
Obesity–An Update on the Basic Pathophysiology and Review of Recent Therapeutic Advances

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
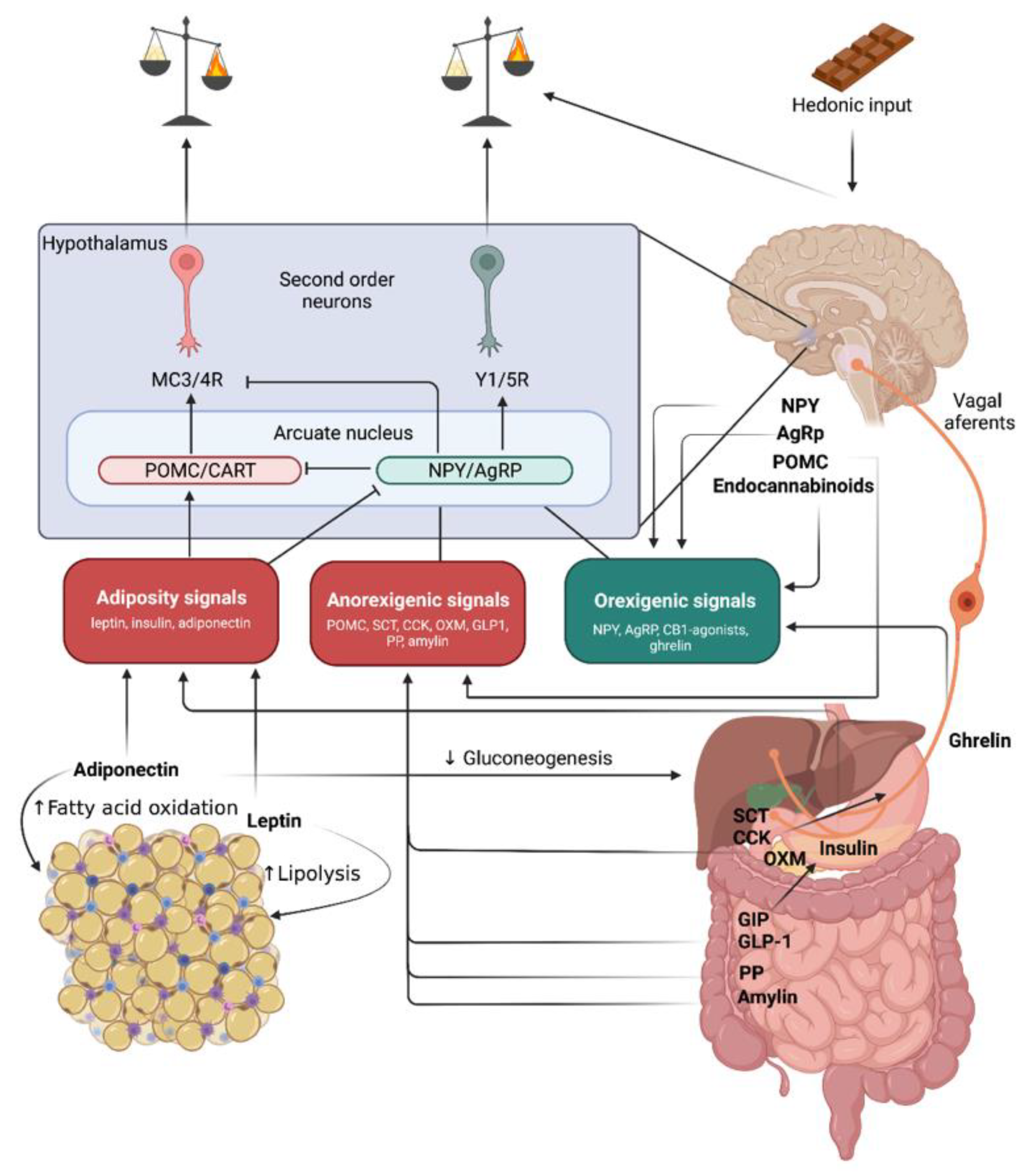
2. Obesity Pathomechanisms
2.1. Molecules Acting on Long-Term Energy Balance
2.1.1. Leptin
2.1.2. Insulin
2.1.3. Proopiomelanocortin (POMC)
2.2. Molecules Leading to Short-Term Positive energy Balance–Orexigenic Stimuli
2.2.1. Neuropeptide Y
2.2.2. Agouti-Related Peptide
2.2.3. Ghrelin
2.2.4. Endocannabinoids
2.3. Molecules Leading to Short-Term Negative Energy Balance–Anorexigenic Stimuli
2.3.1. Secretin
2.3.2. Cholecystokinin
2.3.3. Incretin Hormones
2.3.4. Oxyntomodulin
2.3.5. Polypeptide Fold (PP-Fold) Family
2.3.6. Amylin
2.3.7. Cocaine- and Amphetamine-Regulated Transcript
2.4. White Adipose Tissue Secreted Molecules
2.5. Brown Adipose Tissue (BAT) Secreted Molecules
2.6. Obesity, Mitochondrial Dysfunction and Insulin Resistance–An Interplay
3. Pharmacotherapy Options
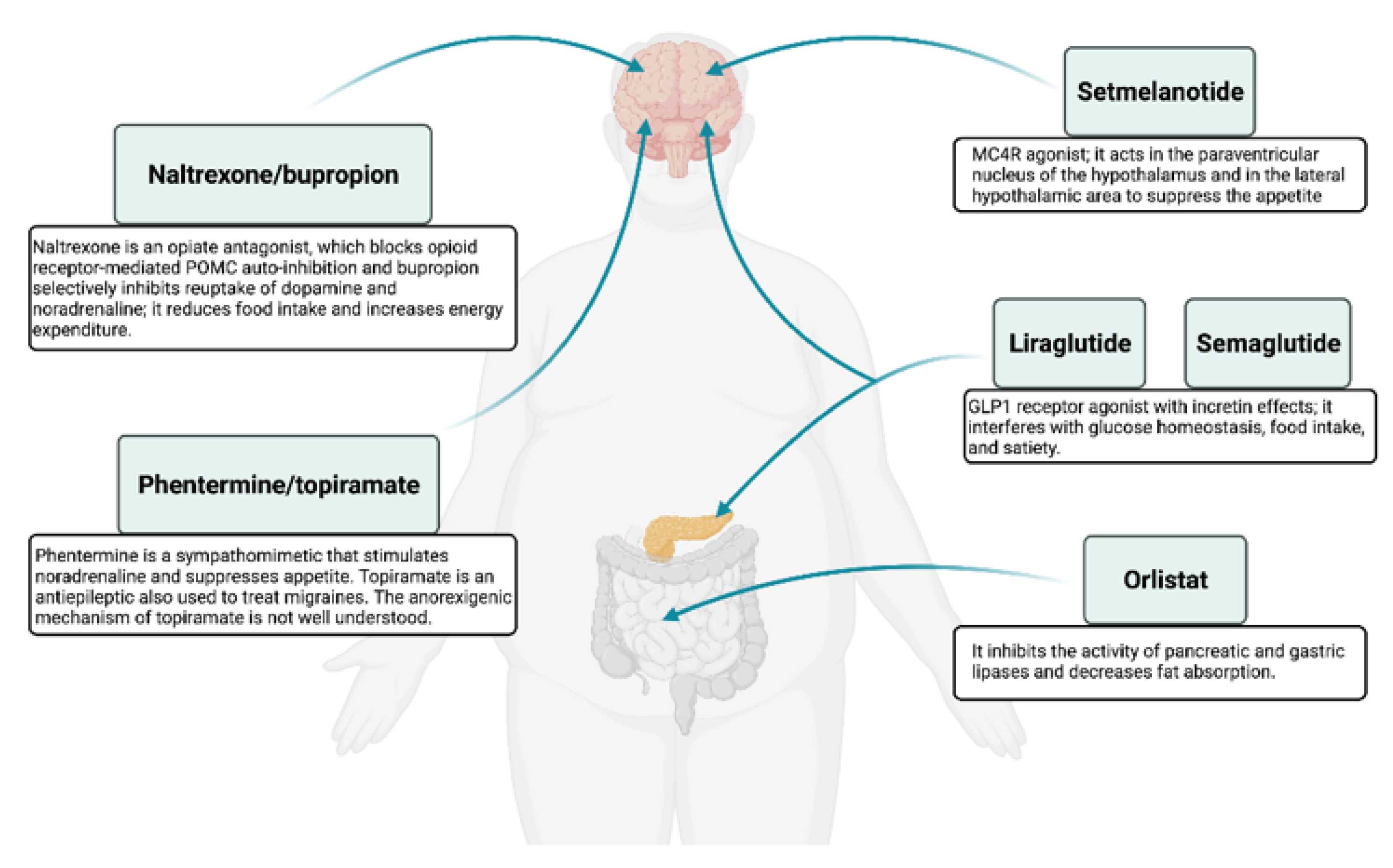
3.1. Approved Anti-Obesity Drugs for Long-Term Weight Management
3.1.1. Orlistat
3.1.2. Naltrexone/Bupropion
3.1.3. Phentermine/Topiramate
3.1.4. Liraglutide
3.1.5. Setmelanotide
3.1.6. Semaglutide
3.2. Other Drugs in Potential Use for Anti-Obesity Treatment
3.2.1. Lorcaserin (Withdrawn from Market in February 2020)
3.2.2. Sodium-Glucose Co-Transporter-2 Inhibitors (Not Indicated for Obesity Alone)
3.3. Cardiovascular Comorbidities Outcomes for Anti-Obesity Medication
3.4. Further Potential Pharmacotherapeutic Targets
3.4.1. Polyagonists of the Incretin System
3.4.2. Amylin Mimetics
3.4.3. Leptin Analogues
3.4.4. Ghrelin Vaccine and Antagonists of Ghrelin and NPY
3.4.5. Cannabinoid Type-1 Receptor Antagonists
3.4.6. Antioxidants
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, E.D.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2019, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Zeng, T.; Lee, K.; Nobis, M.; Loh, K.; Gou, L.; Xia, Z.; Gao, Z.; Bensellam, M.; Hughes, W.; et al. Peripheral-specific Y1 receptor antagonism increases thermogenesis and protects against diet-induced obesity. Nat. Commun. 2021, 12, 2622. [Google Scholar] [CrossRef]
- American Diabetes Association. Prevention or Delay of Type 2 Diabetes: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44, S34–S39. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; McCarthy, W.; Burridge, K.; Tondt, J.; Karjoo, S.; Christensen, S.; Ng, J.; Golden, A.; Davisson, L.R.L. Obesity Algorithm 2021. Available online: https://obesitymedicine.org/wp-content/uploads/2021/01/2021-Obesity-Algorithm.pdf (accessed on 7 August 2021).
