
Kinetic Analysis of the Interaction of Nicking Endonuclease BspD6I with DNA

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
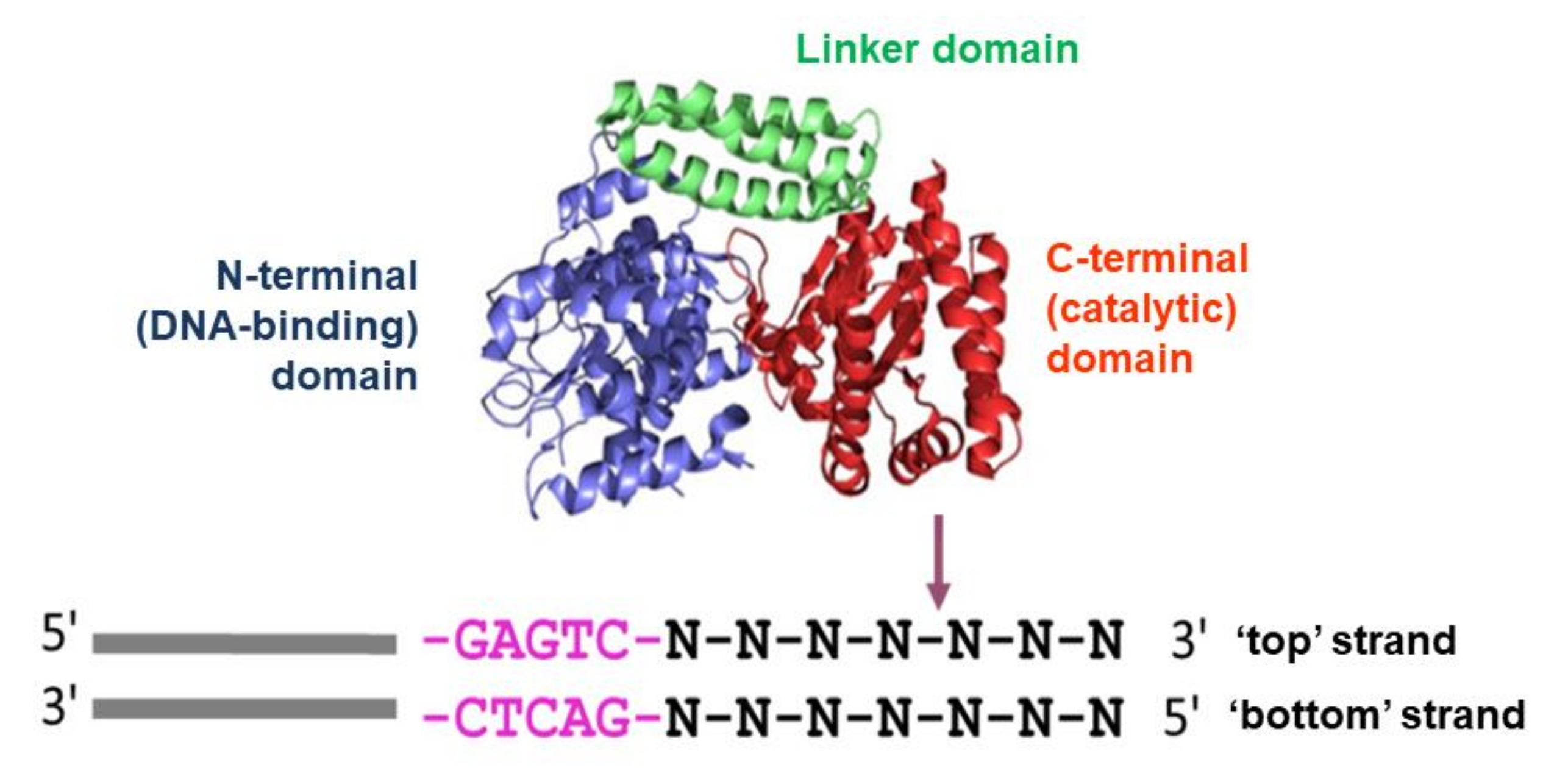
2.1. Proteins and DNA Fragments
2.2. Nicking of DNA Duplex VI-30 by Nt.BspD6I in the Presence of Divalent Metal Ions
2.3. DNA Duplex II-19A Cleavage by Nt.BspD6I for an Analysis of Steady-State Kinetics of the Reaction
2.4. Pre–Steady-State Kinetics of the Interaction of Nt.BspD6I with a DNA Duplex
2.5. Quantitative Analysis of the Results of the Kinetic Experiments Conducted under Pre–Steady-State Conditions
3. Results and Discussion
3.1. Evaluation of the Methods for Assaying the Kinetics of the Nt.BspD6I Interaction with DNA
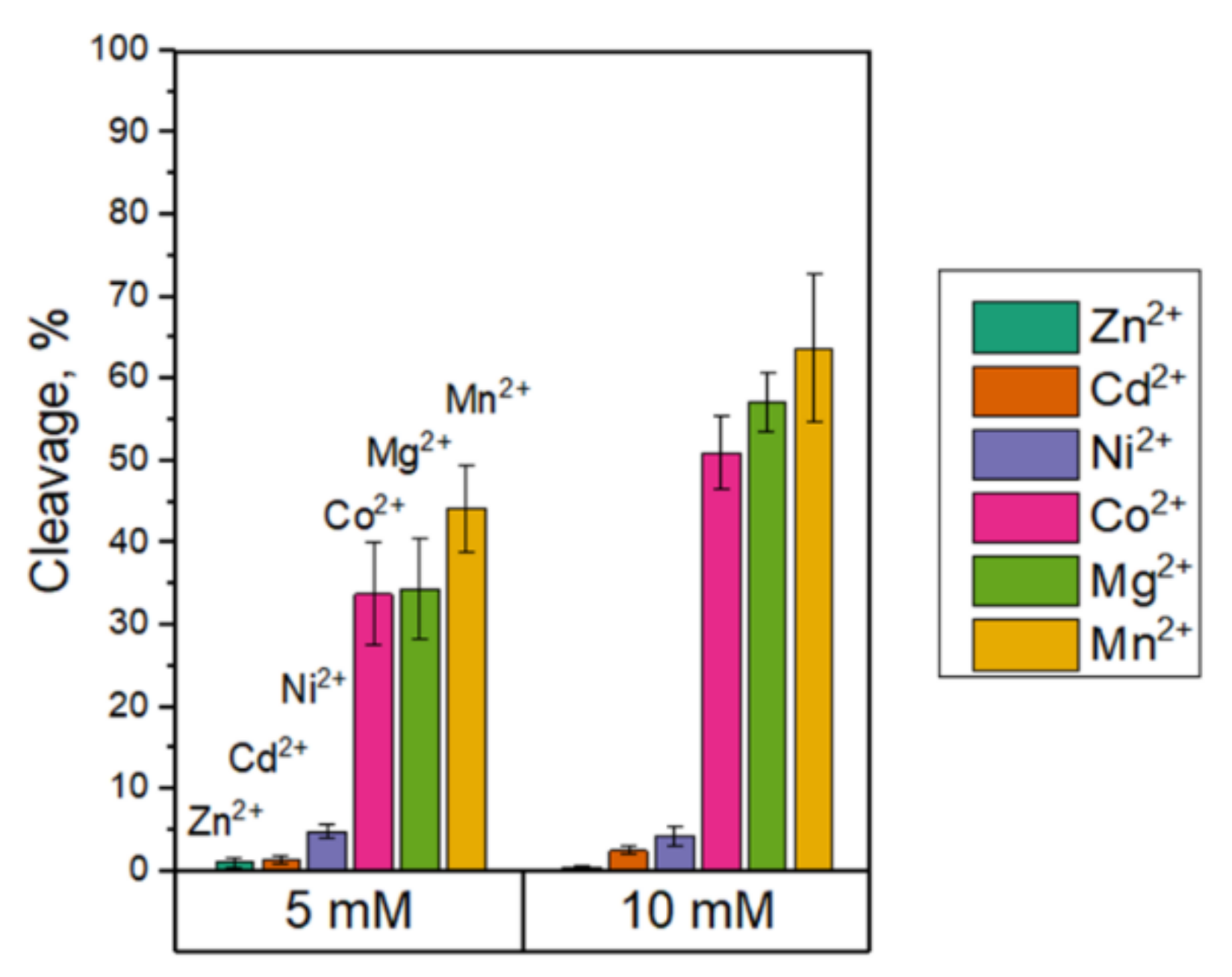
3.2. The Influence of Divalent Metal Cations on the Nt.BspD6I Interaction with DNA
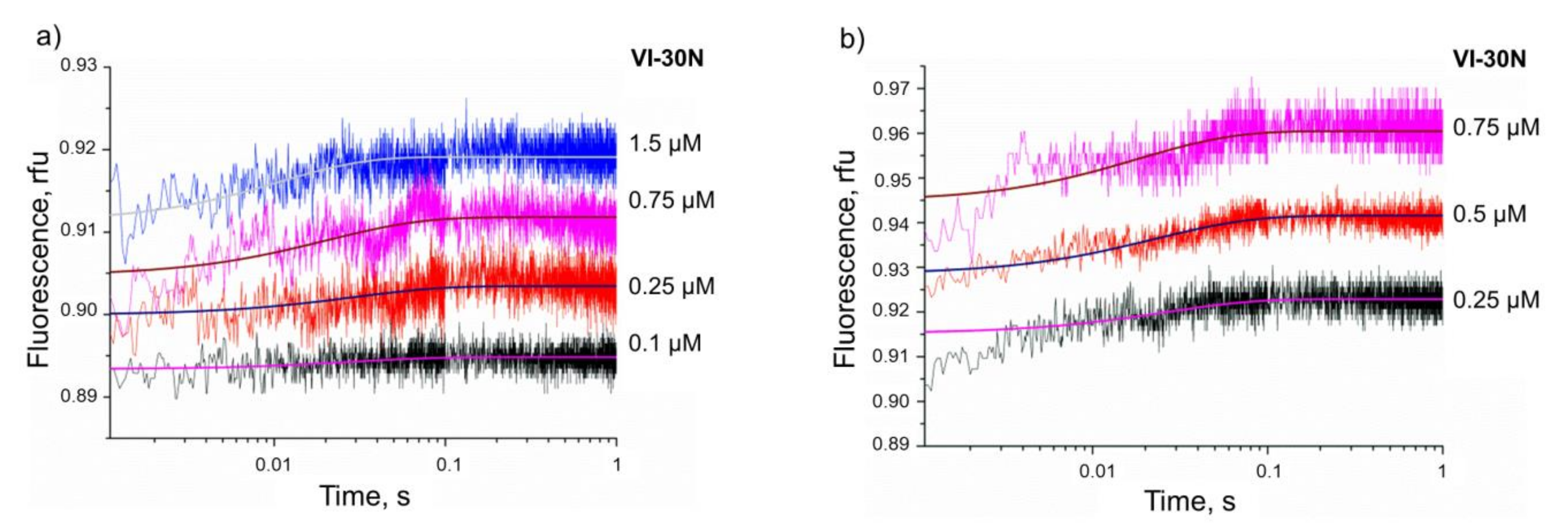
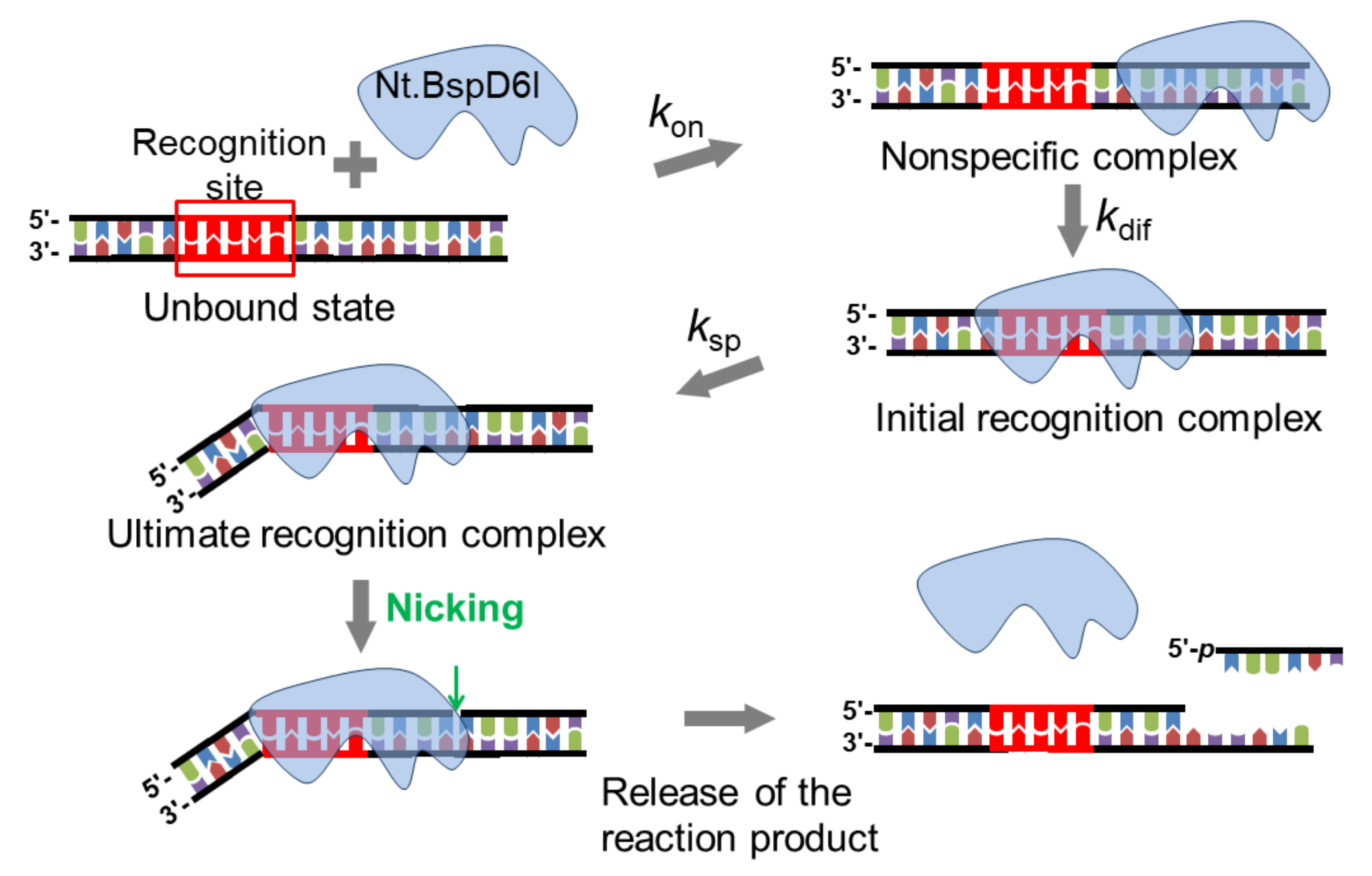
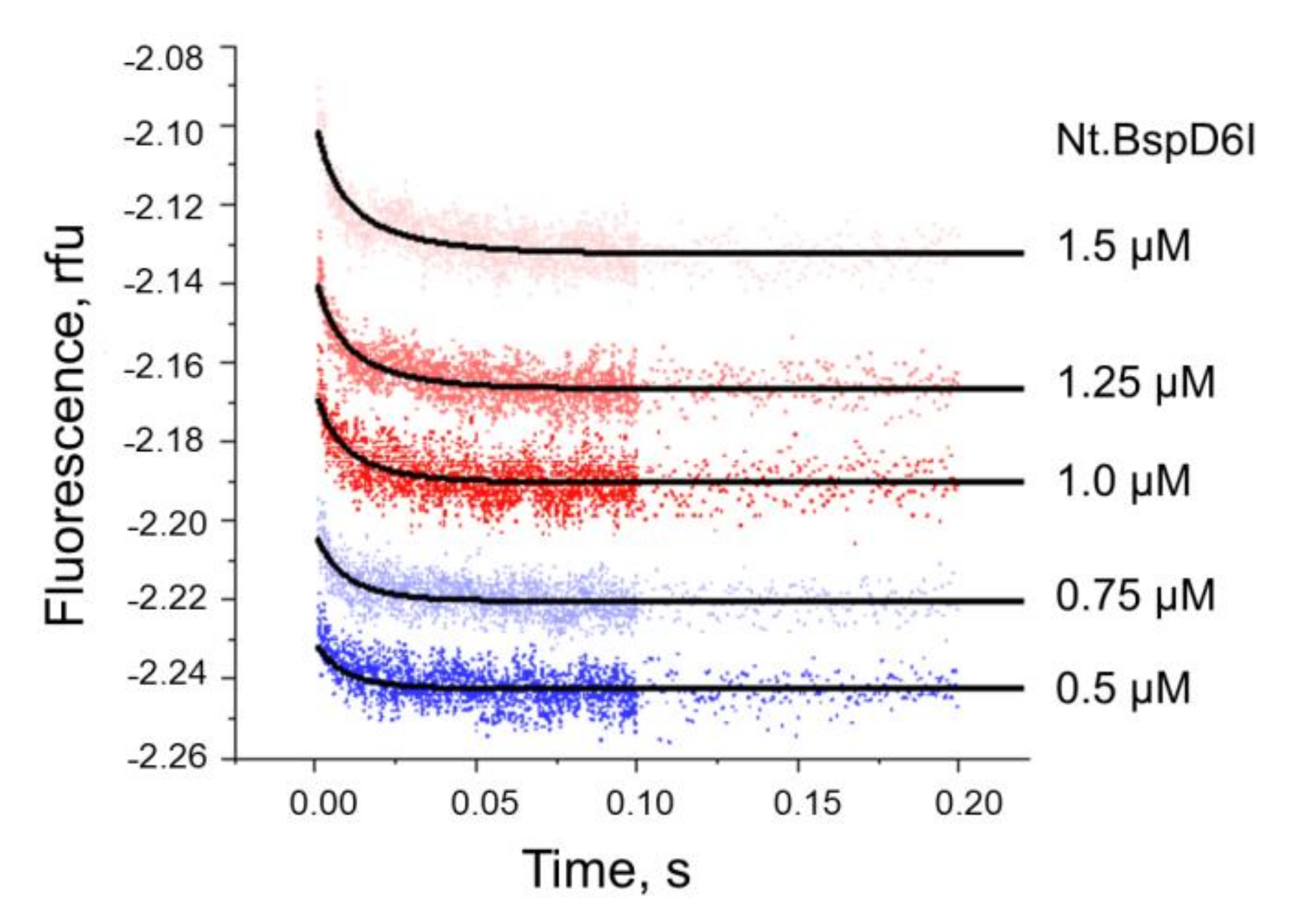
3.3. Kinetics of Non-Specific Nt.BspD6I Binding to DNA
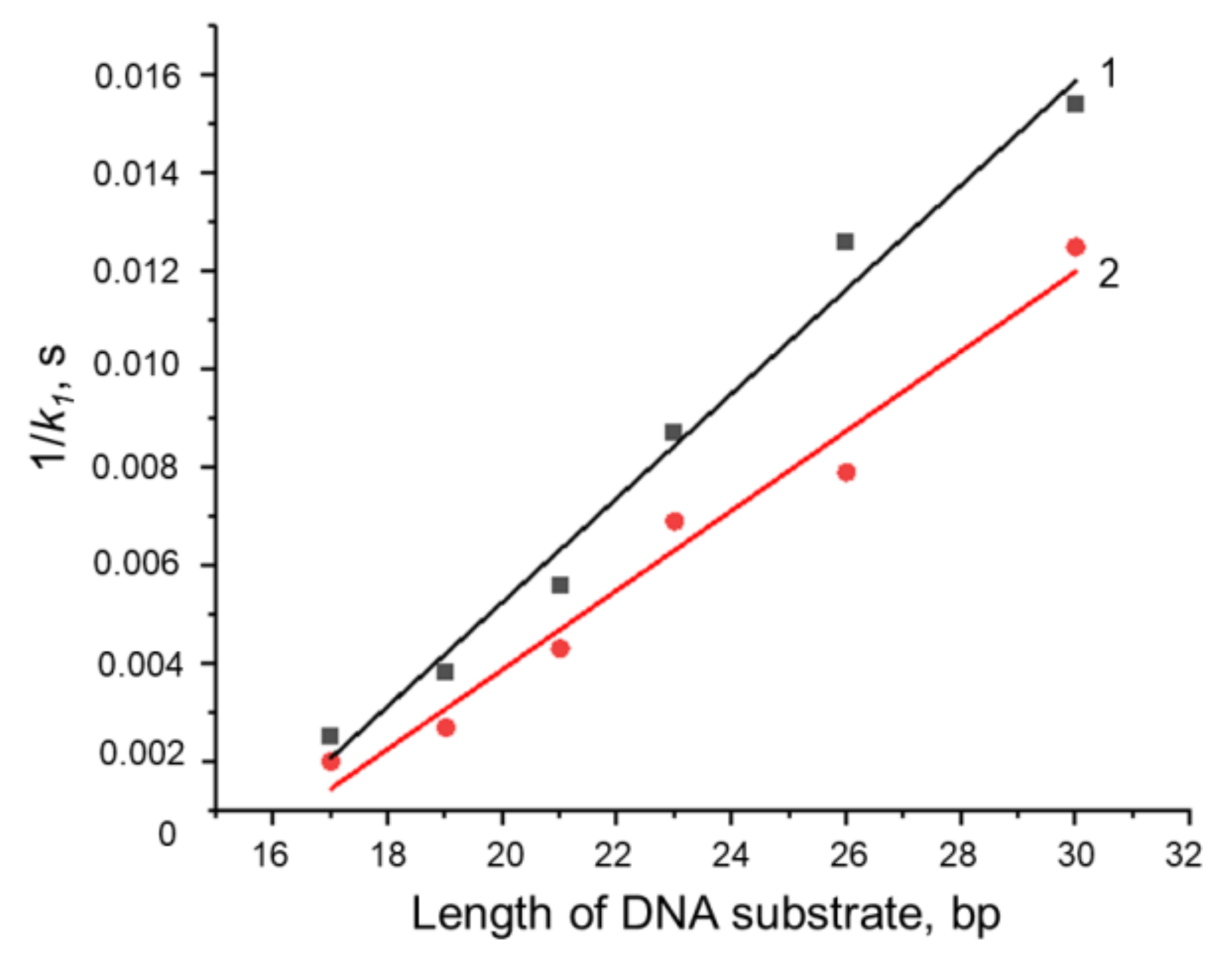
3.4. Formation of a Complex between Nt.BspD6I and DNA Substrates of Different Lengths
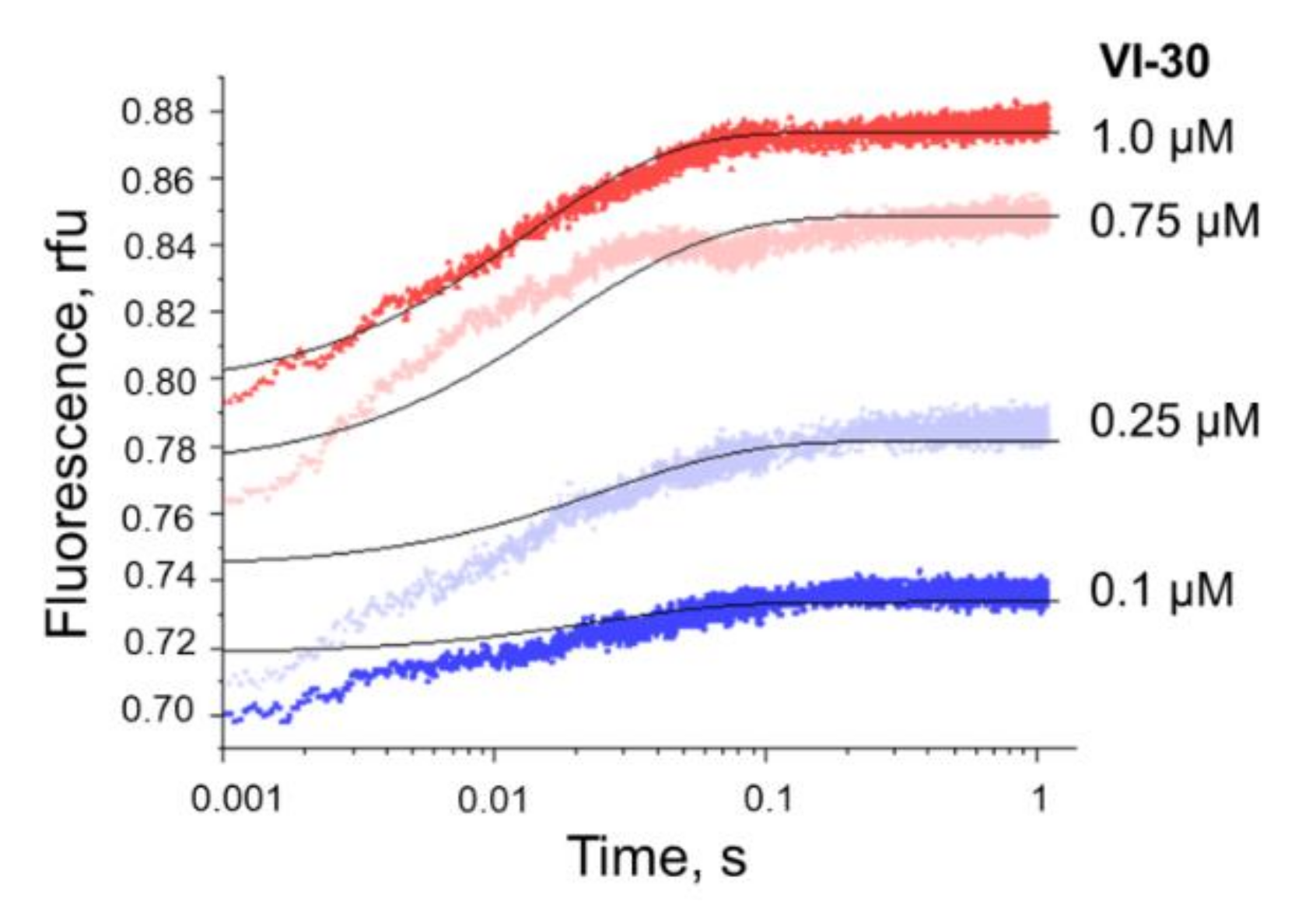
3.5. Kinetics of Specific Interaction of Nt.BspD6I with a DNA Substrate
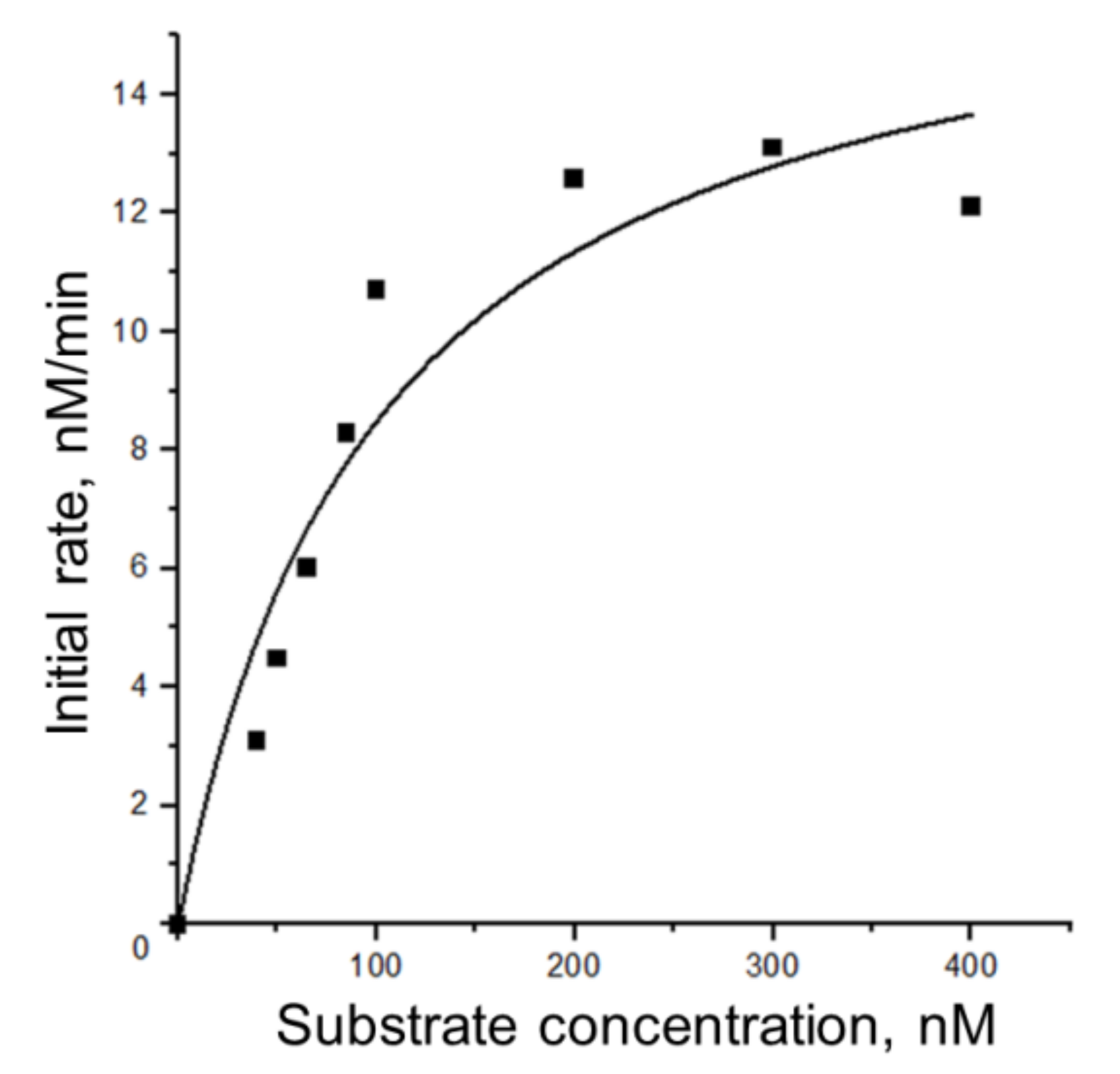
3.5.1. Steady-State Kinetics of Nt.BspD6I Interaction with DNA
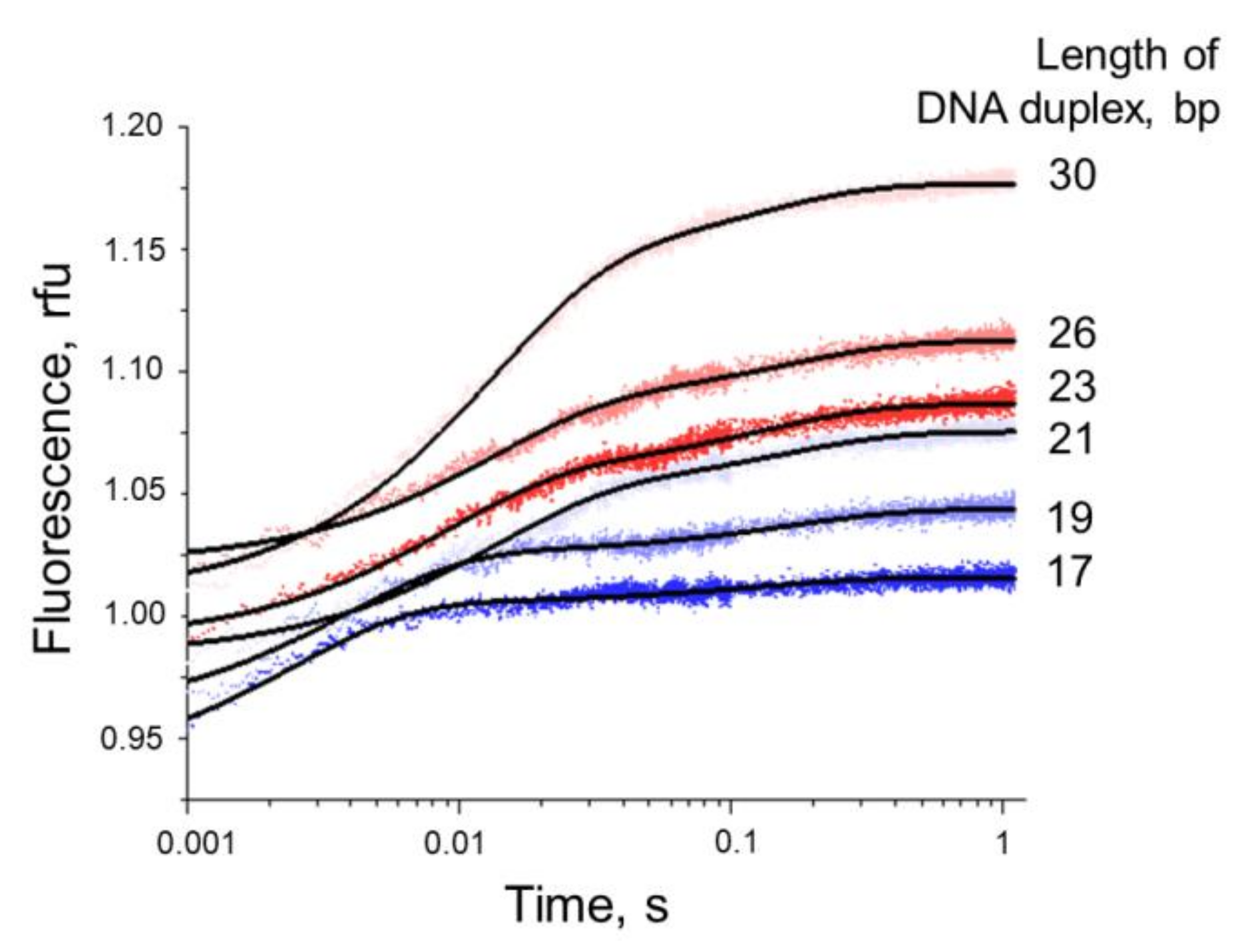
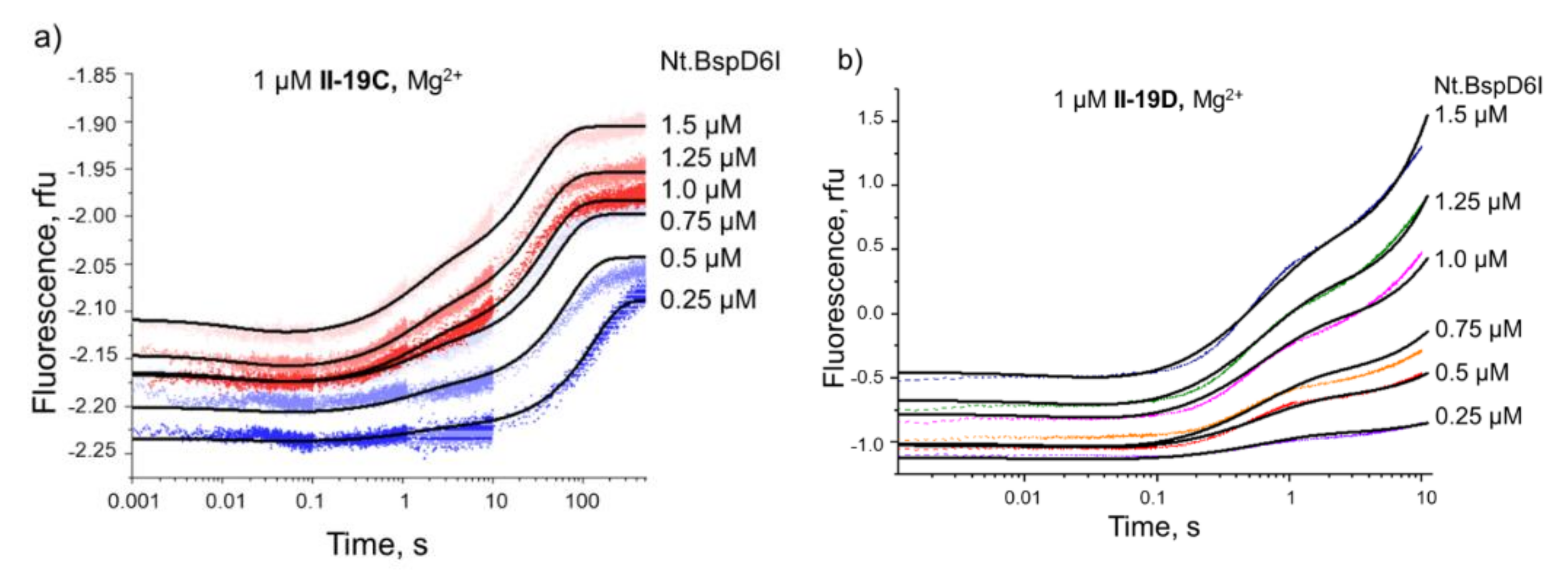
3.5.2. Conformational Dynamics of DNA Substrates during Interaction with Nt.BspD6I
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Abdurashitov, M.A.; Belichenko, O.A.; Shevchenko, A.V.; Degtyarev, S.K.N. BstSE, site-specific nickase from Bacillus stearothermophilus SE-589. Mol. Biol. 1996, 30, 754–758. [Google Scholar]
- Zheleznaya, L.A.; Kachalova, G.S.; Artyukh, R.I.; Yunusova, A.K.; Perevyazova, T.A.; Matvienko, N.I. Nicking endonucleases. Biochemistry 2009, 74, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Abrosimova, L.A.; Kisil, O.V.; Romanova, E.A.; Oretskaya, T.S.; Kubareva, E.A. Nicking Endonucleases as unique tools for biotechnology and gene engineering. Russ. J. Bioorg. Chem. 2019, 45, 303–320. [Google Scholar] [CrossRef]
- Demple, B.; Sung, J.-S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Matveeva, A.G.; Milov, A.D.; Vorobjev, Y.N.; Dzuba, S.A.; Fedorova, O.S.; Kuznetsov, N.A. Substrate specificity of human apurinic/apyrimidinic endonuclease APE1 in the nucleotide incision repair pathway. Nucleic Acids Res. 2018, 46, 11454–11465. [Google Scholar] [CrossRef] [Green Version]
