.J.; Cheong, J.W.; Chung, H.; Jang, J.E.; Kim, Y.; Yang, W.I.; Min, Y.H.; Kim, J.S. Pretreatment Epstein-Barr virus DNA in whole blood is a prognostic marker in peripheral T-cell lymphoma. Oncotarget 2017, 8, 92312–92323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, J.; Emile, J.F.; Mounier, N.; Gisselbrecht, C.; Martin-Garcia, N.; Petrella, T.; Bouabdallah, R.; Berger, F.; Delmer, A.; Coiffier, B.; et al. Prognostic significance of Epstein-Barr virus in nodal peripheral T-cell lymphoma, unspecified: A Groupe d’Etude des Lymphomes de l’Adulte (GELA) study. Blood 2006, 108, 4163–4169. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.J.; Ng, J.C.K.; Huang, L.; Robinson, M.; O’Hara, J.; Wilson, J.A.; Mellor, A.L. The immunotherapeutic role of indoleamine 2,3-dioxygenase in head and neck squamous cell carcinoma: A systematic review. Clin. Otolaryngol. 2021, 46, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Garcia, E.; Lee, E.K.; Lindemann, N.I.; Liu, J.F.; Matulonis, U.A.; Park, P.J.; Konstantinopoulos, P.A. Genomic Determinants of De Novo Resistance to Immune Checkpoint Blockade in Mismatch Repair-Deficient Endometrial Cancer. JCO Precis. Oncol. 2020, 4, 492–497. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Nguyen, A.; Bakhshinyan, D.; Wei, J.; Hare, D.N.; MacNeill, K.L.; Wan, Y.; Oberst, A.; Bramson, J.L.; Nasir, J.A.; et al. De novo necroptosis creates an inflammatory environment mediating tumor susceptibility to immune checkpoint inhibitors. Commun. Biol. 2020, 3, 645. [Google Scholar] [CrossRef]
- Renaud, S.; Lefebvre, A.; Mordon, S.; Morales, O.; Delhem, N. Novel Therapies Boosting T Cell Immunity in Epstein Barr Virus-Associated Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2020, 21, 4292. [Google Scholar] [CrossRef]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Matsuura, H.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. USA 2010, 107, 22641–22646. [Google Scholar] [CrossRef] [Green Version]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Borza, C.M.; Morgan, A.J.; Turk, S.M.; Hutt-Fletcher, L.M. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 2004, 78, 5007–5014. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. EBV Persistence--Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Myburgh, R.; Caduff, N.; Spohn, M.; Kok, Y.L.; Keller, C.W.; Murer, A.; Chatterjee, B.; Ruhl, J.; Engelmann, C.; et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci. Alliance 2020, 3, e202000640. [Google Scholar] [CrossRef]
- Papadopoulos, E.B.; Ladanyi, M.; Emanuel, D.; Mackinnon, S.; Boulad, F.; Carabasi, M.H.; Castro-Malaspina, H.; Childs, B.H.; Gillio, A.P.; Small, T.N.; et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N. Engl. J. Med. 1994, 330, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Munz, C.; Moormann, A. Immune escape by Epstein-Barr virus associated malignancies. Semin. Cancer Biol. 2008, 18, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.L.; Lin, Y.H.; Xiao, H.; Xing, S.; Chen, H.; Chi, P.D.; Zhang, G. Epstein-Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-kappaB pathways: Impairment in T cell functions. J. Virol. 2014, 88, 6660–6671. [Google Scholar] [CrossRef] [Green Version]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Park, H.; Kim, J.; Park, G.; Kim, Y.S.; Kim, S.M.; Kim, D.; Seo, S.K.; Lee, H.K.; Cho, D.; et al. IDO metabolite produced by EBV-transformed B cells inhibits surface expression of NKG2D in NK cells via the c-Jun N-terminal kinase (JNK) pathway. Immunol. Lett. 2011, 136, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Kimbrough, E.O.; Lou, Y. Next generation of immune checkpoint inhibitors and beyond. J. Hematol. Oncol. 