- Centers of Disease Control and Prevention. National Diabetes Statistics Report, 2020; Department of Health and Human Services: Atlanta, GA, USA, 2020. [Google Scholar]
- NCD Risk Factor Collaboration. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Wadden, T.A.; Butryn, M.L.; Hong, P.S.; Tsai, A.G. Behavioral Treatment of Obesity in Patients Encountered in Primary Care Settings. JAMA 2014, 312, 1779–1791. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Neal, B.; Perkovic, V.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Fabbrini, E.; Sun, T.; Li, Q.; et al. Canagliflozin for Primary and Secondary Prevention of Cardiovascular Events. Circulation 2018, 137, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Gadde, K.M.; Martin, C.K.; Berthoud, H.-R.; Heymsfield, S.B. Obesity: Pathophysiology and management. J. Am. Coll. Cardiol. 2018, 71, 69–84. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.; Bonaca, M.P.; Mosenzon, O.; Kato, E.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Udell, J.A.; Yuan, Z.; Rush, T.; Sicignano, N.M.; Galitz, M.; Rosenthal, N. Cardiovascular Outcomes and Risks After Initiation of a Sodium Glucose Cotransporter 2 Inhibitor. Circulation 2018, 137, 1450–1459. [Google Scholar] [CrossRef]
- Crowley, M.J.; McGuire, D.K.; Alexopoulos, A.-S.; Jensen, T.J.; Rasmussen, S.; Saevereid, H.A.; Verma, S.; Buse, J.B. Effects of Liraglutide on Cardiovascular Outcomes in Type 2 Diabetes Patients With and Without Baseline Metformin Use: Post Hoc Analyses of the LEADER Trial. Diabetes Care 2020, 43, e108–e110. [Google Scholar] [CrossRef]
- Xie, J.; Jiang, M.; Li, L. Letter by Xie et al. Regarding Article, “Effects of Liraglutide Versus Placebo on Cardiovascular Events in Patients With Type 2 Diabetes Mellitus and Chronic Kidney Disease: Results From the LEADER Trial”. Circulation 2019, 139, e1015–e1016. [Google Scholar] [CrossRef]
- Bray, G.A.; Frühbeck, G.; Ryan, D.H.; Wilding, J. Management of obesity. Lancet 2016, 387, 1947–1956. [Google Scholar] [CrossRef] [Green Version]
- Gribble, F.M.; O’Rahilly, S. Obesity therapeutics: The end of the beginning. Cell Metab. 2021, 33, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Færch, L.; Jeppesen, O.K.; Pakseresht, A.; Pedersen, S.D.; Perreault, L.; Rosenstock, J.; Shimomura, I.; Viljoen, A.; Wadden, T.A.; et al. Semaglutide 2·4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): A randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet 2021, 397, 971–984. [Google Scholar] [CrossRef]
- Wilding, J.P.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Rubino, D.; Abrahamsson, N.; Davies, M.; Hesse, D.; Greenway, F.L.; Jensen, C.; Lingvay, I.; Mosenzon, O.; Rosenstock, J.; Rubio, M.A.; et al. Effect of Continued Weekly Subcutaneous Semaglutide vs Placebo on Weight Loss Maintenance in Adults With Overweight or Obesity. JAMA 2021, 325, 1414. [Google Scholar] [CrossRef] [PubMed]
- Wadden, T.A.; Bailey, T.S.; Billings, L.K.; Davies, M.; Frias, J.P.; Koroleva, A.; Lingvay, I.; O’Neil, P.M.; Rubino, D.M.; Skovgaard, D.; et al. Effect of Subcutaneous Semaglutide vs Placebo as an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults With Overweight or Obesity. JAMA 2021, 325, 1403. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Approves New Drug Treatment for Chronic Weight Management, First Since 2014. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-chronic-weight-management-first-2014 (accessed on 28 August 2021).
- Marlene Busko. FDA Approves ‘Gamechanger’ Semaglutide for Weight Loss. 2021. Available online: https://www.medscape.com/viewarticle/952441 (accessed on 6 August 2021).
- USFDA. FDA Approves Treatment for Chronic Kidney Disease|FDA. 2021. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-chronic-kidney-disease (accessed on 7 August 2021).
- Lilly, E.; Affairs, M. Breakthrough Results for Jardiance® (Empagliflozin) Confirm EMPEROR-Preserved as First and Only Successful Trial for Heart Failure with Preserved Ejection Fraction. 2021. Available online: www.jardiance.com (accessed on 24 August 2021).
- Hall, K.D.; Sacks, G.; Chandramohan, D.; Chow, C.C.; Wang, Y.C.; Gortmaker, S.L.; Swinburn, B.A. Quantification of the effect of energy imbalance on bodyweight. Lancet 2011, 378, 826–837. [Google Scholar] [CrossRef] [Green Version]
- González-Muniesa, P.; Mártinez-González, M.-A.; Hu, F.B.; Després, J.-P.; Matsuzawa, Y.; Loos, R.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Prim. 2017, 3, 17034. [Google Scholar] [CrossRef]
- Bray, M.S.; Loos, R.; McCaffery, J.; Ling, C.; Franks, P.; Weinstock, G.; Snyder, M.P.; Vassy, J.; Agurs-Collins, T.; The The Conference Working Group. NIH working group report—using genomic information to guide weight management: From universal to precision treatment. Obesity 2016, 24, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Elks, C.E.; Hoed, M.D.; Zhao, J.H.; Sharp, S.J.; Wareham, N.J.; Loos, R.J.F.; Ong, K.K. Variability in the Heritability of Body Mass Index: A Systematic Review and Meta-Regression. Front. Endocrinol. 2012, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Winkler, T.W.; Justice, A.E.; Graff, M.; Barata, L.; Feitosa, M.F.; Chu, S.; Czajkowski, J.; Esko, T.; Fall, T.; Kilpeläinen, T.O.; et al. The Influence of Age and Sex on Genetic Associations with Adult Body Size and Shape: A Large-Scale Genome-Wide Interaction Study. PLoS Genet. 2015, 11, e1005378. [Google Scholar] [CrossRef] [Green Version]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.G.; Leibel, R.L. Lessons from Rodent Models of Obesity. 2000. Available online: http://europepmc.org/books/NBK279123 (accessed on 31 July 2021).