- Zheleznaya, L.A.; Perevyazova, T.A.; Alzhanova, D.V.; Matvienko, N.I. Site-specific nickase from Bacillus species strain D6. Biochemistry 2001, 66, 989–993. [Google Scholar] [CrossRef]
- Yunusova, A.K.; Rogulin, E.A.; Artyukh, R.I.; Zheleznaya, L.A.; Matvienko, N.I. Nickase and a protein encoded by an open reading frame downstream from the nickase BspD6I gene form a restriction endonuclease complex. Biochemistry 2006, 71, 815–820. [Google Scholar] [CrossRef]
- Kachalova, G.; Rogulin, E.; Yunusova, A.; Artyukh, R.; Perevyazova, T.; Matvienko, N. Structural analysis of the heterodimeric type IIS restriction endonuclease R.BspD6I acting as a complex between a monomeric site-specific nickase and a catalytic subunit. J. Mol. Biol. 2008, 384, 489–502. [Google Scholar] [CrossRef]
- Machulin, A.V.; Deryusheva, E.I.; Yunusova, A.K.; Zheleznaya, L.A.; Serdyuk, I.N. Investigation of site-specific DNA binding with nicking endonuclease Nt.BspD6I at single molecule level by atomic force microscopy. Biophysics 2012, 57, 314–317. [Google Scholar] [CrossRef]
- Abrosimova, L.A.; Kubareva, E.A.; Migur, A.Y.; Gavshina, A.V.; Ryazanova, A.Y.; Norkin, M.V.; Perevyazova, T.A.; Wende, W.; Hianik, T.; Zheleznaya, L.A.; et al. Peculiarities of the interaction of the restriction endonuclease BspD6I with DNA containing its recognition site. Biochim. Biophys. Acta 2016, 1864, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Abrosimova, L.A.; Migur, A.Y.; Kubareva, E.A.; Zatsepin, T.S.; Gavshina, A.V.; Yunusova, A.K.; Perevyazova, T.A.; Pingoud, A.; Oretskaya, T.S. A study on endonuclease BspD6I and its stimulus-responsive switching by modified oligonucleotides. PLoS ONE 2018, 13, e0207302. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Tang, S.; Duan, X.; Guan, Y.; Zhao, G. Screening substrate-binding positions by rolling circle amplification suggesting a binding model of Nt.BstNBI. Biochem. J. 2019, 476, 1483–1496. [Google Scholar] [CrossRef]
- Sud’ina, A.E.; Zatsepin, T.S.; Pingoud, V.; Pingoud, A.; Oretskaya, T.S.; Kubareva, E.A. Affinity modification of the restriction endonuclease SsoII by 2′-aldehyde-containing double stranded DNAs. Biochemistry 2005, 70, 941–947. [Google Scholar] [CrossRef]
- Le Hien, T.; Zatsepin, T.S.; Schierling, B.; Volkov, E.M.; Wende, W.; Pingoud, A.; Kubareva, E.A.; Oretskaya, T.S. Restriction endonuclease SsoII with photoregulated activity—A “molecular gate” approach. Bioconjug. Chem. 2011, 22, 1366–1373. [Google Scholar] [CrossRef]
