Mass Spectrometry Based Lipidomics: An Overview of Technological Platforms


Abstract
:1. Introduction
2. Direct Infusion
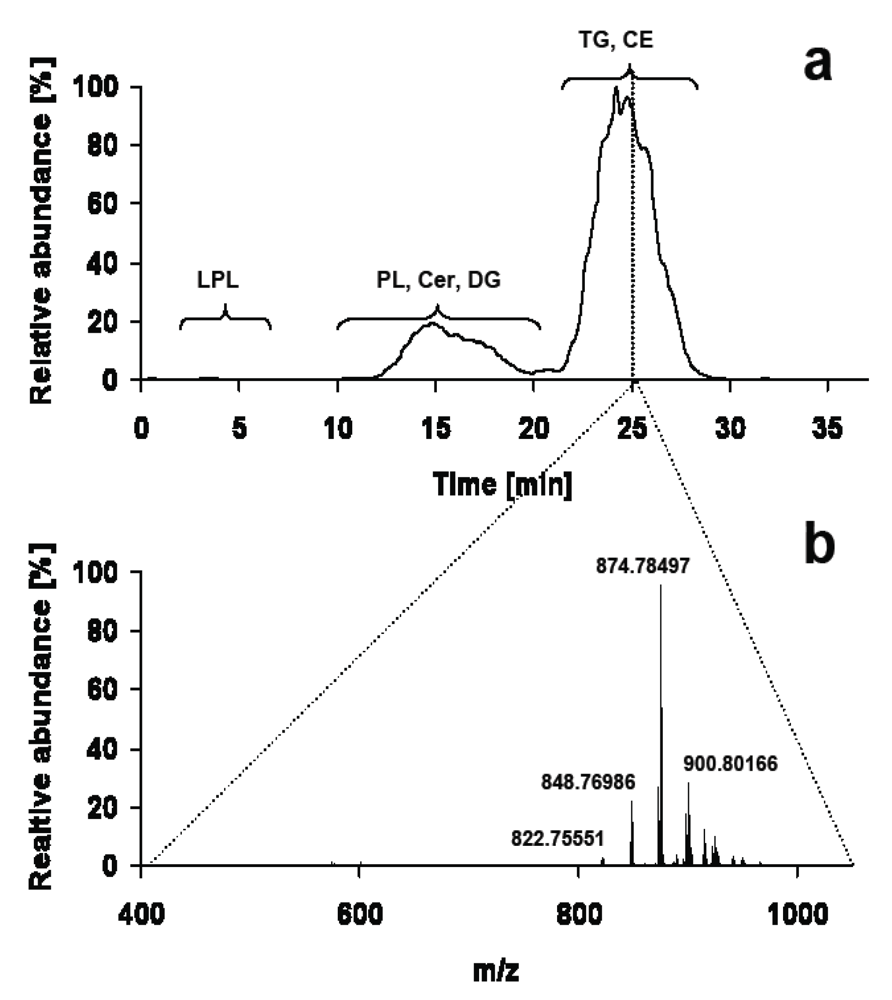
3. LC-MS
3.1. Low Resolution Mass Spectrometry


3.2. High Resolution Mass Spectrometry

3.3. Digging Deeper into Structural Details
4. MALDI-TOF

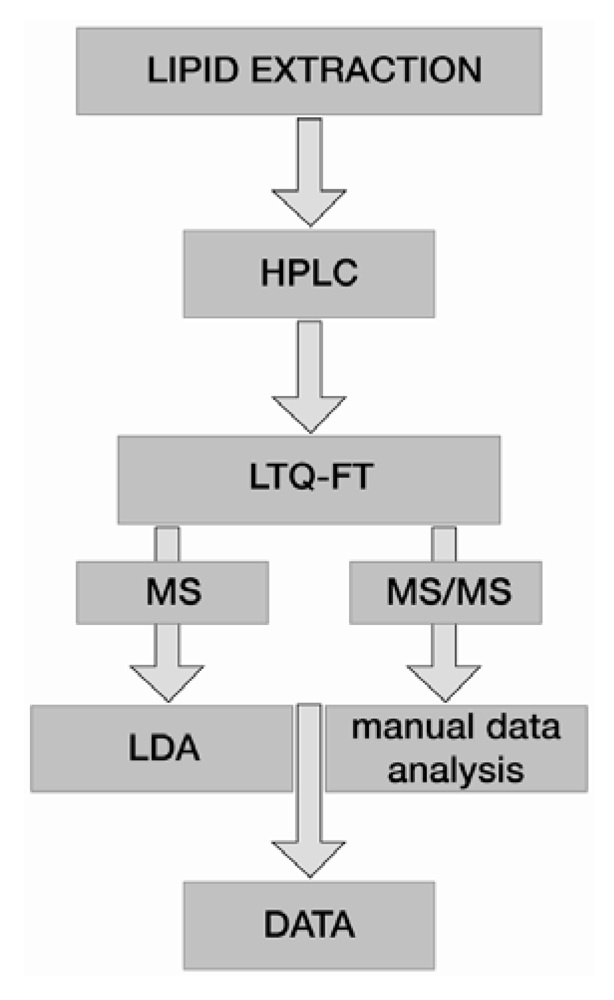
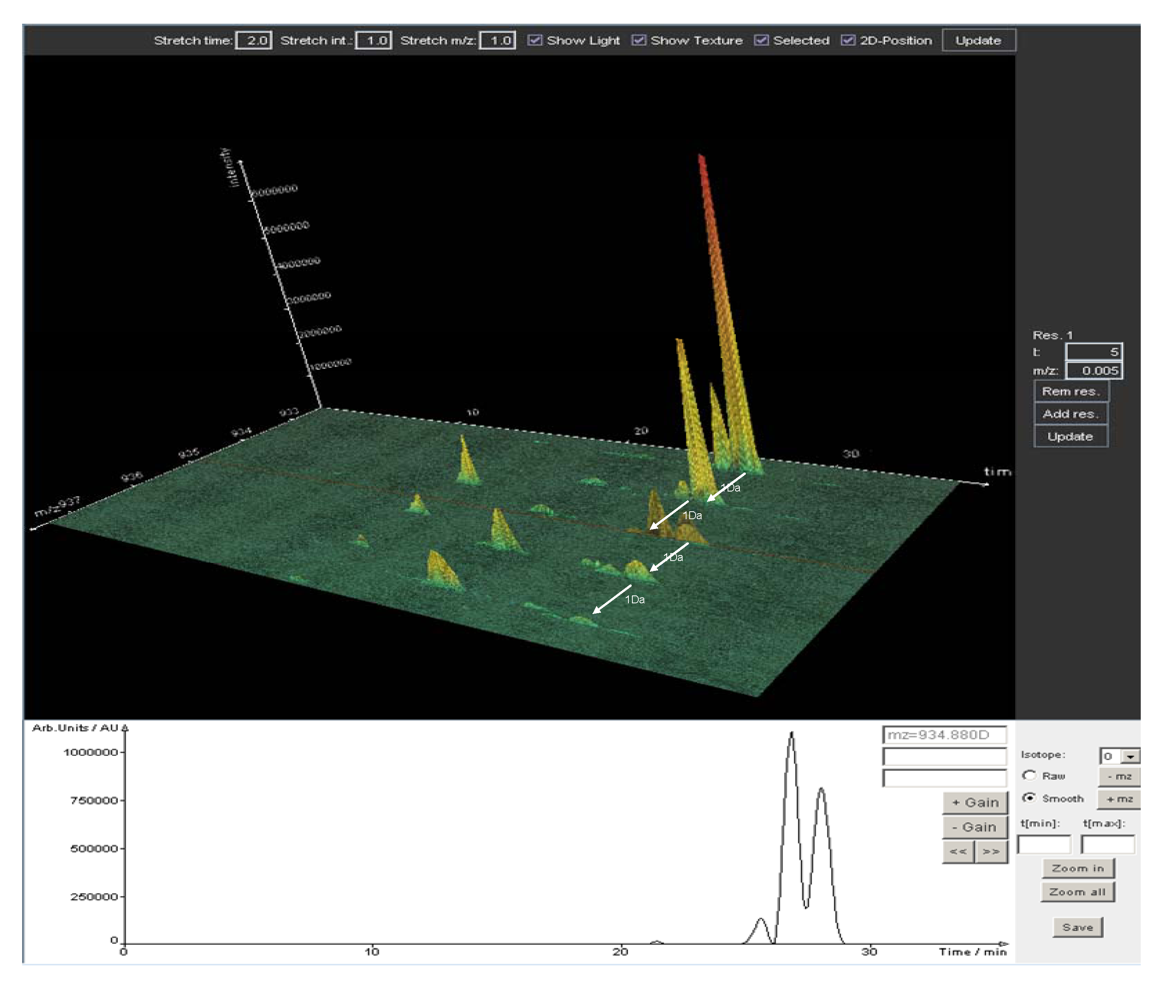
5. Data Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data type | MS vendor restrictions | Recommended instruments | Identification | Quanti- tation | Availability | |
|---|---|---|---|---|---|---|
| Lipid Inspector | shotgun | all possible | triple quadrupole, quadrupole-TOF | MPIS | Yes | upon request |
| Lipid View | shotgun | only AB Sciex | triple quadrupole, quadrupole-TOF | MPIS | Yes | purchase |
| LipidXplorer | shotgun | all possible | orbitrap, quadrupole-TOF | data dependent acquisition | Yes | open source |
| m/z Mine 2 | LC-MS | all possible | quadrupole-TOF | HiRes MS full scan, retention time | Yes | open source |
| Lipid Data Analyzer | LC-MS | all possible | FT-ICR-MS | HiRes MS full scan, retention time | Yes | open source |
6. Conclusions

Acknowledgments
Conflict of Interest
References and Notes
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Eckel, R.; Grundy, S.; Zimmet, P. The metabolic syndrome—Reply. Lancet 2005, 366, 1923–1923. [Google Scholar]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.-L.; Binder, C.J.; Stoeckl, J. Generation and Biological Activities of Oxidized Phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef]
- Wenk, M.R. Lipidomics: New Tools and Applications. Cell 2010, 143, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.F.; Turk, J. Electrospray ionization with low-energy collisionally activated dissociation tandem mass spectrometry of glycerophospholipids: Mechanisms of fragmentation and structural characterization. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2673–2695. [Google Scholar] [CrossRef]
- Han, X.L.; Gross, R.W. Electrospray ionization mass spectroscopic analysis of human erythrocyte plasma membrane phospholipids. Proc. Natl. Acad. Sci. USA 1994, 91, 10635–10639. [Google Scholar] [CrossRef]
- Bruegger, B.; Erben, G.; Sandhoff, R.; Wieland, F.T.; Lehmann, W.D. Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc. Natl. Acad. Sci. USA 1997, 94, 2339–2344. [Google Scholar]