- van der Klaauw, A.; Farooqi, I.S. The Hunger Genes: Pathways to Obesity. Cell 2015, 161, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetherington, A.W.; Ranson, S.W. Hypothalamic lesions and adiposity in the rat. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1940, 78, 149–172. [Google Scholar] [CrossRef]
- Williams, D.M.; Nawaz, A.; Evans, M. Drug Therapy in Obesity: A Review of Current and Emerging Treatments. Diabetes Ther. 2020, 11, 1199–1216. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Kishi, T.; Lee, C.E.; Choi, B.J.; Fang, H.; Hollenberg, A.N.; Drucker, D.J.; Elmquist, J.K. Glucagon-Like Peptide-1-Responsive Catecholamine Neurons in the Area Postrema Link Peripheral Glucagon-Like Peptide-1 with Central Autonomic Control Sites. J. Neurosci. 2003, 23, 2939–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badman, M.K. The Gut and Energy Balance: Visceral Allies in the Obesity Wars. Science 2005, 307, 1909–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqi, I. Monogenic human obesity syndromes. In Progress in Brain Research; Elsevier BV: Amsterdam, The Netherlands, 2006; Volume 153, pp. 119–125. [Google Scholar]
- Myers, M.G. Leptin Keeps Working, Even in Obesity. Cell Metab. 2015, 21, 791–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced Weight Loss With Pramlintide/Metreleptin: An Integrated Neurohormonal Approach to Obesity Pharmacotherapy. Obesity 2009, 17, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Fryk, E.; Olausson, J.; Mossberg, K.; Strindberg, L.; Schmelz, M.; Brogren, H.; Gan, L.-M.; Piazza, S.; Provenzani, A.; Becattini, B.; et al. Hyperinsulinemia and insulin resistance in the obese may develop as part of a homeostatic response to elevated free fatty acids: A mechanistic case-control and a population-based cohort study. EBioMedicine 2021, 65, 103264. [Google Scholar] [CrossRef] [PubMed]
- Klem, M.; Wing, R.R.; McGuire, M.T.; Seagle, H.M.; Hill, J.O. A descriptive study of individuals successful at long-term maintenance of substantial weight loss. Am. J. Clin. Nutr. 1997, 66, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Ebbeling, C.B.; Swain, J.F.; Feldman, H.A.; Wong, W.W.; Hachey, D.L.; Garcia-Lago, E.; Ludwig, D.S. Effects of Dietary Composition on Energy Expenditure During Weight-Loss Maintenance. JAMA 2012, 307, 2627–2634. [Google Scholar] [CrossRef] [Green Version]
- Morton, G.J.; Meek, T.H.; Schwartz, M.W. Neurobiology of food intake in health and disease. Nat. Rev. Neurosci. 2014, 15, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Fenselau, H.; Campbell, J.N.; Verstegen, A.M.; Madara, J.C.; Xu, J.; Shah, B.P.; Resch, J.; Yang, Z.; Mandelblat-Cerf, Y.; Livneh, Y.; et al. A rapidly acting glutamatergic ARC→PVH satiety circuit postsynaptically regulated by α-MSH. Nat. Neurosci. 2017, 20, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, G.; Schwartz, M. The NPY/AgRP neuron and energy homeostasis. Int. J. Obes. 2001, 25, S56–S62. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.E.; Kitlinska, J.B.; Tilan, J.U.; Li, L.; Baker, S.B.; Johnson, M.D.; Lee, E.; Burnett, M.S.; Fricke, S.T.; Kvetnansky, R.; et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 2007, 13, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X.; Mozzoli, M.; Ryan, I. Effect of fasting on serum leptin in normal human subjects. J. Clin. Endocrinol. Metab. 1996, 81, 3419–3423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krashes, M.J.; Koda, S.; Ye, C.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Investig. 2011, 121, 1424–1428. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lin, Y.-C.; Zimmerman, C.; Essner, R.A.; Knight, Z.A. Hunger neurons drive feeding through a sustained, positive reinforcement signal. eLife 2016, 5, e18640. [Google Scholar] [CrossRef]
- Betley, J.N.; Xu, S.; Cao, Z.F.H.; Gong, R.; Magnus, C.J.; Yu, Y.; Sternson, S.M. Neurons for hunger and thirst transmit a negative-valence teaching signal. Nat. Cell Biol. 2015, 521, 180–185. [Google Scholar] [CrossRef]
- Cummings, D.E.; Purnell, J.Q.; Frayo, R.S.; Schmidova, K.; Wisse, B.E.; Weigle, D.S. A Preprandial Rise in Plasma Ghrelin Levels Suggests a Role in Meal Initiation in Humans. Diabetes 2001, 50, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Nogueiras, R.; Andermann, M.; Andrews, Z.B.; Anker, S.; Argente, J.; Batterham, R.; Benoit, S.; Bowers, C.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nat. Cell Biol. 2001, 409, 194–198. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Wang, W.; Tao, Y.-X. Ghrelin Receptor Mutations and Human Obesity. In Progress in Molecular Biology and Translational Science; Elsevier BV: Amsterdam, The Netherlands, 2016; Volume 140, pp. 131–150. [Google Scholar]
- Holst, B. Ghrelin receptor mutations—too little height and too much hunger. J. Clin. Investig. 2006, 116, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ahmed, S.; Smith, R.G. Deletion of Ghrelin Impairs neither Growth nor Appetite. Mol. Cell. Biol. 2003, 23, 7973–7981. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, P.; Zheng, H.; Smith, R.G. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 4679–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, D.E.; Weigle, D.S.; Frayo, R.S.; Breen, P.A.; Ma, M.K.; Dellinger, E.P.; Purnell, J.Q. Plasma Ghrelin Levels after Diet-Induced Weight Loss or Gastric Bypass Surgery. N. Engl. J. Med. 2002, 346, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Neary, N.M.; Small, C.J.; Wren, A.M.; Lee, J.L.; Druce, M.R.; Palmieri, C.; Frost, G.S.; Ghatei, M.A.; Coombes, R.C.; Bloom, S.R. Ghrelin Increases Energy Intake in Cancer Patients with Impaired Appetite: Acute, Randomized, Placebo-Controlled Trial. J. Clin. Endocrinol. Metab. 2004, 89, 2832–2836. [Google Scholar] [CrossRef]
- Kirkham, T.C. Endocannabinoids in the regulation of appetite and body weight. Behav. Pharmacol. 2005, 16, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Pharmacological Actions of Cannabinoids. Handb. Exp. Pharmacol. 2005, 168, 1–51. [Google Scholar] [CrossRef]
- Richey, J.M.; Woolcott, O. Revisiting the Endocannabinoid System and Its Therapeutic Potential in Obesity and Associated Diseases. Curr. Diabetes Rep. 2017, 17, 99. [Google Scholar] [CrossRef]
- Trillou, C.R.; Delgorge, C.; Menet, C.; Arnone, M.; Soubrié, P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int. J. Obes. 2004, 28, 640–648. [Google Scholar] [CrossRef] [Green Version]
- Nogueiras, R.; Veyrat-Durebex, C.; Suchanek, P.M.; Klein, M.; Tschöp, J.; Caldwell, C.; Woods, S.C.; Wittmann, G.; Watanabe, M.; Liposits, Z.; et al. Peripheral, but Not Central, CB1 Antagonism Provides Food Intake-Independent Metabolic Benefits in Diet-Induced Obese Rats. Diabetes 2008, 57, 2977–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, R.; Wang, L.; Chow, B.K.C. Central Control of Feeding Behavior by the Secretin, PACAP, and Glucagon Family of Peptides. Front. Endocrinol. 2017, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Schnabl, K.; Gabler, S.-M.; Willershäuser, M.; Reber, J.; Karlas, A.; Laurila, S.; Lahesmaa, M.; U-Din, M.; Bast-Habersbrunner, A.; et al. Secretin-Activated Brown Fat Mediates Prandial Thermogenesis to Induce Satiation. Cell 2018, 175, 1561–1574.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.R. Gastrointestinal Phisiology, 9th ed.; Elsevier: Philadelphia, PA, USA, 2019. [Google Scholar]
- Muurahainen, N.; Kissileff, H.R.; Derogatis, A.J.; Pi-Sunyer, F.X. Effects of cholecystokinin-octapeptide (CCK-8) on food intake and gastric emptying in man. Physiol. Behav. 1988, 44, 645–649. [Google Scholar] [CrossRef]
- Grill, H.J.; Hayes, M.R. Hindbrain Neurons as an Essential Hub in the Neuroanatomically Distributed Control of Energy Balance. Cell Metab. 2012, 16, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Roman, C.W.; Derkach, V.A.; Palmiter, R.D. Genetically and functionally defined NTS to PBN brain circuits mediating anorexia. Nat. Commun. 2016, 7, 11905. [Google Scholar] [CrossRef]
- Campos, C.A.; Bowen, A.; Schwartz, M.W.; Palmiter, R.D. Parabrachial CGRP Neurons Control Meal Termination. Cell Metab. 2016, 23, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. 1), 5–21. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, T.J.; McIntosh, C.H.; Pederson, R.A. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995, 136, 3585–3596. [Google Scholar] [CrossRef]
- Larsen, P.J.; Fledelius, C.; Knudsen, L.B.; Tang-Christensen, M. Systemic administration of the long-acting GLP-1 derivative NN2211 induces lasting and reversible weight loss in both normal and obese rats. Diabetes 2001, 50, 2530–2539. [Google Scholar] [CrossRef] [Green Version]
- Meeran, K.; O’Shea, D.; Edwards, C.M.B.; Turton, M.D.; Heath, M.M.; Gunn, I.; Abusnana, S.; Rossi, M.; Small, C.J.; Goldstone, A.P.; et al. Repeated Intracerebroventricular Administration of Glucagon-Like Peptide-1-(7–36) Amide or Exendin-(9–39) Alters Body Weight in the Rat**This work was supported by the United Kingdom Medical Research Council. Endocrinology 1999, 140, 244–250. [Google Scholar] [CrossRef]
- Scrocchi, L.; Brown, T.; MacLusky, N.; Brubaker, P.; Auerbach, A.; Joyner, A.; Drucker, D. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon–like peptide 1 receptor gene. Nat. Med. 1996, 2, 1254–1258. [Google Scholar] [CrossRef]
- Miyawaki, K.; Yamada, Y.; Ban, N.; Ihara, Y.; Tsukiyama, K.; Zhou, H.; Fujimoto, S.; Oku, A.; Tsuda, K.; Toyokuni, S.; et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 2002, 8, 738–742. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity. 2021. Available online: https://www.who.int/health-topics/obesity (accessed on 8 June 2021).