- Fersht, A. Structure and Mechanism of Action of Enzymes; Mir: Moscow, Russia, 1980; p. 432. [Google Scholar]
- Lorsch, J.R. Practical steady-state enzyme kinetics. Methods Enzymol. 2014, 536, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamics of damaged DNA binding and catalysis by human AP endonuclease 1. Acta Naturae 2016, 8, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Kuzmic, P. DynaFit—A software package for enzymology. Methods Enzymol. 2009, 467, 247–280. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Vorobjev, Y.N.; Krasnoperov, L.N.; Fedorova, O.S. Thermodynamics of the multi-stage DNA lesion recognition and repair by formamidopyrimidine-DNA glycosylase using pyrrolocytosine fluorescence—Stopped-flow pre-steady-state kinetics. Nucleic Acids Res. 2012, 40, 7384–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, N.A.; Koval, V.V.; Zharkov, D.O.; Vorobjev, Y.N.; Nevinsky, G.A.; Douglas, K.T.; Fedorova, O.S. Pre-steady-state kinetic study of substrate specificity of Escherichia coli formamidopyrimidine—DNA glycosylase. Biochemistry 2007, 46, 424–435. [Google Scholar] [CrossRef]
- Pernstich, C.; Halford, S.E. Illuminating the reaction pathway of the FokI restriction endonuclease by fluorescence resonance energy transfer. Nucleic Acids Res. 2012, 40, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, G.S.; Sessions, R.B.; Erskine, S.G.; Halford, S.E. DNA cleavage by the EcoRV restriction endonuclease: Roles of divalent metal ions in specificity and catalysis. J. Mol. Biol. 1999, 288, 87–103. [Google Scholar] [CrossRef]
- Pingoud, A.; Wilson, G.G.; Wende, W. Type II restriction endonucleases—A historical perspective and more. Nucleic Acids Res. 2014, 42, 7489–7527. [Google Scholar] [CrossRef]
- Zheleznaya, L.A.; Perevyazova, T.A.; Zheleznyakova, E.N.; Matvienko, N.I. Some properties of site-specific nickase BspD6I and the possibility of its use in hybridization analysis of DNA. Biochemistry 2002, 67, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; p. 954. [Google Scholar] [CrossRef]
- Alves, J.; Urbanke, C.; Fliess, A.; Maass, G.; Pingoud, A. Fluorescence stopped-flow kinetics of the cleavage of synthetic oligodeoxynucleotides by the EcoRI restriction endonuclease. Biochemistry 1989, 28, 7879–7888. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, G.S.; Vipond, I.B.; Halford, S.E. Rapid reaction analysis of the catalytic cycle of the EcoRV restriction endonuclease. Biochemistry 1995, 34, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Stanford, N.P.; Halford, S.E.; Baldwin, G.S. DNA cleavage by the EcoRV restriction endonuclease: pH dependence and proton transfers in catalysis. J. Mol. Biol. 1999, 288, 105–116. [Google Scholar] [CrossRef]