2021, 14, 45. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Plimack, E.R.; Procopio, G.; McDermott, D.F.; et al. Nivolumab versus everolimus in patients with advanced renal cell carcinoma: Updated results with long-term follow-up of the randomized, open-label, phase 3 CheckMate 025 trial. Cancer 2020, 126, 4156–4167. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Reck, M.; Ciuleanu, T.E.; Lee, J.S.; Schenker, M.; Audigier-Valette, C.; Zurawski, B.; Linardou, H.; Otterson, G.A.; Salman, P.; Nishio, M.; et al. First-Line Nivolumab Plus Ipilimumab Versus Chemotherapy in Advanced NSCLC with 1% or Greater Tumor PD-L1 Expression: Patient-Reported Outcomes from CheckMate 227 Part 1. J. Thorac. Oncol. 2021, 16, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Choe, J.Y.; Yun, J.Y.; Jeon, Y.K.; Kim, S.H.; Park, G.; Huh, J.R.; Oh, S.; Kim, J.E. Indoleamine 2,3-dioxygenase (IDO) is frequently expressed in stromal cells of Hodgkin lymphoma and is associated with adverse clinical features: A retrospective cohort study. BMC Cancer 2014, 14, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, C.; Fabiani, B.; Do, C.; Tchernonog, E.; Cartron, G.; Gravelle, P.; Amara, N.; Malot, S.; Palisoc, M.M.; Copie-Bergman, C.; et al. Immune-checkpoint expression in Epstein-Barr virus positive and negative plasmablastic lymphoma: A clinical and pathological study in 82 patients. Haematologica 2016, 101, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.; Patel, S.P.; Kurzrock, R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol. 2017, 14, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Hyeon, J.; Cho, I.; Ko, Y.H.; Kim, W.S. Comparison of Efficacy of Pembrolizumab between Epstein-Barr VirusPositive and Negative Relapsed or Refractory Non-Hodgkin Lymphomas. Cancer Res. Treat. 2019, 51, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef]
- Panda, A.; Mehnert, J.M.; Hirshfield, K.M.; Riedlinger, G.; Damare, S.; Saunders, T.; Kane, M.; Sokol, L.; Stein, M.N.; Poplin, E.; et al. Immune Activation and Benefit from Avelumab in EBV-Positive Gastric Cancer. J. Natl. Cancer Inst. 2018, 110, 316–320. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W. Expression of PD-L1 in EBV-associated malignancies. Int. Immunopharmacol. 2021, 95, 107553. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Padget, M.R.; Fujii, R.; Schlom, J.; Hodge, J.W. Differential combination immunotherapy requirements for inflamed (warm) tumors versus T cell excluded (cool) tumors: Engage, expand, enable, and evolve. J. Immunother. Cancer 2021, 9, e001691. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A. Tryptophan metabolism in alcoholism. Nutr. Res. Rev. 2002, 15, 123–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Indoleamine 2,3-dioxygenase depletes tryptophan, activates general control non-derepressible 2 kinase and down-regulates key enzymes involved in fatty acid synthesis in primary human CD4+ T cells. Immunology 2015, 146, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Mellor, A.L.; Chandler, P.; Baban, B.; Hansen, A.M.; Marshall, B.; Pihkala, J.; Waldmann, H.; Cobbold, S.; Adams, E.; Munn, D.H. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int. Immunol. 