- Novo Nordisk. Novo Nordisk Files for US FDA Regulatory Approval of Once- Weekly Semaglutide 2.4 mg for Weight Management. 2020. Available online: https://www.globenewswire.com/news-release/2020/12/04/2139776/0/en/Novo-Nordisk-files-for-US-FDA-regulatory-approval-of-once-weekly-semaglutide-2-4-mg-for-weight-management.html (accessed on 8 June 2021).
- Cohen, M.A.; Ellis, S.M.; Le Roux, C.; Batterham, R.; Park, A.; Patterson, M.M.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Oxyntomodulin Suppresses Appetite and Reduces Food Intake in Humans. J. Clin. Endocrinol. Metab. 2003, 88, 4696–4701. [Google Scholar] [CrossRef] [PubMed]
- Dakin, C.L.; Gunn, I.; Small, C.J.; Edwards, C.M.B.; Hay, D.L.; Smith, D.M.; Ghatei, M.A.; Bloom, S.R. Oxyntomodulin Inhibits Food Intake in the Rat. Endocrinology 2001, 142, 4244–4250. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A. Action and therapeutic potential of oxyntomodulin. Mol. Metab. 2014, 3, 241–251. [Google Scholar] [CrossRef]
- Trapp, S.; Richards, J.E. The gut hormone glucagon-like peptide-1 produced in brain: Is this physiologically relevant? Curr. Opin. Pharmacol. 2013, 13, 964–969. [Google Scholar] [CrossRef] [Green Version]
- Dakin, C.L.; Small, C.J.; Batterham, R.L.; Neary, N.M.; Cohen, M.A.; Patterson, M.; Ghatei, M.A.; Bloom, S.R. Peripheral Oxyntomodulin Reduces Food Intake and Body Weight Gain in Rats. Endocrinology 2004, 145, 2687–2695. [Google Scholar] [CrossRef] [Green Version]
- Baggio, L.L.; Huang, Q.; Brown, T.; Drucker, D.J. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology 2004, 127, 546–558. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Kosinski, J.R.; Lao, J.; Shen, X.; Petrov, A.; Chicchi, G.G.; Eiermann, G.J.; Pocai, A. Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. Am. J. Physiol. Metab. 2012, 303, E265–E271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosinski, J.R.; Hubert, J.; Carrington, P.E.; Chicchi, G.G.; Mu, J.; Miller, C.; Cao, J.; Bianchi, E.; Pessi, A.; Sinharoy, R.; et al. The Glucagon Receptor Is Involved in Mediating the Body Weight-Lowering Effects of Oxyntomodulin. Obesity 2012, 20, 1566–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, A.; Lovshin, J.A.; Baggio, L.L.; Drucker, D.J. The Glucagon-Like Peptide-1 Receptor Agonist Oxyntomodulin Enhances β-Cell Function but Does Not Inhibit Gastric Emptying in Mice. Endocrinology 2008, 149, 5670–5678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, S.S.; Shankar, R.R.; Mixson, L.A.; Miller, D.L.; Pramanik, B.; O’Dowd, A.K.; Williams, D.M.; Frederick, C.B.; Beals, C.R.; Stoch, S.A.; et al. Native Oxyntomodulin Has Significant Glucoregulatory Effects Independent of Weight Loss in Obese Humans With and Without Type 2 Diabetes. Diabetes 2018, 67, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Field, B.; Chaudhri, O.B.; Bloom, S.R. Bowels control brain: Gut hormones and obesity. Nat. Rev. Endocrinol. 2010, 6, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, E.; Sundler, F. Distribution of pancreatic polypeptide and peptide YY. Peptides 2002, 23, 251–261. [Google Scholar] [CrossRef]
- Schaper, S.J.; Hofmann, T.; Wölk, E.; Weibert, E.; Rose, M.; Stengel, A. Pancreatic Polypeptide but Not Other Members of the Neuropeptide Y Family Shows a Moderate Association With Perceived Anxiety in Obese Men. Front. Hum. Neurosci. 2020, 14, 578578. [Google Scholar] [CrossRef]
- Shi, Y.-C.; Lin, Z.; Lau, J.; Zhang, H.; Yagi, M.; Kanzler, I.; Sainsbury, A.; Herzog, H.; Lin, S. PYY3-36 and pancreatic polypeptide reduce food intake in an additive manner via distinct hypothalamic dependent pathways in mice. Obesity 2013, 21, E669–E678. [Google Scholar] [CrossRef] [Green Version]
- Ghamari-Langroudi, M.; Colmers, W.F.; Cone, R.D. PYY3–36 inhibits the action potential firing activity of POMC neurons of arcuate nucleus through postsynaptic Y2 receptors. Cell Metab. 2005, 2, 191–199. [Google Scholar] [CrossRef] [Green Version]
- McTigue, D.M.; Hermann, G.E.; Rogers, R.C. Effect of pancreatic polypeptide on rat dorsal vagal complex neurons. J. Physiol. 1997, 499, 475–483. [Google Scholar] [CrossRef]
- Batterham, R.; Cohen, M.A.; Ellis, S.M.; Le Roux, C.; Withers, D.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Inhibition of Food Intake in Obese Subjects by Peptide YY3–36. N. Engl. J. Med. 2003, 349, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Batterham, R.; Le Roux, C.; Cohen, M.A.; Park, A.J.; Ellis, S.M.; Patterson, M.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Pancreatic Polypeptide Reduces Appetite and Food Intake in Humans. J. Clin. Endocrinol. Metab. 2003, 88, 3989–3992. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, C.L.; Baile, C.A. Obese mice and the satiety effects of cholecystokinin, bombesin and pancreatic polypeptide. Physiol. Behav. 1981, 26, 433–437. [Google Scholar] [CrossRef]
- Marco, J.; Zulueta, M.A.; Correas, I.; Villanueva, M.L. Reduced Pancreatic Polypeptide Secretion in Obese Subjects. J. Clin. Endocrinol. Metab. 1980, 50, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Baltazi, M.; Katsiki, N.; Savopoulos, C.; Iliadis, F.; Koliakos, G.; Hatzitolios, A.I. Plasma neuropeptide Y (NPY) and alpha-melanocyte stimulating hormone (a-MSH) levels in patients with or without hypertension and/or obesity: A pilot study. Am. J. Cardiovasc. Dis. 2011, 1, 48–59. [Google Scholar]
- Butler, P.C.; Chou, J.; Carter, W.B.; Wang, Y.-N.; Bu, B.-H.; Chang, D.; Chang, J.-K.; Rizza, R.A. Effects of Meal Ingestion on Plasma Amylin Concentration in NIDDM and Nondiabetic Humans. Diabetes 1990, 39, 752–756. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Kelly, L.; Gergi, I.; Vieweg, P.; Heiman, M.; Greengard, P.; Friedman, J.M. Hypothalamic Amylin Acts in Concert with Leptin to Regulate Food Intake. Cell Metab. 2015, 22, 1059–1067. [Google Scholar] [CrossRef] [Green Version]
- Mietlicki-Baase, E.G.; Reiner, D.J.; Cone, J.; Olivos, D.R.; McGrath, L.E.; Zimmer, D.J.; Roitman, M.F.; Hayes, M.R. Amylin Modulates the Mesolimbic Dopamine System to Control Energy Balance. Neuropsychopharmacology 2015, 40, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Whiting, L.; McCutcheon, J.; Boyle, C.N.; Roitman, M.F.; Lutz, T.A. The area postrema (AP) and the parabrachial nucleus (PBN) are important sites for salmon calcitonin (sCT) to decrease evoked phasic dopamine release in the nucleus accumbens (NAc). Physiol. Behav. 2017, 176, 9–16. [Google Scholar] [CrossRef]