- Schwarz, F.W.; Tóth, J.; Aelst, K.; Cui, G.; Clausing, S.; Szczelkun, M.D.; Seidel, R. The helicase-like domains of type III restriction enzymes trigger long-range diffusion along DNA. Science 2013, 340, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Hiller, D.A.; Fogg, J.M.; Martin, A.M.; Beechem, J.M.; Reich, N.O.; Perona, J.J. Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence. Biochemistry 2003, 42, 14375–14385. [Google Scholar] [CrossRef]
- Hiller, D.A.; Rodriguez, A.M.; Perona, J.J. Non-cognate enzyme–DNA complex: Structural and kinetic analysis of EcoRV endonuclease bound to the EcoRI recognition site GAATTC. J. Mol. Biol. 2005, 354, 121–136. [Google Scholar] [CrossRef]
- Raper, A.T.; Stephenson, A.A.; Suo, Z. Functional insights revealed by the kinetic mechanism of CRISPR/Cas9. J. Am. Chem. Soc. 2018, 140, 2971–2984. [Google Scholar] [CrossRef]
- Alekseeva, I.V.; Kuznetsova, A.A.; Bakman, A.S.; Fedorova, O.S.; Kuznetsov, N.A. The role of active-site amino acid residues in the cleavage of DNA and RNA substrates by human apurinic/apyrimidinic endonuclease APE1. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129718. [Google Scholar] [CrossRef] [PubMed]
- Polisky, B.; Greene, P.; Garfin, D.E.; McCarthy, B.J.; Goodman, H.M.; Boyer, H.W. Specificity of substrate recognition by the EcoRI restriction endonuclease. Proc. Natl. Acad. Sci. USA 1975, 72, 3310–3314. [Google Scholar] [CrossRef] [Green Version]
- Monakhova, M.V.; Penkina, A.I.; Pavlova, A.V.; Lyaschuk, A.M.; Kucherenko, V.V.; Alexeevski, A.V.; Lunin, V.G.; Friedhoff, P.; Klug, G.; Oretskaya, T.S.; et al. Endonuclease activity of MutL protein of the Rhodobacter sphaeroides mismatch repair system. Biochemistry 2018, 83, 281–293. [Google Scholar] [CrossRef]
- Chandrashekaran, S.; Saravanan, M.; Radha, D.R.; Nagaraja, V. Ca2+-mediated site-specific DNA cleavage and suppression of promiscuous activity of KpnI restriction endonuclease. J. Biol. Chem. 2004, 279, 49736–49740. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, M.; Vasu, K.; Ghosh, S.; Nagaraja, V. Dual role for Zn2+ in maintaining structural integrity and inducing DNA sequence specificity in a promiscuous endonuclease. J. Biol. Chem. 2007, 282, 32320–32326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O.S. Effects of mono- and divalent metal ions on DNA binding and catalysis of human apurinic/apyrimidinic endonuclease 1. Mol. BioSyst. 