- Han, X.; Yang, K.; Gross, R.W. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev. 2011, in press. [Google Scholar]
- Yang, K.; Zhao, Z.D.; Gross, R.W.; Han, X.L. Systematic analysis of choline-containing phospholipids using multi-dimensional mass spectrometry-based shotgun lipidomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2924–2936. [Google Scholar] [CrossRef]
- Han, X.L. Characterization and direct quantitation of ceramide molecular species from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal. Biochem. 2002, 302, 199–212. [Google Scholar] [CrossRef]
- Koivusalo, M.; Haimi, P.; Heikinheimo, L.; Kostiainen, R.; Somerharju, P. Quantitative determination of phospholipid compositions by ESI-MS: effects of acyl chain length, unsaturation, and lipid concentration on instrument response. J. Lipid Res. 2001, 42, 663–672. [Google Scholar]
- Han, X.L.; Yang, J.Y.; Cheng, H.; Ye, H.P.; Gross, R.W. Toward fingerprinting cellular lipidomes directly from biological samples by two-dimensional electrospray ionization mass spectrometry. Anal. Biochem. 2004, 330, 317–331. [Google Scholar]
- Han, X.L.; Gross, R.W. Shotgun lipidomics: multidimensional MS analysis of cellular lipidomes. Expert Rev. Proteomics 2005, 2, 253–264. [Google Scholar] [CrossRef]
- Han, X.L.; Gross, R.W. Quantitative analysis and molecular species fingerprinting of triacylglyceride molecular species directly from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal. Biochem. 2001, 295, 88–100. [Google Scholar] [CrossRef]
- Wiesner, P.; Leidl, K.; Boettcher, A.; Schmitz, G.; Liebisch, G. Lipid profiling of FPLC-separated lipoprotein fractions by electrospray ionization tandem mass spectrometry. J. Lipid Res. 2009, 50, 574–585. [Google Scholar]
- Liebisch, G.; Lieser, B.; Rahtenberg, J.; Drobnik, W.; Schmitz, G. High-throughput quantification of phosphatidylcholine and sphingomyelin by electrospray ionization tandem mass spectrometry coupled with isotope corrections algorithim (vol 1686, pg 108, 2004). Biochim. Biophys. Acta 1734, 1734, 86–89. [Google Scholar]
- Liebisch, G.; Drobnik, W.; Lieser, B.; Schmitz, G. High-throughput quantification of lysophosphatidylcholine by electrospray ionization tandem mass spectrometry. Clin. Chem. 2002, 48, 2217–2224. [Google Scholar]
- Liebisch, G.; Drobnik, W.; Reil, M.; Trumbach, B.; Arnecke, R.; Olgemoller, B.; Roscher, A.; Schmitz, G. Quantitative measurement of different ceramide species from crude cellular extracts by electrospray ionization tandem mass spectrometry (ESI-MS/MS). J. Lipid Res. 1999, 40, 1539–1546. [Google Scholar]