- Mollet, A.; Gilg, S.; Riediger, T.; Lutz, T.A. Infusion of the amylin antagonist AC 187 into the area postrema increases food intake in rats. Physiol. Behav. 2004, 81, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Riediger, T.; Schmid, H.A.; Lutz, T.; Simon, E. Amylin potently activates AP neurons possibly via formation of the excitatory second messenger cGMP. Am. J. Physiol. Integr. Comp. Physiol. 2001, 281, R1833–R1843. [Google Scholar] [CrossRef] [PubMed]
- Potes, C.S.; Boyle, C.N.; Wookey, P.J.; Riediger, T.; Lutz, T.A. Involvement of the extracellular signal-regulated kinase 1/2 signaling pathway in amylin’s eating inhibitory effect. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R340–R351. [Google Scholar] [CrossRef] [PubMed]
- Boyle, C.N.; Lutz, T.A.; Le Foll, C. Amylin—Its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol. Metab. 2018, 8, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mietlicki-Baase, E.G.; Rupprecht, L.E.; Olivos, D.R.; Zimmer, D.J.; Alter, M.D.; Pierce, R.C.; Schmidt, H.D.; Hayes, M.R. Amylin Receptor Signaling in the Ventral Tegmental Area is Physiologically Relevant for the Control of Food Intake. Neuropsychopharmacology 2013, 38, 1685–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, D.J.; Mietlicki-Baase, E.G.; Olivos, D.R.; McGrath, L.E.; Zimmer, D.J.; Koch-Laskowski, K.; Krawczyk, J.; Turner, C.A.; Noble, E.; Hahn, J.; et al. Amylin Acts in the Lateral Dorsal Tegmental Nucleus to Regulate Energy Balance Through Gamma-Aminobutyric Acid Signaling. Biol. Psychiatry 2017, 82, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.; Kumar, S.; Singh, U.; Kumar, V.; Lechan, R.M.; Singru, P.S. Cocaine- and amphetamine-regulated transcript peptide (CART) in the brain of zebra finch, Taeniopygia guttata: Organization, interaction with neuropeptide Y, and response to changes in energy status. J. Comp. Neurol. 2016, 524, 3014–3041. [Google Scholar] [CrossRef] [PubMed]
- Sathanoori, R.; Olde, B.; Erlinge, D.; Göransson, O.; Wierup, N. Cocaine- and Amphetamine-regulated Transcript (CART) Protects Beta Cells against Glucotoxicity and Increases Cell Proliferation. J. Biol. Chem. 2013, 288, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A Novel Serum Protein Similar to C1q, Produced Exclusively in Adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef] [Green Version]
- Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Adiponectin: Action, regulation and association to insulin sensitivity. Obes. Rev. 2005, 6, 13–21. [Google Scholar] [CrossRef]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhao, S.; Zhang, Z.; Zhu, Y.; Gliniak, C.M.; Vishvanath, L.; An, Y.A.; Wang, M.-Y.; Deng, Y.; Zhu, Q.; et al. Adiponectin preserves metabolic fitness during aging. eLife 2021, 10, e65108. [Google Scholar] [CrossRef] [PubMed]
- Fisman, E.Z.; Tenenbaum, A. Adiponectin: A manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc. Diabetol. 2014, 13, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Takahashi, N.; Hileman, S.M.; Patel, H.R.; Berg, A.H.; Pajvani, U.B.; Scherer, P.E.; Ahima, R.S. Adiponectin acts in the brain to decrease body weight. Nat. Med. 2004, 10, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Mcternan, P.G.; Kumar, S. Role of resistin in obesity, insulin resistance and Type II diabetes. Clin. Sci. 2005, 109, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nat. Cell Biol. 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Hivert, M.-F.; Sullivan, L.; Fox, C.S.; Nathan, D.M.; D’Agostino, R.B.; Wilson, P.W.F.; Meigs, J.B. Associations of Adiponectin, Resistin, and Tumor Necrosis Factor-α with Insulin Resistance. J. Clin. Endocrinol. Metab. 2008, 93, 3165–3172. [Google Scholar] [CrossRef]
- Zaidi, S.I.Z.; Shirwany, T.A.K. Relationship of Serum Resistin with Insulin Resistance and Obesity. J. Ayub Med. Coll. Abbottabad 2015, 27, 552–555. Available online: https://pubmed.ncbi.nlm.nih.gov/26721005/ (accessed on 4 August 2021).
- Gerber, M.; Boettner, A.; Seidel, B.; Lammert, A.; Bar, J.; Schuster, E.; Thiery, J.; Kiess, W.; Kratzsch, J. Serum Resistin Levels of Obese and Lean Children and Adolescents: Biochemical Analysis and Clinical Relevance. J. Clin. Endocrinol. Metab. 2005, 90, 4503–4509. [Google Scholar] [CrossRef] [Green Version]
- Bu, J.; Feng, Q.; Ran, J.; Li, Q.; Mei, G.; Zhang, Y. Visceral fat mass is always, but adipokines (adiponectin and resistin) are diversely associated with insulin resistance in Chinese type 2 diabetic and normoglycemic subjects. Diabetes Res. Clin. Pr. 2012, 96, 163–169. [Google Scholar] [CrossRef]
- Su, K.-Z.; Li, Y.-R.; Zhang, D.; Yuan, J.-H.; Zhang, C.-S.; Liu, Y.; Song, L.-M.; Lin, Q.; Li, M.-W.; Dong, J. Relation of Circulating Resistin to Insulin Resistance in Type 2 Diabetes and Obesity: A Systematic Review and Meta-Analysis. Front. Physiol. 2019, 10, 1399. [Google Scholar] [CrossRef] [Green Version]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef]
- Min, S.Y.; Kady, J.; Nam, M.; Rojas-Rodriguez, R.; Berkenwald, A.; Kim, J.H.; Noh, H.-L.; Kim, J.; Cooper, M.P.; Fitzgibbons, T.P.; et al. Human ’brite/beige’ adipocytes develop from capillary networks, and their implantation improves metabolic homeostasis in mice. Nat. Med. 2016, 22, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, S.; You, Y.; Meng, M.; Zheng, Z.; Dong, M.; Lin, J.; Zhao, Q.; Zhang, C.; Yuan, X.; et al. Brown Adipose Tissue Transplantation Reverses Obesity in Ob/Ob Mice. Endocrinology 2015, 156, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Ghorbani, M.; Claus, T.H.; Himms-Hagen, J. Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a β3-adrenoceptor agonist. Biochem. Pharmacol. 1997, 54, 121–131. [Google Scholar] [CrossRef]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kayahara, T.; Kameya, T.; Kawai, Y.; Iwanaga, T.; Saito, M. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 2013, 123, 3404–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-Activated Brown Adipose Tissue in Healthy Men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warwick, P.M.; Busby, R. Influence of mild cold on 24 h energy expenditure in ‘normally’ clothed adults. Br. J. Nutr. 1990, 63, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, K.; Tseng, Y.-H. Brown adipose tissue. Adipocyte 2012, 1, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arch, J.R.; Wilson, S. Prospects for beta 3-adrenoceptor agonists in the treatment of obesity and diabetes. Int. J. Obes. Relat. Metab. Disord. 1996, 20, 191–199. Available online: https://pubmed.ncbi.nlm.nih.gov/8653138/ (accessed on 14 September 2021).