2016, 12, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- Bowen, L.M.; Dupureur, C.M. Investigation of restriction enzyme cofactor requirements: A relationship between metal ion properties and sequence specificity. Biochemistry 2003, 42, 12643–12653. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.D.; Halford, S.E. Discrimination between DNA sequences by the EcoRV restriction endonuclease. Biochemistry 1989, 28, 6198–6207. [Google Scholar] [CrossRef]
- Jen-Jacobson, L.; Kurpiewski, M.; Lesser, D.; Grable, J.; Boyer, H.W.; Rosenberg, J.M.; Greene, P.J. Coordinate ion pair formation between EcoRI endonuclease and DNA. J. Biol. Chem. 1983, 258, 14638–14646. [Google Scholar] [CrossRef]
- Erskine, S.G.; Halford, S.E. Reactions of the EcoRV restriction endonuclease with fluorescent oligodeoxynucleotides: Identical equilibrium constants for binding to specific and non-specific DNA. J. Mol. Biol. 1998, 275, 759–772. [Google Scholar] [CrossRef]
- Zebala, J.F.; Choi, J.; Barany, F. Characterization of steady-state, single-turnover and binding kinetics of the TaqI restriction endonuclease. J. Biol. Chem. 1992, 267, 8097–8105. [Google Scholar] [CrossRef]
- Siksnys, V.; Pleckaityte, M. Catalytic and binding properties of restriction endonuclease Cfr9I. Eur. J. Biochem. 1993, 217, 411–419. [Google Scholar] [CrossRef]
- Kong, H.; Roemer, S.E.; Waite-Ress, P.A.; Benner, J.S.; Wilson, G.G.; Nwanko, D.O. Characterization of BcgI, a new kind of restriction-modication system. J. Biol. Chem. 1994, 269, 683–690. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Santagata, S.; Aggarwal, A.K. FokI requires two specific DNA sites for cleavage. J. Mol. Biol. 2001, 309, 69–78. [Google Scholar] [CrossRef]
- Sekerina, S.A.; Grishin, A.V.; Ryazanova, A.Y.; Artiukh, R.I.; Rogulin, E.A.; Iunusova, A.K.; Oretskaia, T.S.; Zheleznaia, L.A.; Kubareva, E.A. Oligomerization of site-specific nicking endonuclease BspD6I at high protein concentrations. Russ. J. Bioorg. Chem. 2012, 38, 376–382. [Google Scholar] [CrossRef]
- Martin, A.M.; Horton, N.C.; Lusetti, S.; Reich, N.O.; Perona, J.J. Divalent metal dependence of site-specific DNA binding by EcoRV endonuclease. Biochemistry 1999, 38, 8430–8439. [Google Scholar] [CrossRef]
- Engler, L.E.; Welch, K.K.; Jen-Jacobson, L. Specific binding by EcoRV endonuclease to its DNA recognition site GATATC. J. Mol. Biol. 1997, 269, 82–101. [Google Scholar] [CrossRef]