- Liebisch, G.; Binder, M.; Schifferer, R.; Langmann, T.; Schulz, B.; Schmitz, G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta 1761, 121–128. [Google Scholar]
- Guan, X.L.; He, X.; Ong, W.Y.; Yeo, W.K.; Shui, G.H.; Wenk, M.R. Non-targeted profiling of lipids during kainate-induced neuronal injury. FASEB J. 2006, 20, 1152–1161. [Google Scholar]
- Schwudke, D.; Oegema, J.; Burton, L.; Entchev, E.; Hannich, J.T.; Ejsing, C.S.; Kurzchalia, T.; Shevchenko, A. Lipid profiling by multiple precursor and neutral loss scanning driven by the data-dependent acquisition. Anal. Chem. 2006, 78, 585–595. [Google Scholar] [CrossRef]
- Ejsing, C.S.; Duchoslav, E.; Sampaio, J.; Simons, K.; Bonner, R.; Thiele, C.; Ekroos, K.; Shevchenko, A. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal. Chem. 2006, 78, 6202–6214. [Google Scholar]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar]
- Kalvodova, L.; Sampaio, J.L.; Cordo, S.; Ejsing, C.S.; Shevchenko, A.; Simons, K. The Lipidomes of Vesicular Stomatitis Virus, Semliki Forest Virus, and the Host Plasma Membrane Analyzed by Quantitative Shotgun Mass Spectrometry. J. Virol. 2009, 83, 7996–8003. [Google Scholar]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2011, 108, 1903–1907. [Google Scholar]
- Stahlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Borén, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2664–2672. [Google Scholar] [CrossRef]
- Schwudke, D.; Hannich, J.T.; Surendranath, V.; Grimard, V.; Moehring, T.; Burton, L.; Kurzchalia, T.; Shevchenko, A. Top-down lipidomic screens by multivariate analysis of high-resolution survey mass spectra. Anal. Chem. 2007, 79, 4083–4093. [Google Scholar] [CrossRef]
- Schuhmann, K.; Herzog, R.; Schwudke, D.; Metelmann-Strupat, W.; Bornstein, S.R.; Shevchenko, A. Bottom-Up Shotgun Lipidomics by Higher Energy Collisional Dissociation on LTQ Orbitrap Mass Spectrometers. Anal. Chem. 2011, 83, 5480–5487. [Google Scholar]
- Graessler, J.; Schwudke, D.; Schwarz, P.E.H.; Herzog, R.; Shevchenko, A.; Bornstein, S.R. Top-Down Lipidomics Reveals Ether Lipid Deficiency in Blood Plasma of Hypertensive Patients. PLoS One 2009, 4, 13. [Google Scholar]
- Esch, S.W.; Tamura, P.; Sparks, A.A.; Roth, M.R.; Devaiah, S.P.; Heinz, E.; Wang, X.; Williams, T.D.; Welti, R. Rapid characterization of the fatty acyl composition of complex lipids by collision-induced dissociation time-of-flight mass spectrometry. J. Lipid Res. 2007, 48, 235–241. [Google Scholar]