2004, 16, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Reznik, E.; Luna, A.; Aksoy, B.A.; Liu, E.M.; La, K.; Ostrovnaya, I.; Creighton, C.J.; Hakimi, A.A.; Sander, C. A Landscape of Metabolic Variation across Tumor Types. Cell Syst. 2018, 6, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Ball, H.J.; Sanchez-Perez, A.; Weiser, S.; Austin, C.J.; Astelbauer, F.; Miu, J.; McQuillan, J.A.; Stocker, R.; Jermiin, L.S.; Hunt, N.H. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 2007, 396, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Smith, C.; DuHadaway, J.B.; Chandler, P.; Baban, B.; Merlo, L.M.; Pigott, E.; Keough, M.P.; Rust, S.; Mellor, A.L.; et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int. Immunol. 2014, 26, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lob, S.; Konigsrainer, A.; Zieker, D.; Brucher, B.L.; Rammensee, H.G.; Opelz, G.; Terness, P. IDO1 and IDO2 are expressed in human tumors: Levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 2009, 58, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H. Blocking IDO activity to enhance anti-tumor immunity. Front. Biosci. (Elite Ed.) 2012, 4, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e487. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.P.; Ferreira, A.C.; Pinho, M.P.; de Moraes, C.J.; Bergami-Santos, P.C.; Barbuto, J.A. Tolerogenic IDO(+) Dendritic Cells Are Induced by PD-1-Expressing Mast Cells. Front. Immunol. 2016, 7, 9. [Google Scholar] [CrossRef]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef]
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; Plaen, E.; van den Eynde, B.J. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol. Res. 2017, 5, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.J.; Sharma, M.D.; Chandler, P.R.; Duhadaway, J.B.; Everhart, M.E.; Johnson, B.A., III; Kahler, D.J.; Pihkala, J.; Soler, A.P.; Munn, D.H.; et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc. Natl. Acad. Sci. USA 2008, 105, 17073–17078. [Google Scholar] [CrossRef] [Green Version]
- Wingender, G.; Garbi, N.; Schumak, B.; Jungerkes, F.; Endl, E.; von Bubnoff, D.; Steitz, J.; Striegler, J.; Moldenhauer, G.; Tuting, T.; et al. Systemic application of CpG-rich DNA suppresses adaptive T cell immunity via induction of IDO. Eur. J. Immunol. 2006, 36, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Nouel, A.; Pochard, P.; Simon, Q.; Segalen, I.; le Meur, Y.; Pers, J.O.; Hillion, S. B-Cells induce regulatory T cells through TGF-beta/IDO production in A CTLA-4 dependent manner. J. Autoimmun. 2015, 59, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chamuleau, M.E.D.; van de Loosdrecht, A.A.; Hess, C.J.; Janssen, J.J.W.M.; Zevenbergen, A.; Delwel, R.; Valk, P.J.M.; Lowenberg, B.; Ossenkoppele, G.J. High INDO (Indoleamine 2,3-Dioxygenase) mRNA Level in Blasts of Acute Myeloid Leukemic Patients Predicts Poor Clinical Outcome. Blood 2007, 110, 4297. [Google Scholar] [CrossRef]
- Liu, H.; Shen, Z.; Wang, Z.; Wang, X.; Zhang, H.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Increased expression of IDO associates with poor postoperative clinical outcome of patients with gastric adenocarcinoma. Sci. Rep. 2016, 6, 21319. [Google Scholar] [CrossRef] [Green Version]
- Mitra, D.; Horick, N.K.; Brackett, D.G.; Mouw, K.W.; Hornick, J.L.; Ferrone, S.; Hong, T.S.; Mamon, H.; Clark, J.W.; Parikh, A.R.; et al. High IDO1 Expression Is Associated with Poor Outcome in Patients with Anal Cancer Treated with Definitive Chemoradiotherapy. Oncologist 2019, 24, e275–e283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wu, J.; Shen, H.; Wang, J. The prognostic value of IDO expression in solid tumors: A systematic review and meta-analysis. BMC Cancer 2020, 20, 471. [Google Scholar] [CrossRef]