- Villarroya, F.; Cereijo, R.; Villarroya, J.; Giralt, M. Brown adipose tissue as a secretory organ. Nat. Rev. Endocrinol. 2017, 13, 26–35. [Google Scholar] [CrossRef]
- Villarroya, J.; Cereijo, R.; Gavaldà-Navarro, A.; Peyrou, M.; Giralt, M.; Villarroya, F. New insights into the secretory functions of brown adipose tissue. J. Endocrinol. 2019, 243, R19–R27. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Linderman, J.D.; Smith, S.; Brychta, R.J.; Wang, J.; Idelson, C.; Perron, R.M.; Werner, C.D.; Phan, G.Q.; Kammula, U.S.; et al. Irisin and FGF21 Are Cold-Induced Endocrine Activators of Brown Fat Function in Humans. Cell Metab. 2014, 19, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Hondares, E.; Iglesias, R.; Giralt, A.; Gonzalez, F.J.; Giralt, M.; Mampel, T.; Villarroya, F. Thermogenic Activation Induces FGF21 Expression and Release in Brown Adipose Tissue. J. Biol. Chem. 2011, 286, 12983–12990. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Kleiner, S.; Douris, N.; Fox, E.C.; Mepani, R.J.; Verdeguer, F.; Wu, J.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E.; et al. FGF21 regulates PGC-1 and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012, 26, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Planavila, A.; Redondo, I.; Hondares, E.; Vinciguerra, M.; Munts, C.; Iglesias, R.; Gabrielli, L.A.; Sitges, M.; Giralt, M.; Van Bilsen, M.; et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat. Commun. 2013, 4, 2019. [Google Scholar] [CrossRef]
- Lee, P.; Brychta, R.J.; Linderman, J.; Smith, S.; Chen, K.Y.; Celi, F.S. Mild cold exposure modulates fibroblast growth factor 21 (FGF21) diurnal rhythm in humans: Relationship between FGF21 levels, lipolysis, and cold-induced thermogenesis. J. Clin. Endocrinol. Metab. 2013, 98, E98–E102. [Google Scholar] [CrossRef] [Green Version]
- Cereijo, R.; Gavaldà-Navarro, A.; Cairó, M.; López, T.P.Q.; Villarroya, J.; Morón-Ros, S.; Sánchez-Infantes, D.; Peyrou, M.; Iglesias, R.; Mampel, T.; et al. CXCL14, a Brown Adipokine that Mediates Brown-Fat-to-Macrophage Communication in Thermogenic Adaptation. Cell Metab. 2018, 28, 750–763.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.-H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Whittle, A.J.; Carobbio, S.; Martins, L.; Slawik, M.; Hondares, E.; Vázquez, M.J.; Morgan, D.; Csikasz, R.I.; Gallego, R.; Rodriguez-Cuenca, S.; et al. BMP8B Increases Brown Adipose Tissue Thermogenesis through Both Central and Peripheral Actions. Cell 2012, 149, 871–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, S.; Straub, L.; Balaz, M.; Sun, W.; Varga, L.; Stefanicka, P.; Profant, M.; Simon, E.; Neubauer, H.; Ukropcova, B.; et al. Bmp4 Promotes a Brown to White-like Adipocyte Shift. Cell Rep. 2016, 16, 2243–2258. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.R.; Long, J.Z.; White, J.P.; Svensson, K.J.; Lou, J.; Lokurkar, I.; Jedrychowski, M.P.; Ruas, J.; Wrann, C.D.; Lo, J.C.; et al. Meteorin-like Is a Hormone that Regulates Immune-Adipose Interactions to Increase Beige Fat Thermogenesis. Cell 2014, 157, 1279–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralisch, S.; Hoffmann, A.; Kratzsch, J.; Blüher, M.; Stumvoll, M.; Fasshauer, M.; Ebert, T. The brown-fat-secreted adipokine neuregulin 4 is decreased in gestational diabetes mellitus. Diabetes Metab. 2017, 44, 150–154. [Google Scholar] [CrossRef]
- Chiu-Tsao, S.-T.; Ho, Y.; Shankar, R.; Wang, L.; Harrison, L.B. Energy dependence of response of new high sensitivity radiochromic films for megavoltage and kilovoltage radiation energies. Med. Phys. 2005, 32, 3350–3354. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, F.; Arroyave, F. Control of Adipose Cell Browning and Its Therapeutic Potential. Metabolites 2020, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.; Jokinen, R.; Rissanen, A.; Pietiläinen, K.H. White adipose tissue mitochondrial metabolism in health and in obesity. Obes. Rev. 2020, 21, e12958. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nat. Cell Biol. 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.N.; Heilbronn, L.H.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N.L.-M. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Jankovic, A.; Korac, A.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Daiber, A.; Korac, B. Redox implications in adipose tissue (dys)function—A new look at old acquaintances. Redox Biol. 2015, 6, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef] [PubMed]
- Desogus, D.; Menon, V.; Singhal, R.; Oyebode, O. An Examination of Who Is Eligible and Who Is Receiving Bariatric Surgery in England: Secondary Analysis of the Health Survey for England Dataset. Obes. Surg. 2019, 29, 3246–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.; Sadeghirad, B.; Ball, G.D.C.; Da Costa, B.R.; Hitchcock, C.L.; Svendrovski, A.; Kiflen, R.; Quadri, K.; Kwon, H.Y.; Karamouzian, M.; et al. Comparison of dietary macronutrient patterns of 14 popular named dietary programmes for weight and cardiovascular risk factor reduction in adults: Systematic review and network meta-analysis of randomised trials. BMJ 2020, 369, m696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hvizdos, K.M.; Markham, A. Orlistat. Drugs 1999, 58, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Singh, R. Pharmacotherapy in obesity: A systematic review and meta-analysis of randomized controlled trials of anti-obesity drugs. Expert Rev. Clin. Pharmacol. 2020, 13, 53–64. [Google Scholar] [CrossRef]
- Torgerson, J.S.; Hauptman, J.; Boldrin, M.N.; Sjöström, L. XENical in the Prevention of Diabetes in Obese Subjects (XENDOS) Study: A randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2003, 27, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Muls, E.; Kolanowski, J.; Scheen, A.; Van Gaal, L. The effects of orlistat on weight and on serum lipids in obese patients with hypercholesterolemia: A randomized, double-blind, placebo-controlled, multicentre study. Int. J. Obes. 2001, 25, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, G.; Apovian, C.M. Current pharmacotherapy for obesity. Nat. Rev. Endocrinol. 2018, 14, 12–24. [Google Scholar] [CrossRef]
- Sherman, M.M.; Ungureanu, S.; Rey, J.A. Naltrexone/bupropion ER (Contrave): Newly approved treatment option for chronic weight management in obese adults. Pharm. Ther. 2016, 41, 164. [Google Scholar]
- Hollander, P.; Gupta, A.K.; Plodkowski, R.; Greenway, F.; Bays, H.; Burns, C.; Klassen, P.; Fujioka, K.; COR-Diabetes Study Group. Effects of Naltrexone Sustained- Release/Bupropion Sustained-Release Combination Therapy on Body Weight and Glycemic Parameters in Overweight and Obese Patients With Type 2 Diabetes. Diabetes Care 2013, 36, 4022–4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apovian, C.M.; Aronne, L.; Rubino, D.; Still, C.; Wyatt, H.; Burns, C.; Kim, D.; Dunayevich, E.; COR-II Study Group. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity 2013, 21, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Wadden, T.A.; Foreyt, J.P.; Foster, G.D.; Hill, J.O.; Klein, S.; O’Neil, P.; Perri, M.; Pi-Sunyer, F.X.; Rock, C.L.; Erickson, J.S.; et al. Weight Loss With Naltrexone SR/Bupropion SR Combination Therapy as an Adjunct to Behavior Modification: The COR-BMOD Trial. Obesity 2011, 19, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Greenway, F.L.; Fujioka, K.; Plodkowski, R.A.; Mudaliar, S.; Guttadauria, M.; Erickson, J.; Kim, D.D.; Dunayevich, E. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2010, 376, 595–605. [Google Scholar] [CrossRef]
- Calderone, A.; Calabro, P.F.; Lippi, C.; Jaccheri, R.; Vitti, J.; Santini, F. Psychopathological Behaviour and Cognition in Morbid Obesity. Recent Pat. Endocr. Metab. Immune Drug Discov. 2017, 10, 112–118. [Google Scholar] [CrossRef]
- CHMP. Committee for Medicinal Products for Human Use (CHMP) Assessment Report—Qsiva. 2013. Available online: www.ema.europa.eu (accessed on 6 August 2021).