- Whitehouse, C.M.; Dreyer, R.N.; Yamashita, M.; Fenn, J.B. Electrospray Interface for Liquid Chromatographs and Mass Spectrometers. Anal. Chem. 1985, 57, 675–679. [Google Scholar]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar]
- Andreyev, A.Y.; Fahy, E.; Guan, Z.Q.; Kelly, S.; Li, X.A.; McDonald, J.G.; Milne, S.; Myers, D.; Park, H.; Ryan, A.; et al. Subcellular organelle lipidomics in TLR-4-activated macrophages. J. Lipid Res. 2010, 51, 2785–2797. [Google Scholar]
- Shui, G.; Guan, X.L.; Low, C.P.; Chua, G.H.; Goh, J.S.Y.; Yang, H.; Wenk, M.R. Toward one step analysis of cellular lipidomes using liquid chromatography coupled with mass spectrometry: application to Saccharomyces cerevisiae and Schizosaccharomyces pombe lipidomics. Mol. Biosyst. 2010, 6, 1008–1017. [Google Scholar] [CrossRef]
- Masoodi, M.; Eiden, M.; Koulman, A.; Spaner, D.; Volmer, D.A. Comprehensive Lipidomics Analysis of Bioactive Lipids in Complex Regulatory Networks. Anal. Chem. 2010, 82, 8176–8185. [Google Scholar]
- Scherer, M.; Bottcher, A.; Schmitz, G.; Liebisch, G. Sphingolipid profiling of human plasma and FPLC-separated lipoprotein fractions by hydrophilic interaction chromatography tandem mass spectrometry. Biochim. Biophys. Acta 1811, 68–75. [Google Scholar]
- Merrill, A.H.; Stokes, T.H.; Momin, A.; Park, H.; Portz, B.J.; Kelly, S.; Wang, E.; Sullards, M.C.; Wang, M.D. Sphingolipidomics: A valuable tool for understanding the roles of sphingolipids in biology and disease. J. Lipid Res. 2009, 50, S97–S102. [Google Scholar]
- Retra, K.; Bleijerveld, O.B.; van Gesteil, R.A.; Tielens, A.G.M.; van Hellemond, J.J.; Brouwers, J.F. A simple and universal method for the separation and identification of phospholipid molecular species. Rapid Commun. Mass Spectrom. 2008, 22, 1853–1862. [Google Scholar]
- Clark, J.; Anderson, K.E.; Juvin, V.; Smith, T.S.; Karpe, F.; Wakelam, M.J.O.; Stephens, L.R.; Hawkins, P.T. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods 2011, 8, 267–272. [Google Scholar] [CrossRef]
- Karu, K.; Turton, J.; Wang, Y.Q.; Griffiths, W.J. Nano-liquid chromatography-tandem mass spectrometry analysis of oxysterols in brain: Monitoring of cholesterol autoxidation. Chem. Phys. Lipids 2011, 164, 411–424. [Google Scholar] [CrossRef]