- Panda, A.; Ganesan, S. Genomic and Immunologic Correlates of Indoleamine 2,3-Dioxygenase Pathway Expression in Cancer. Front. Genet. 2021, 12, 706435. [Google Scholar] [CrossRef]
- Dai, C.; Geng, R.; Wang, C.; Wong, A.; Qing, M.; Hu, J.; Sun, Y.; Lo, A.W.; Li, J. Concordance of immune checkpoints within tumor immune contexture and their prognostic significance in gastric cancer. Mol. Oncol. 2016, 10, 1551–1558. [Google Scholar] [CrossRef] [Green Version]
- Derks, S.; Liao, X.; Chiaravalli, A.M.; Xu, X.; Camargo, M.C.; Solcia, E.; Sessa, F.; Fleitas, T.; Freeman, G.J.; Rodig, S.J.; et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget 2016, 7, 32925–32932. [Google Scholar] [CrossRef]
- Ball, H.J.; Jusof, F.F.; Bakmiwewa, S.M.; Hunt, N.H.; Yuasa, H.J. Tryptophan-catabolizing enzymes—party of three. Front. Immunol. 2014, 5, 485. [Google Scholar] [CrossRef] [Green Version]
- Burassakarn, A.; Srisathaporn, S.; Pientong, C.; Wongjampa, W.; Vatanasapt, P.; Patarapadungkit, N.; Ekalaksananan, T. Exosomes-carrying Epstein-Barr virus-encoded small RNA-1 induces indoleamine 2,3-dioxygenase expression in tumor-infiltrating macrophages of oral squamous-cell carcinomas and suppresses T-cell activity by activating RIG-I/IL-6/TNF-alpha pathway. Oral Oncol. 2021, 117, 105279. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, T.I.; Son, S.A.; Lee, H.W. Immunohistochemical Features of Indoleamine 2,3-Dioxygenase (IDO) in Various Types of Lymphoma: A Single Center Experience. Diagnostics 2020, 10, 275. [Google Scholar] [CrossRef]
- Morscio, J.; Dierickx, D.; Ferreiro, J.F.; Herreman, A.; van Loo, P.; Bittoun, E.; Verhoef, G.; Matthys, P.; Cools, J.; Wlodarska, I.; et al. Gene expression profiling reveals clear differences between EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am. J. Transplant. 2013, 13, 1305–1316. [Google Scholar] [CrossRef]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.; Raffeld, M.; Jaffe, E.S. EBV-positive large B-cell lymphomas in young patients: A nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Ben-Haj-Ayed, A.; Moussa, A.; Ghedira, R.; Gabbouj, S.; Miled, S.; Bouzid, N.; Tebra-Mrad, S.; Bouaouina, N.; Chouchane, L.; Zakhama, A.; et al. Prognostic value of indoleamine 2,3-dioxygenase activity and expression in nasopharyngeal carcinoma. Immunol. Lett. 2016, 169, 23–32. [Google Scholar] [CrossRef]
- Muraro, E.; Vaccher, E.; Furlan, C.; Fratta, E.; Fanetti, G.; Fae, D.A.; Martorelli, D.; Cangemi, M.; Polesel, J.; Navarria, F.; et al. Predictive Value of CD8 Expression and FoxP3 Methylation in Nasopharyngeal Carcinoma Patients Treated with Chemoradiotherapy in a Non-endemic Area. Pathol. Oncol. Res. 2020, 26, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhang, Y.; Jiang, W.; Chen, Y.P.; Xu, S.Y.; Liu, N.; Zhao, Y.; Li, L.; Lei, Y.; Hong, X.H.; et al. Development and validation of an immune checkpoint-based signature to predict prognosis in nasopharyngeal carcinoma using computational pathology analysis. J. Immunother. Cancer 2019, 7, 298. [Google Scholar] [CrossRef]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Wada, R.; Yajima, N.; Imaizumi, T.; Yagihashi, S. Tumor microenvironment and RIG-I signaling molecules in Epstein Barr virus-positive and -negative classical Hodgkin lymphoma of the elderly. J. Clin. Exp. Hematop. 