- Horton, J.R.; Zhang, X.; Maunus, R.; Yang, Z.; Wilson, G.G.; Roberts, R.J.; Cheng, X. DNA nicking by HinP1I endonuclease: Bending, base flipping and minor groove expansion. Nucleic Acids Res. 2006, 34, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedhoff, P.; Thomas, E.; Pingoud, A. Tyr212: A key residue involved in strand discrimination by the DNA mismatch repair endonuclease MutH. J. Mol. Biol. 2003, 325, 285–297. [Google Scholar] [CrossRef]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10570–10575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perona, J.J. Type II restriction endonucleases. Methods 2002, 28, 353–364. [Google Scholar] [CrossRef]
- Von Hippel, P.H.; Berg, O.G. Facilitated target location in biological systems. J. Biol. Chem. 1989, 264, 675–678. [Google Scholar] [CrossRef]
- Shimamoto, N. One-dimensional diffusion of proteins along DNA. Its biological and chemical significance revealed by single-molecule measurements. J. Biol. Chem. 1999, 274, 15293–15296. [Google Scholar] [CrossRef] [Green Version]
- Ehbrecht, H.J.; Pingoud, A.; Urbanke, C.; Maass, G.; Gualerzi, C. Linear diffusion of restriction endonucleases on DNA. J. Biol. Chem. 1985, 260, 6160–6166. [Google Scholar] [CrossRef]
- Shvets, A.A.; Kochugaeva, M.P.; Kolomeisky, A.B. Mechanisms of protein search for targets on DNA: Theoretical insights. Molecules 2018, 23, 2106. [Google Scholar] [CrossRef] [Green Version]
- Mechetin, G.V.; Zharkov, D.O. Mechanisms of diffusional search for specific targets by DNA-dependent proteins. Biochemistry 2014, 79, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Mirny, L.; Slutsky, M.; Wunderlich, Z.; Tafvizi, A.; Leith, J.; Kosmrlj, A. How a protein searches for its site on DNA: The mechanism of facilitated diffusion. J. Phys. A Math. Theor. 2009, 42, 434013. [Google Scholar] [CrossRef] [Green Version]
- Slutsky, M.; Mirny, L.A. Kinetics of protein-DNA interaction: Facilitated target location in sequence-dependent potential. Biophys. J. 2004, 87, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, L.W.; Benseler, F.; Graeser, E.; Piel, N.; Scholtissek, S. Effects of functional group changes in the EcoRI recognition site on the cleavage reaction catalyzed by the endonuclease. Biochemistry 1987, 26, 7238–7245. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.S.; Eis, P.S.; Blumeyer, K.; Fearon, K.; Millar, D.P. Real time kinetics of restriction endonuclease cleavage monitored by fluorescence resonance energy transfer. Nucleic Acids Res. 1994, 22, 3155–3159. [Google Scholar] [CrossRef] [Green Version]