- Nakanishi, H.; Iida, Y.; Shimizu, T.; Taguchi, R. Analysis of oxidized phosphatidylcholines as markers for oxidative stress, using multiple reaction monitoring with theoretically expanded data sets with reversed-phase liquid chromatography/tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1366–1374. [Google Scholar] [CrossRef]
- Pettitt, T.R.; Dove, S.K.; Lubben, A.; Calaminus, S.D.J.; Wakelam, M.J.O. Analysis of intact phosphoinositides in biological samples. J. Lipid Res. 2006, 47, 1588–1596. [Google Scholar] [CrossRef]
- Hutchins, P.M.; Barkley, R.M.; Murphy, R.C. Separation of cellular nonpolar neutral lipids by normal-phase chromatography and analysis by electrospray ionization mass spectrometry. J. Lipid Res. 2008, 49, 804–813. [Google Scholar] [CrossRef]
- Murphy, R.C.; Leiker, T.J.; Barkley, R.M. Glycerolipid and cholesterol ester analyses in biological samples by mass spectrometry. Biochim. Biophys. Acta 1811, 776–783. [Google Scholar]
- Ikeda, K.; Oike, Y.; Shimizu, T.; Taguchi, R. Global analysis of triacylglycerols including oxidized molecular species by reverse-phase high resolution LC/ESI-QTOF MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2639–2647. [Google Scholar] [CrossRef]
- Nygren, H.; Seppanen-Laakso, T.; Castillo, S.; Hyotylainen, T.; Oresic, M. Liquid Chromatography-Mass Spectrometry (LC-MS)-Based Lipidomics for Studies of Body Fluids and Tissues. In Metabolic Profiling: Methods and Protocols; Metz, T., Ed.; Springer: New York, NY, USA, 2011. [Google Scholar]
- Shui, G.H.; Bendt, A.K.; Pethe, K.; Dick, T.; Wenk, M.R. Sensitive profiling of chemically diverse bioactive lipids. J. Lipid Res. 2007, 48, 1976–1984. [Google Scholar] [CrossRef]
- Fauland, A.; Kofeler, H.; Trotzmuller, M.; Knopf, A.; Hartler, J.; Eberl, A.; Chitraju, C.; Lankmayr, E.; Spener, F. A comprehensive method for lipid profiling by liquid chromatography-ion cyclotron resonance mass spectrometry. J. Lipid Res. 2011, 52, 2314–2322. [Google Scholar] [CrossRef]
- Hu, C.X.; van Dommelen, J.; van der Heijden, R.; Spijksma, G.; Reijmers, T.H.; Wang, M.; Slee, E.; Lu, X.; Xu, G.W.; van der Greef, J.; et al. RPLC-Ion-Trap-FTMS Method for Lipid Profiling of Plasma: Method Validation and Application to p53 Mutant Mouse Model. J. Proteome Res. 2008, 7, 4982–4991. [Google Scholar]