- Antel, J.; Hebebrand, J. Weight-Reducing Side Effects of the Antiepileptic Agents Topiramate and Zonisamide. Sphingolipids Dis. 2012, 209, 433–466. [Google Scholar] [CrossRef]
- Gadde, K.M.; Allison, D.; Ryan, D.; Peterson, C.A.; Troupin, B.; Schwiers, M.L.; Day, W.W. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 1341–1352. [Google Scholar] [CrossRef]
- Allison, D.B.; Gadde, K.M.; Garvey, W.T.; Peterson, C.A.; Schwiers, M.L.; Najarian, T.; Tam, P.Y.; Troupin, B.; Day, W.W. Controlled-Release Phentermine/Topiramate in Severely Obese Adults: A Randomized Controlled Trial (EQUIP). Obesity 2012, 20, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Garvey, W.T.; Ryan, D.; Look, M.; Gadde, K.M.; Allison, D.; Peterson, C.A.; Schwiers, M.; Day, W.W.; Bowden, C.H. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): A randomized, placebo-controlled, phase 3 extension study. Am. J. Clin. Nutr. 2011, 95, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelstein, E.A.; Kruger, E.; Karnawat, S. Cost-Effectiveness Analysis of Qsymia for Weight Loss. Pharmacoeconomics 2014, 33, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grilo, C.M.; Reas, D.L.; Mitchell, J.E. Combining Pharmacological and Psychological Treatments for Binge Eating Disorder: Current Status, Limitations, and Future Directions. Curr. Psychiatry Rep. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Stanford, F.C.; Alfaris, N.; Gomez, G.; Ricks, E.; Shukla, A.P.; Corey, K.E.; Pratt, J.S.; Pomp, A.; Rubino, F.; Aronne, L.J. The utility of weight loss medications after bariatric surgery for weight regain or inadequate weight loss: A multi-center study. Surg. Obes. Relat. Dis. 2017, 13, 491–500. [Google Scholar] [CrossRef]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.L.; Halpern, A.; Krempf, M.; Lau, D.C.; Le Roux, C.; Ortiz, R.V.; Jensen, C.B.; et al. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef]
- Davies, M.J.; Bergenstal, R.; Bode, B.; Kushner, R.F.; Lewin, A.; Skjøth, T.V.; Andreasen, A.H.; Jensen, C.B.; DeFronzo, R.A. Efficacy of Liraglutide for Weight Loss Among Patients With Type 2 Diabetes. JAMA 2015, 314, 687–699. [Google Scholar] [CrossRef] [Green Version]
- le Roux, C.W.; Astrup, A.; Fujioka, K.; Greenway, F.; Lau, D.C.W.; Van Gaal, L.; Ortiz, R.V.; Wilding, J.P.H.; Skjøth, T.V.; Manning, L.S.; et al. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: A randomised, double-blind trial. Lancet 2017, 389, 1399–1409. [Google Scholar] [CrossRef] [Green Version]
- Wadden, T.A.; Hollander, P.; Klein, S.; Niswender, K.; Woo, V.; Hale, P.M.; Aronne, L. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: The SCALE Maintenance randomized study. Int. J. Obes. 2013, 37, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, M. Thyroid safety in patients treated with liraglutide. J. Endocrinol. Investig. 2013, 36, 140–145. [Google Scholar] [CrossRef]
- Funch, D.; Gydesen, H.; Tornøe, K.; Major-Pedersen, A.; Chan, K. A prospective, claims-based assessment of the risk of pancreatitis and pancreatic cancer with liraglutide compared to other antidiabetic drugs. Diabetes, Obes. Metab. 2013, 16, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Chai, S.; Zhao, X.; Ji, L. Risk of Malignant Neoplasia with Glucagon-Like Peptide-1 Receptor Agonist Treatment in Patients with Type 2 Diabetes: A Meta-Analysis. J. Diabetes Res. 2019, 2019, 1534365. [Google Scholar] [CrossRef]
- FDA Approves First Treatment for Weight Management for People with Certain Rare Genetic Conditions|FDA. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-first-treatment-weight-management-people-certain-rare-genetic-conditions (accessed on 6 August 2021).
- EMA. Imcivree|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/imcivree (accessed on 6 August 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Collet, T.-H.; Dubern, B.; Mokrosinski, J.; Connors, H.; Keogh, J.M.; de Oliveira, E.M.; Henning, E.; Poitou-Bernert, C.; Oppert, J.-M.; Tounian, P.; et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. 2017, 6, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Setmelanotide (RM-493), Melanocortin-4 Receptor (MC4R) Agonist, in Bardet-Biedl Syndrome (BBS) and Alström Syndrome (AS) Patients with Moderate to Severe Obesity—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03746522 (accessed on 6 August 2021).
- Long Term Extension Trial of Setmelanotide—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03651765 (accessed on 6 August 2021).
- Rasmussen, M.F. The development of oral semaglutide, an oral GLP-1 analog, for the treatment of type 2 diabetes. Diabetol. Int. 2020, 11, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Prescription. Indications and Usage for Durlaza. 2013. Available online: www.fda.gov/medwatch (accessed on 5 August 2021).
- O’Neil, P.; Smith, S.R.; Weissman, N.J.; Fidler, M.C.; Sanchez, M.; Zhang, J.; Raether, B.; Anderson, C.M.; Shanahan, W.R. Randomized Placebo-Controlled Clinical Trial of Lorcaserin for Weight Loss in Type 2 Diabetes Mellitus: The BLOOM-DM Study. Obesity 2012, 20, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.R.; Weissman, N.J.; Anderson, C.M.; Sanchez, M.; Chuang, E.; Stubbe, S.; Bays, H.; Shanahan, W.R. Multicenter, Placebo-Controlled Trial of Lorcaserin for Weight Management. N. Engl. J. Med. 2010, 363, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, M.C.; Sanchez, M.; Raether, B.; Weissman, N.J.; Smith, S.R.; Shanahan, W.R.; Anderson, C.M. A One-Year Randomized Trial of Lorcaserin for Weight Loss in Obese and Overweight Adults: The BLOSSOM Trial. J. Clin. Endocrinol. Metab. 2011, 96, 3067–3077. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.W.Y.; He, Y.; Chui, C.S.L.; Wong, A.Y.S.; Lau, W.; Wong, I.C.K. Efficacy and safety of lorcaserin in obese adults: A meta-analysis of 1-year randomized controlled trials (RCTs) and narrative review on short-term RCTs. Obes. Rev. 2013, 14, 383–392. [Google Scholar] [CrossRef]
- Cowie, M.R.; Fisher, M. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- Himsworth, H.P. The relation of glycosuria to glycaemia and the determination of the renal threshold for glucose. Biochem. J. 1931, 25, 1128–1146. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of Human Sodium Glucose Transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [Green Version]
- Van Bommel, E.J.; Muskiet, M.; Tonneijck, L.; Kramer, M.H.; Nieuwdorp, M.; Van Raalte, D.H. SGLT2 Inhibition in the Diabetic Kidney—From Mechanisms to Clinical Outcome. Clin. J. Am. Soc. Nephrol. 2017, 12, 700–710. [Google Scholar] [CrossRef]
- Nelinson, D.S.; Sosa, J.M.; Chilton, R.J. SGLT2 inhibitors: A narrative review of efficacy and safety. J. Osteopat. Med. 2021, 121, 229–239. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- OECD. Obesity Update 2017. Diabetologe 2017, 13, 331–341. Available online: www.oecd.org/health/obesity-update.htm (accessed on 12 August 2021).