- Marras, S.A.E.; Kramer, F.R.; Tyagi, S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002, 30, e122. [Google Scholar] [CrossRef] [Green Version]
- Allemann, R.K.; Egli, M. DNA recognition and bending. Chem. Biol. 1997, 4, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Bulygin, A.A.; Kuznetsova, A.A.; Vorobjev, Y.N.; Fedorova, O.S.; Kuznetsov, N.A. The role of active-site plasticity in damaged-nucleotide recognition by human apurinic/apyrimidinic endonuclease APE1. Molecules 2020, 25, 3940. [Google Scholar] [CrossRef] [PubMed]
- Timofeyeva, N.A.; Koval, V.V.; Knorre, D.G.; Zharkov, D.O.; Saparbaev, M.K.; Ishchenko, A.A.; Fedorova, O.S. Conformational dynamics of human AP endonuclease in base excision and nucleotide incision repair pathways. J. Biomol. Struct. Dyn. 2009, 26, 637–652. [Google Scholar] [CrossRef]
- Kanazhevskaya, L.Y.; Koval, V.V.; Zharkov, D.O.; Strauss, P.R.; Fedorova, O.S. Conformational transitions in human AP endonuclease 1 and its active site mutant during abasic site repair. Biochemistry 2010, 49, 6451–6461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monakhova, M.V.; Milakina, M.A.; Trikin, R.M.; Oretskaya, T.S.; Kubareva, E.A. Functional specifics of the MutL protein of the DNA mismatch repair system in different organisms. Rus. J. Bioorg. Chem. 2020, 46, 875–890. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
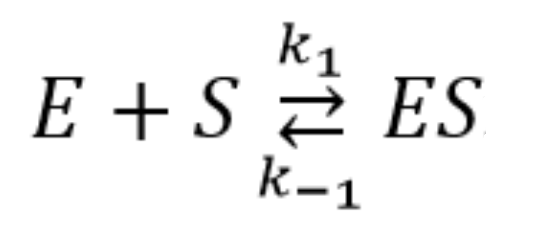{kind=link}
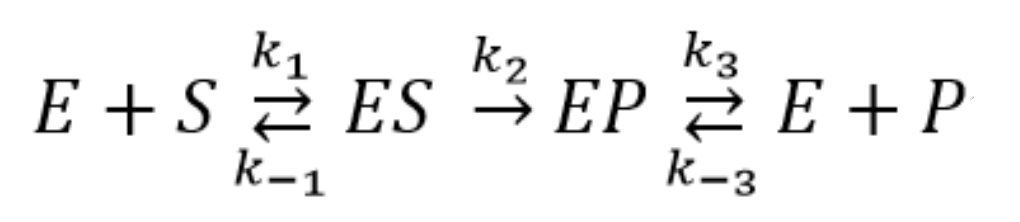{kind=link}
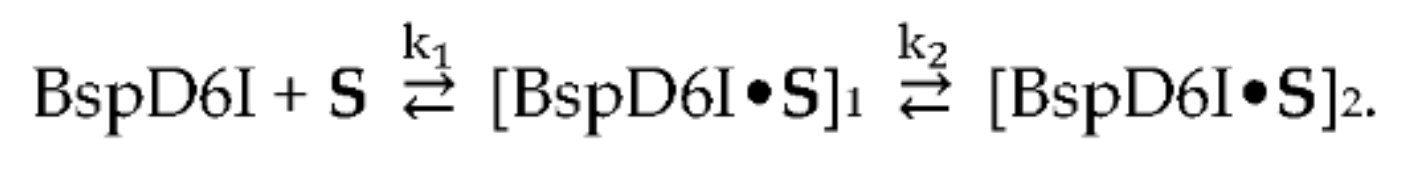{kind=link}
| DNA Duplex | Duplex Structure 5′→3′ 3′→5′ | Number of bpa in Duplex | Number of bp after Cleavage Site |
| I-17 | GGTCTCGAGTCTTCT↓CAb CCAGAGCTCAGAAGA-GT | 17 | 2 |
| II-19 | GGTCTCGAGTCTTCT↓CAAG CCAGAGCTCAGAAGA-GTTC | 19 | 4 |
| II-19A | FAM-GGTCTCGAGTCTTCT↓CAAGc CCAGAGCTCAGAAGA-GTTC | 19 | 4 |
| II-19B | GGTCTCGAGTCTTCT↓CAAG-BHQ1d CCAGAGCTCAGAAGA-GTTC | 19 | 4 |
| II-19C | FAM-GGTCTCGAGTCTTCT↓CAAG-BHQ1 CCAGAGCTCAGAAGA-GTTC | 19 | 4 |
| II-19D | GGTCTCGAGTCTTCT↓CAAG-BHQ1 CCAGAGCTCAGAAGA-GTTC-FAM | 19 | 4 |
| III-21 | GGTCTCGAGTCTTCT↓CAAGGT CCAGAGCTCAGAAGA-GTTCCA | 21 | 6 |
| IV-23 | GGTCTCGAGTCTTCT↓CAAGGTAC CCAGAGCTCAGAAGA-GTTCCATG | 23 | 8 |
| V-26 | CGTGGTCTCGAGTCTTCT↓CAAGGTAC GCACCAGAGCTCAGAAGA-GTTCCATG | 26 | 8 |
| VI-30 | GCGTGGTCTCGAGTCTTCT↓CAAGGTACCTG CGCACCAGAGCTCAGAAGA-GTTCCATGGAC | 30 | 11 |