- Hein, E.M.; Blank, L.M.; Heyland, J.; Baumbach, J.I.; Schmid, A.; Hayen, H. Glycerophospholipid profiling by high-performance liquid chromatography/mass spectrometry using exact mass measurements and multi-stage mass spectrometric fragmentation experiments in parallel. Rapid Commun. Mass Spectrom. 2009, 23, 1636–1646. [Google Scholar] [CrossRef]
- Ogiso, H.; Suzuki, T.; Taguchi, R. Development of a reverse-phase liquid chromatography electrospray ionization mass spectrometry method for lipidomics, improving detection of phosphatidic acid and phosphatidylserine. Anal. Biochem. 2008, 375, 124–131. [Google Scholar]
- Taguchi, R. Global and targeted methods for lipidomics by mass spectrometry. Neurosci. Res. 2008, 61, S20–S20. [Google Scholar]
- Sato, Y.; Nakamura, T.; Aoshima, K.; Oda, Y. Quantitative and Wide-Ranging Profiling of Phospholipids in Human Plasma by Two-dimensional Liquid Chromatography/Mass Spectrometry. Anal. Chem. 2010, 82, 9858–9864. [Google Scholar]
- Koulman, A.; Woffendin, G.; Narayana, V.K.; Welchman, H.; Crone, C.; Volmer, D.A. High-resolution extracted ion chromatography, a new tool for metabolomics and lipidomics using a second-generation orbitrap mass spectrometer. Rapid Commun. Mass Spectrom. 2009, 23, 1411–1418. [Google Scholar] [CrossRef]
- Holcapek, M.; Dvorakova, H.; Lisa, M.; Giron, A.J.; Sandra, P.; Cvacka, J. Regioisomeric analysis of triacylglycerols using silver-ion liquid chromatography atmospheric pressure chemical ionization mass spectrometry: Comparison of five different mass analyzers. J. Chromatogr. A 1217, 8186–8194. [Google Scholar]
- Lisa, M.; Cifkova, E.; Holcapek, M. Lipidomic profiling of biological tissues using off-line two-dimensional high-performance liquid chromatography mass spectrometry. J. Chromatogr. A 1218, 5146–5156. [Google Scholar]
- Lee, S.H.; Blair, I.A. Targeted chiral lipidomics analysis of bioactive eicosanoid lipids in cellular systems. BMB Rep. 2009, 42, 401–410. [Google Scholar] [CrossRef]
- Brown, H.A.; Murphy, R.C. Working towards an exegesis for lipids in biology. Nat. Chem. Biol. 2009, 5, 602–606. [Google Scholar]
- Thomas, M.C.; Mitchell, T.W.; Harman, D.G.; Deeley, J.M.; Nealon, J.R.; Blanksby, S.J. Ozone-induced dissociation: Elucidation of double bond position within mass-selected lipid ions. Anal. Chem. 2008, 80, 303–311. [Google Scholar] [CrossRef]
- Ellis, S.R.; Wu, C.P.; Deeley, J.M.; Zhu, X.J.; Truscott, R.J.W.; Panhuis, M.I.H.; Cooks, R.G.; Mitchell, T.W.; Blanksby, S.J. Imaging of Human Lens Lipids by Desorption Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 2095–2104. [Google Scholar] [CrossRef]