- Katzmann, J.L.; Mason, A.M.; März, W.; Kleber, M.E.; Niessner, A.; Blüher, M.; Speer, T.; Laufs, U. Genetic Variation in Sodium-glucose Cotransporter 2 and Heart Failure. Clin. Pharmacol. Ther. 2021, 110, 149–158. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Rocca, H.-P.B.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Frías, J.P.; Davies, M.J.; Rosenstock, J.; Manghi, F.C.P.; Landó, L.F.; Bergman, B.K.; Liu, B.; Cui, X.; Brown, K. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Nahra, R.; Wang, T.; Gadde, K.M.; Oscarsson, J.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Effects of Cotadutide on Metabolic and Hepatic Parameters in Adults With Overweight or Obesity and Type 2 Diabetes: A 54-Week Randomized Phase 2b Study. Diabetes Care 2021, 44, 1433–1442. [Google Scholar] [CrossRef]
- Sánchez-Garrido, M.A.; Brandt, S.J.; Clemmensen, C.; Müller, T.D.; DiMarchi, R.D.; Tschöp, M.H. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 2017, 60, 1851–1861. [Google Scholar] [CrossRef]
- Brandt, S.J.; Kleinert, M.; Tschöp, M.; Müller, T. Are peptide conjugates the golden therapy against obesity? J. Endocrinol. 2018, 238, R109–R119. [Google Scholar] [CrossRef]
- Chapman, I.; Parker, B.; Doran, S.; Feinle-Bisset, C.; Wishart, J.; Strobel, S.; Wang, Y.; Burns, C.; Lush, C.; Weyer, C.; et al. Effect of pramlintide on satiety and food intake in obese subjects and subjects with type 2 diabetes. Diabetologia 2005, 48, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymsfield, S.B.; Greenberg, A.S.; Fujioka, K.; Dixon, R.M.; Kushner, R.; Hunt, T.; Lubina, J.A.; Patane, J.; Self, B.; Hunt, P.; et al. Recombinant Leptin for Weight Loss in Obese and Lean Adults. JAMA 1999, 282, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla, E.P.; Iwasaki, S.; Moss, J.A.; Chang, J.; Otsuji, J.; Inoue, K.; Meijler, M.M.; Janda, K.D. Vaccination against weight gain. Proc. Natl. Acad. Sci. USA 2006, 103, 13226–13231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esler, W.P.; Rudolph, J.; Claus, T.H.; Tang, W.; Barucci, N.; Brown, S.-E.; Bullock, W.; Daly, M.; DeCarr, L.; Li, Y.; et al. Small-Molecule Ghrelin Receptor Antagonists Improve Glucose Tolerance, Suppress Appetite, and Promote Weight Loss. Endocrinology 2007, 148, 5175–5185. [Google Scholar] [CrossRef] [Green Version]
- Zjacic-Rotkvic, V.; Altabas, V. Anti-ghrelin antibodies in appetite suppression: Recent advances in obesity pharmacotherapy. ImmunoTargets Ther. 2015, 4, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erondu, N.; Gantz, I.; Musser, B.; Suryawanshi, S.; Mallick, M.; Addy, C.; Cote, J.; Bray, G.; Fujioka, K.; Bays, H.; et al. Neuropeptide Y5 receptor antagonism does not induce clinically meaningful weight loss in overweight and obese adults. Cell Metab. 2006, 4, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Després, J.-P.; Golay, A.; Sjöström, L. Effects of Rimonabant on Metabolic Risk Factors in Overweight Patients with Dyslipidemia. N. Engl. J. Med. 2005, 353, 2121–2134. [Google Scholar] [CrossRef] [Green Version]
- Sam, A.H.; Salem, V.; Ghatei, M.A. Rimonabant: From RIO to Ban. J. Obes. 2011, 2011, 432607. [Google Scholar] [CrossRef]
- Abdali, D.; Samson, S.E.; Grover, A.K. How Effective Are Antioxidant Supplements in Obesity and Diabetes? Med Princ. Pr. 2015, 24, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ojeda, F.J.; Olza, J.; Gil, Á.; Aguilera, C.M. Oxidative Stress and Inflammation in Obesity and Metabolic Syndrome. In Obesity; Academic Press: Cambridge, MA, USA, 2018; pp. 1–15. [Google Scholar]
- Hosseini, B.; Saedisomeolia, A.; Allman-Farinelli, M. Association between Antioxidant Intake/Status and Obesity: A Systematic Review of Observational Studies. Biol. Trace Elem. Res. 2017, 175, 287–297. [Google Scholar] [CrossRef]
- Tun, S.; Spainhower, C.J.; Cottrill, C.L.; Lakhani, H.V.; Pillai, S.S.; Dilip, A.; Chaudhry, H.; Shapiro, J.I.; Sodhi, K. Therapeutic Efficacy of Antioxidants in Ameliorating Obesity Phenotype and Associated Comorbidities. Front. Pharmacol. 2020, 11, 1234. [Google Scholar] [CrossRef]
- World Health Organization. Controlling the Global Obesity Epidemic. 2021. Available online: http://www.who.int/nutrition/topics/obesity/en/ (accessed on 22 August 2021).
- Kleinert, M.; Clemmensen, C.; Hofmann, S.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Duc, D.; Lin, C.-C.; Popkova, Y.; Yang, Z.; Akhil, V.; Çakir, M.V.; Grunewald, S.; Simon, J.-C.; Dietz, A.; Dannenberger, D.; et al. Reduced lipolysis in lipoma phenocopies lipid accumulation in obesity. Int. J. Obes. 2021, 45, 565–576. [Google Scholar] [CrossRef]
- Kässner, F.; Kirstein, A.; Händel, N.; Schmid, G.L.; Landgraf, K.; Berthold, A.; Tannert, A.; Schaefer, M.; Wabitsch, M.; Kiess, W.; et al. A new human adipocyte model with PTEN haploinsufficiency. Adipocyte 2020, 9, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ojeda, F.J.; Rupérez, A.I.; Gomez-Llorente, C.; Gil, A.; Aguilera, C.M. Cell Models and Their Application for Studying Adipogenic Differentiation in Relation to Obesity: A Review. Int. J. Mol. Sci. 2016, 17, 1040. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug | Mean Weight Loss at ≥1 year, Placebo-Subtracted | Side Effects | Precaution |
|---|---|---|---|
| Phentermine/topiramate | 9.8 kg | insomnia, dizziness, paresthesia, depression, anxiety, memory problems | abrupt withdrawal of topiramate increases the risk of seizures |
| Naltrexone/bupropion | 4.4 kg | nausea, constipation, headaches, vomiting, dizziness, dry mouth | not recommended for patients with seizures, drug addiction, bulimia, anorexia nervosa or in combination with opiates |
| Liraglutide | 5.3–5.9 kg | nausea, diarrhoea, constipation, vomiting, dyspepsia, abdominal pain | contraindicated in patients with a family or personal history of medullary thyroid carcinoma or with MEN2 syndrome (rats and mice developed thyroid C-cell carcinomas; unclear implication for humans) |
| Semaglutide | 6.6–15.8 kg | nausea, diarrhea, vomiting, constipation, abdominal pain, headache, fatigue, dyspepsia, dizziness, hypoglycemia for diabetic patients, flatulence, gastroenteritis | potential risk of thyroid C-cell tumors. It is contraindicated for patients with a personal or family history of medullary thyroid carcinoma or MEN2. |
| Orlistat | 3.1 kg | vitamin deficiency, steatorrhea, fecal urgency, fecal incontinence | daily multivitamin intake is recommended because of malabsorption of fat-soluble vitamins |
| Setmelanotide | 2.6 kg in MC4R deficiency, 51 and 20.5 kg in 2 patients with POMC deficiency | injection site reactions, skin hyperpigmentation, headache, nausea, diarrhea, abdominal pain | approved for monogenic forms of obesity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gjermeni, E.; Kirstein, A.S.; Kolbig, F.; Kirchhof, M.; Bundalian, L.; Katzmann, J.L.; Laufs, U.; Blüher, M.; Garten, A.; Le Duc, D. Obesity–An Update on the Basic Pathophysiology and Review of Recent Therapeutic Advances. Biomolecules 2021, 11, 1426. https://doi.org/10.3390/biom11101426
Gjermeni E, Kirstein AS, Kolbig F, Kirchhof M, Bundalian L, Katzmann JL, Laufs U, Blüher M, Garten A, Le Duc D. Obesity–An Update on the Basic Pathophysiology and Review of Recent Therapeutic Advances. Biomolecules. 2021; 11(10):1426. https://doi.org/10.3390/biom11101426
Chicago/Turabian StyleGjermeni, Erind, Anna S. Kirstein, Florentien Kolbig, Michael Kirchhof, Linnaeus Bundalian, Julius L. Katzmann, Ulrich Laufs, Matthias Blüher, Antje Garten, and Diana Le Duc. 2021. "Obesity–An Update on the Basic Pathophysiology and Review of Recent Therapeutic Advances" Biomolecules 11, no. 10: 1426. https://doi.org/10.3390/biom11101426