2014, 54, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus miR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Kolakofsky, D.; Kowalinski, E.; Cusack, S. A structure-based model of RIG-I activation. RNA 2012, 18, 2118–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Magri, G.; Pende, D.; Angulo, A.; Lopez-Botet, M. Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood 2010, 115, 5170–5179. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.S.; Chen, S.; Zhang, M.J.; Chan, J.Y.; Gao, W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef] [Green Version]
- Kenison, J.E.; Wang, Z.; Yang, K.; Snyder, M.; Quintana, F.J.; Sherr, D.H. The aryl hydrocarbon receptor suppresses immunity to oral squamous cell carcinoma through immune checkpoint regulation. Proc. Natl. Acad. Sci. USA 2021, 118, e2012692118. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280. [Google Scholar] [CrossRef] [Green Version]
- Gonin, J.; Larousserie, F.; Bastard, C.; Picquenot, J.M.; Couturier, J.; Radford-Weiss, I.; Dietrich, C.; Brousse, N.; Vacher-Lavenu, M.C.; Devergne, O. Epstein-Barr virus-induced gene 3 (EBI3): A novel diagnosis marker in Burkitt lymphoma and diffuse large B-cell lymphoma. PLoS ONE 2011, 6, e24617. [Google Scholar] [CrossRef]
- Niedobitek, G.; Pazolt, D.; Teichmann, M.; Devergne, O. Frequent expression of the Epstein-Barr virus (EBV)-induced gene, EBI3, an IL-12 p40-related cytokine, in Hodgkin and Reed-Sternberg cells. J. Pathol. 2002, 198, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Nishino, R.; Takano, A.; Oshita, H.; Ishikawa, N.; Akiyama, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; Tsuchiya, E.; Kohno, N.; et al. Identification of Epstein-Barr virus-induced gene 3 as a novel serum and tissue biomarker and a therapeutic target for lung cancer. Clin. Cancer Res. 2011, 17, 6272–6286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horlad, H.; Ma, C.; Yano, H.; Pan, C.; Ohnishi, K.; Fujiwara, Y.; Endo, S.; Kikukawa, Y.; Okuno, Y.; Matsuoka, M.; et al. An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016, 107, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Larousserie, F.; Bardel, E.; Pflanz, S.; Arnulf, B.; Lome-Maldonado, C.; Hermine, O.; Bregeaud, L.; Perennec, M.; Brousse, N.; Kastelein, R.; et al. Analysis of interleukin-27 (EBI3/p28) expression in Epstein-Barr virus- and human T-cell leukemia virus type 1-associated lymphomas: Heterogeneous expression of EBI3 subunit by tumoral cells. Am. J. Pathol. 2005, 166, 1217–1228. [Google Scholar] [CrossRef]
- Kyi, C.; Postow, M.A. Immune checkpoint inhibitor combinations in solid tumors: Opportunities and challenges. Immunotherapy 2016, 8, 821–837. [Google Scholar] [CrossRef] [Green Version]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Eroglu, Z.; Kim, D.W.; Wang, X.; Camacho, L.H.; Chmielowski, B.; Seja, E.; Villanueva, A.; Ruchalski, K.; Glaspy, J.A.; Kim, K.B.; et al. Long term survival with cytotoxic T lymphocyte-associated antigen 4 blockade using tremelimumab. Eur. J. Cancer 2015, 51, 2689–2697. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Carlo-Stella, C. Are EBV-related and EBV-unrelated Hodgkin lymphomas different with regard to susceptibility to checkpoint blockade? Blood 2018, 132, 17–22. [Google Scholar] [CrossRef]
- Daud, A.; Saleh, M.N.; Hu, J.; Bleeker, J.S.; Riese, M.J.; Meier, R.; Zhou, L.; Serbest, G.; Lewis, K.D. Epacadostat plus nivolumab for advanced melanoma: Updated phase 2 results of the ECHO-204 study. J. Clin. Oncol. 2018, 36, 9511. [Google Scholar] [CrossRef]