- Shaner, R.L.; Allegood, J.C.; Park, H.; Wang, E.; Kelly, S.; Haynes, C.A.; Sullards, M.C.; Merrill, A.H., Jr. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 2009, 50, 1692–1707. [Google Scholar] [CrossRef]
- Fuchs, B.; Suss, R.; Schiller, J. An update of MALDI-TOF mass spectrometry in lipid research. Prog. Lipid Res. 2010, 49, 450–475. [Google Scholar] [CrossRef]
- Petkovic, M.; Schiller, J.; Muller, M.; Benard, S.; Reichl, S.; Arnold, K.; Arnhold, J. Detection of individual phospholipids in lipid mixtures by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry: Phosphatidylcholine prevents the detection of further species. Anal. Biochem. 2001, 289, 202–216. [Google Scholar] [CrossRef]
- Luftmann, H.; Aranda, M.; Morlock, G.E. Automated interface for hyphenation of planar chromatography with mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 3772–3776. [Google Scholar] [CrossRef]
- Dreisewerd, K.; Muthing, J.; Rohlfing, A.; Meisen, I.; Vukelic, Z.; Peter-Katalinic, J.; Hillenkamp, F.; Berkenkamp, S. Analysis of gangliosides directly from thin-layer chromatography plates by infrared matrix-assisted laser desorption/ionization orthogonal time-of-flight mass spectrometry with a glycerol matrix. Anal. Chem. 2005, 77, 4098–4107. [Google Scholar]
- Stuebiger, G.; Pittenauer, E.; Belgacem, O.; Rehulka, P.; Widhalm, K.; Allmaier, G. Analysis of human plasma lipids and soybean lecithin by means of high-performance thin-layer chromatography and matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 2711–2723. [Google Scholar] [CrossRef]
- Souady, J.; Soltwisch, J.; Dreisewerd, K.; Haier, J.; Peter-Katalinic, J.; Muething, J. Structural Profiling of Individual Glycosphingolipids in a Single Thin-Layer Chromatogram by Multiple Sequential Immunodetection Matched with Direct IR-MALDI-o-TOF Mass Spectrometry. Anal. Chem. 2009, 81, 9481–9492. [Google Scholar]
- Muesken, A.; Souady, J.; Dreisewerd, K.; Zhang, W.; Distler, U.; Peter-Katalinic, J.; Miller-Podraza, H.; Karch, H.; Muething, J. Application of thin-layer chromatography/infrared matrix-assisted laser desorption/ionization orthogonal time-of-flight mass spectrometry to structural analysis of bacteria-binding glycosphingolipids selected by affinity detection. Rapid Commun. Mass Spectrom. 2010, 24, 1032–1038. [Google Scholar] [CrossRef]