| VI-30N | GTATGAAGCTAGAGCCAGGTTGGCAGCATC CATACTTCGATCTCGGTCCAACCGTCGTAG | 30 | - |
| Constants | VI-30N, Ca2+ | VI-30N, Mg2+ | VI-30, EDTA |
|---|---|---|---|
| k1, μM−1 × s−1 | 63 ± 3 | 68 ± 6 | 83 ± 2 |
| k−1, s−1 | 3.5 ± 0.7 | 4.6 ± 0.9 | 1.1 ± 0.2 |
| Kd, nM | 60 ± 20 | 70 ± 20 | 13 ± 3 |
| Length of DNA Duplex, bp | Number of bp Downstream of Cleavage Site | Mg2+ | Ca2+ | ||
|---|---|---|---|---|---|
| k1, s−1 | k2, s−1 | k1, s−1 | k2, s−1 | ||
| 17 | 2 | 490 ± 6 | 128 ± 2 | 405 ± 6 | 8.1 ± 0.3 |
| 19 | 4 | 370 ± 4 | 28 ± 1 | 261 ± 4 | 6.2 ± 0.2 |
| 21 | 6 | 232 ± 4 | 21 ± 1 | 179 ± 4 | 15 ± 0.5 |
| 23 | 8 | 145 ± 2 | 10.0 ± 0.2 | 115 ± 2 | 7.7 ± 0.2 |
| 26 | 8 | 126 ± 1 | 7.0 ± 0.2 | 80 ± 1 | 6.4 ± 0.2 |
| 30 | 11 | 80 ± 1 | 10.0 ± 0.2 | 65 ± 1 | 7.0 ± 0.2 |
| Enzyme | Substrate Length, bp | Incubation Temperature, °C | KM, μM | kcat, s−1 | Reference |
|---|---|---|---|---|---|
| Nt.BspD6I | 19 | 37 | 0.10 ± 0.03 | 0.05 ± 0.01 | present study |
| R.EcoRV | 12 | 25 | 0.58 ± 0.06 | 0.70 ± 0.03 | [27] |
| R.EcoRI | 12 | 15 | 0.13 ± 0.09 | 0.011 ± 0.003 | [61] |
| R.PaeR7 | 19 | 37 | 0.072 ± 0.008 | 0.30 ± 0.02 | [62] |
| Constants * | II-19C, Ca2+ | II-19C, Mg2+ | II-19D, Mg2+ |
|---|---|---|---|
| k1, μM−1 × s−1 | 120 ± 5 | 125 ± 7 | 24.3 ± 0.4 |
| k−1, s−1 | 1.1 ± 0.8 | 20 ± 1 | 4.0 ± 0.1 |
| Kd = k−1/k1, nM | 9 ± 7 | 160 ± 20 | 165 ± 7 |
| k2 (kcat), s−1 | 0.06 ± 0.01 | 0.06 ± 0.01 | |
| k3, s−1 | 0.62 ± 0.01 | 1.48 ± 0.01 | |
| k−3, μM−1 × s−1 | 0.041 ± 0.001 | 0.34 ± 0.01 | |
| KM = (k−1+ k2)/k1, μM | 0.16 | 0.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrosimova, L.A.; Kuznetsov, N.A.; Astafurova, N.A.; Samsonova, A.R.; Karpov, A.S.; Perevyazova, T.A.; Oretskaya, T.S.; Fedorova, O.S.; Kubareva, E.A. Kinetic Analysis of the Interaction of Nicking Endonuclease BspD6I with DNA. Biomolecules 2021, 11, 1420. https://doi.org/10.3390/biom11101420
Abrosimova LA, Kuznetsov NA, Astafurova NA, Samsonova AR, Karpov AS, Perevyazova TA, Oretskaya TS, Fedorova OS, Kubareva EA. Kinetic Analysis of the Interaction of Nicking Endonuclease BspD6I with DNA. Biomolecules. 2021; 11(10):1420. https://doi.org/10.3390/biom11101420
Chicago/Turabian StyleAbrosimova, Liudmila A., Nikita A. Kuznetsov, Natalia A. Astafurova, Anastasiia R. Samsonova, Andrey S. Karpov, Tatiana A. Perevyazova, Tatiana S. Oretskaya, Olga S. Fedorova, and Elena A. Kubareva. 2021. "Kinetic Analysis of the Interaction of Nicking Endonuclease BspD6I with DNA" Biomolecules 11, no. 10: 1420. https://doi.org/10.3390/biom11101420