- Gangadhar, T.C.; Schneider, B.J.; Bauer, T.M.; Wasser, J.S.; Spira, A.I.; Patel, S.P.; Balmanoukian, A.S.; Bauml, J.; Schmidt, E.V.; Zhao, Y.; et al. Efficacy and safety of epacadostat plus pembrolizumab treatment of NSCLC: Preliminary phase I/II results of ECHO-202/KEYNOTE-037. J. Clin. Oncol. 2017, 35, 9014. [Google Scholar] [CrossRef]
- Gibney, G.; Hamid, O.; Lutzky, J.; Olszanski, A.; Gangadhar, T.; Gajewski, T.; Chmielowski, B.; Hanks, B.; Boasberg, P.; Zhao, Y.; et al. 511 Updated results from a phase 1/2 study of epacadostat (INCB024360) in combination with ipilimumab in patients with metastatic melanoma. Eur. J. Cancer 2015, 51, S106–S107. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients with Advanced Solid Tumors: Phase I Results from a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Larkin, J.; Ascierto, P.A.; Long, G.V.; Hassel, J.C.; Schadendorf, D.; Hodi, F.S.; Lebbé, C.; Grob, J.J.; Grossmann, K.; et al. Characterization of complete responses (CRs) in patients with advanced melanoma (MEL) who received the combination of nivolumab (NIVO) and ipilimumab (IPI), NIVO or IPI alone. Ann. Oncol. 2017, 28, v428. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Zakharia, Y.; Rixe, O.; Ward, J.H.; Drabick, J.J.; Shaheen, M.F.; Milhem, M.M.; Munn, D.; Kennedy, E.P.; Vahanian, N.N.; Link, C.J.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 2018, 36, 9512. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Endo, R.; Nakamura, T.; Kawakami, K.; Sato, Y.; Harashima, H. The silencing of indoleamine 2,3-dioxygenase 1 (IDO1) in dendritic cells by siRNA-loaded lipid nanoparticles enhances cell-based cancer immunotherapy. Sci. Rep. 2019, 9, 11335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. A new cancer immunotherapy suffers a setback. Science 2018, 360, 588. [Google Scholar] [CrossRef] [PubMed]
- Svane, I.M.; Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Andersen, M.H. LBA48 Clinical efficacy and immunity of combination therapy with nivolumab and IDO/PD-L1 peptide vaccine in patients with metastatic melanoma: A phase I/II trial. Ann. Oncol. 2020, 31, S1176. [Google Scholar] [CrossRef]


{kind=link}
| EBV Latency Pattern | Viral Expression Pattern | EBV-Associated Malignancies |
|---|---|---|
| III | EBNA-1–6, LMP1, LMP2A-B, EBER, BART | PTLD, DLBCL |
| II | EBNA-1, LMP1, LMP2A, EBER, BART | HL, NHL, GC, ENKTCL, DLBCL, PBL and NPC |
| I | EBNA1, EBER, BART | BL, PL |
| 0 | EBER, BART | - |
| EBV-Associated Malignancies | IDO Status | Reference |
|---|---|---|
| HL | Enhanced when compared to EBV(−) | [44] |
| DLBCL | Detection/enhanced when compared to EBV(−) | [92,93,94] |
| EBV(+)GC | Overexpression | [87] |
| NPC | Detection/overexpression | [33,95,96,97] |
| PL | Expression was similar to EBV(−)PL | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawada, L.; Vallinoto, A.C.R.; Brasil-Costa, I. Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus. Biomolecules 2021, 11, 1792. https://doi.org/10.3390/biom11121792
Sawada L, Vallinoto ACR, Brasil-Costa I. Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus. Biomolecules. 2021; 11(12):1792. https://doi.org/10.3390/biom11121792
Chicago/Turabian StyleSawada, Leila, Antonio Carlos Rosário Vallinoto, and Igor Brasil-Costa. 2021. "Regulation of the Immune Checkpoint Indoleamine 2,3-Dioxygenase Expression by Epstein–Barr Virus" Biomolecules 11, no. 12: 1792. https://doi.org/10.3390/biom11121792