- Rohlfing, A.; Muthing, J.; Pohlentz, G.; Distler, U.; Peter-Katalinic, J.; Berkenkamp, S.; Dreisewerd, K. IR-MALDI-MS analysis of HPTLC—Separated phospholipid mixtures directly from the TLC plate. Anal. Chem. 2007, 79, 5793–5808. [Google Scholar]
- Cheng, C.F.; Pittenauer, E.; Gross, M.L. Charge-remote fragmentations are energy-dependent processes. J. Am. Soc. Mass Spectrom. 1998, 9, 840–844. [Google Scholar] [CrossRef]
- Pittenauer, E.; Allmaier, G. A universal product ion nomenclature for M-H (-), M+H (+) and M+nNa-(n-1)H (+) (n=1-3) glycerophospholipid precursor ions based on high-energy CID by MALDI-TOF/RTOF mass spectrometry. Int. J. Mass Spectrom. 2011, 301, 90–101. [Google Scholar] [CrossRef]
- Pittenauer, E.; Allmaier, G. The Renaissance of High-Energy CID for Structural Elucidation of Complex Lipids: MALDI-TOF/RTOF-MS of Alkali Cationized Triacylglycerols. J. Am. Soc. Mass Spectrom. 2009, 20, 1037–1047. [Google Scholar] [CrossRef]
- Herzog, R.; Schwudke, D.; Schuhmann, K.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011, 12, 1–25. [Google Scholar]
- Katajamaa, M.; Miettinen, J.; Oresic, M. MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 2006, 22, 634–636. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010, 11, 1–11. [Google Scholar]
- Pietilainen, K.H.; Sysi-Aho, M.; Rissanen, A.; Seppanen-Laakso, T.; Yki-Jarvinen, H.; Kaprio, J.; Oresic, M. Acquired Obesity Is Associated with Changes in the Serum Lipidomic Profile Independent of Genetic Effects—A Monozygotic Twin Study. PLoS One 2007, 2, 14. [Google Scholar]
- Hartler, J.; Trötzmüller, M.; Chitraju, C.; Spener, F.; Köfeler, H.C.; Thallinger, G.G. Lipid Data Analyzer: Unattended Identification and Quantitation of Lipids in LC-MS Data. Bioinformatics 2011, 27, 572–577. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Köfeler, H.C.; Fauland, A.; Rechberger, G.N.; Trötzmüller, M. Mass Spectrometry Based Lipidomics: An Overview of Technological Platforms. Metabolites 2012, 2, 19-38. https://doi.org/10.3390/metabo2010019
Köfeler HC, Fauland A, Rechberger GN, Trötzmüller M. Mass Spectrometry Based Lipidomics: An Overview of Technological Platforms. Metabolites. 2012; 2(1):19-38. https://doi.org/10.3390/metabo2010019
Chicago/Turabian StyleKöfeler, Harald C., Alexander Fauland, Gerald N. Rechberger, and Martin Trötzmüller. 2012. "Mass Spectrometry Based Lipidomics: An Overview of Technological Platforms" Metabolites 2, no. 1: 19-38. https://doi.org/10.3390/metabo2010019