

Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies
 ,
,  ,
,  , , and
, , and 
Abstract
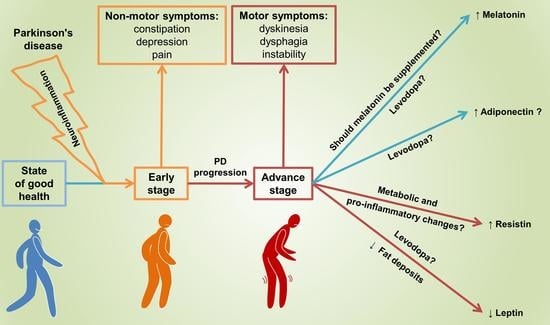
:
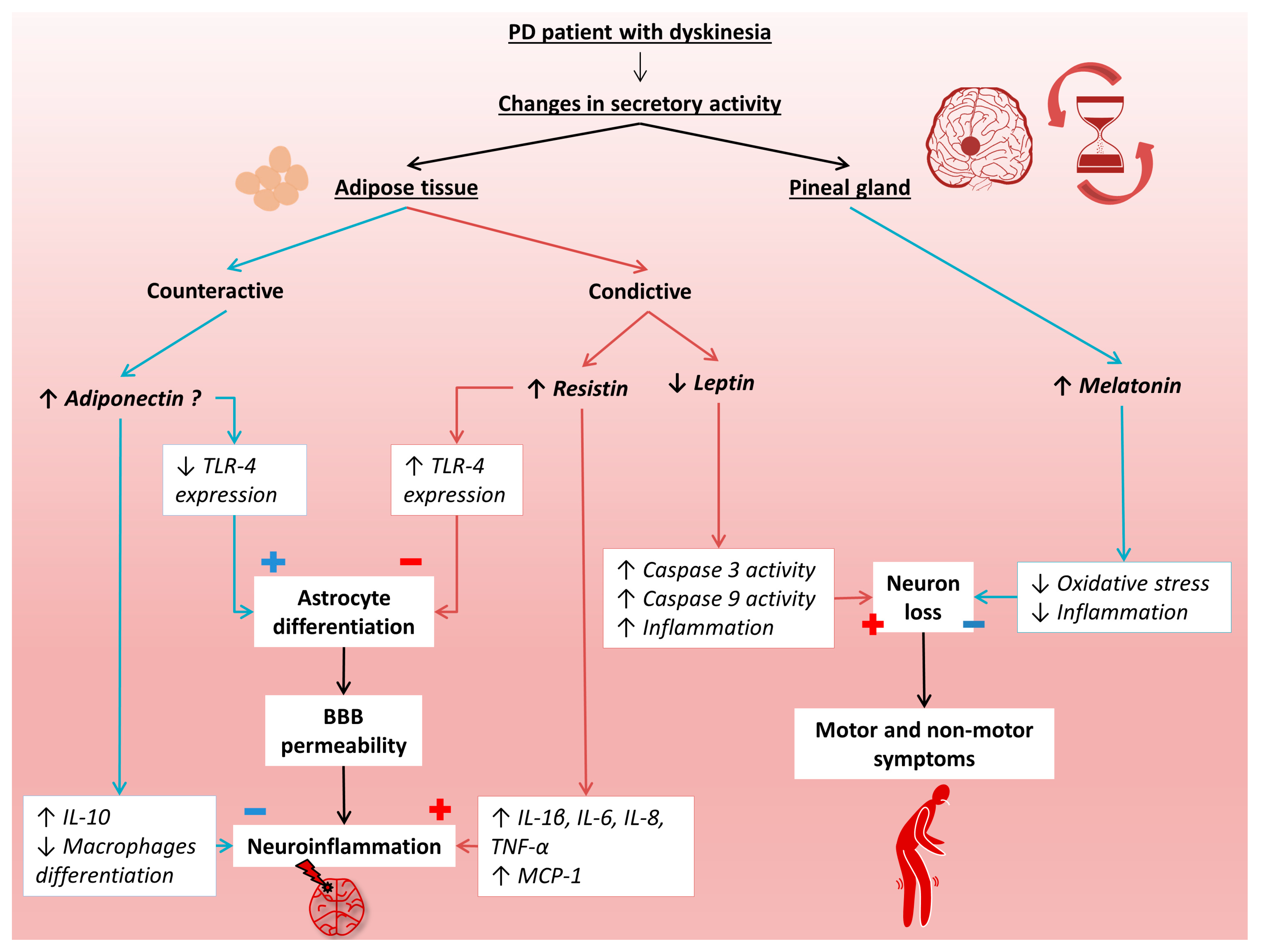
1. Introduction
2. Material and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armstrong, M.J.; Okun, M.S. Time for a New Image of Parkinson Disease. JAMA Neurol. 2020, 77, 1345. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease. JAMA 2020, 323, 548. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Gustafson, B.; Jack, M.M.; Cushman, S.W.; Smith, U. Adiponectin Gene Activation by Thiazolidinediones Requires PPARγ2, but Not C/EBPα—Evidence for Differential Regulation of the AP2 and Adiponectin Genes. Biochem. Biophys. Res. Commun. 2003, 308, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. Alpha-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Salari, M.; Barzegar, M.; Etemadifar, M.; Mirmosayyeb, O. Serum Leptin Levels in Iranian Patients with Parkinson’s Disease. Iran. J. Neurol. 2018, 17, 71–77. [Google Scholar]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Daneshvar Kakhaki, R.; Ostadmohammadi, V.; Kouchaki, E.; Aghadavod, E.; Bahmani, F.; Tamtaji, O.R.; Reiter, R.J.; Mansournia, M.A.; Asemi, Z. Melatonin Supplementation and the Effects on Clinical and Metabolic Status in Parkinson’s Disease: A Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Neurol. Neurosurg. 2020, 195, 105878. [Google Scholar] [CrossRef]
- Ahn, J.H.; Kim, M.; Park, S.; Jang, W.; Park, J.; Oh, E.; Cho, J.W.; Kim, J.S.; Youn, J. Prolonged-Release Melatonin in Parkinson’s Disease Patients with a Poor Sleep Quality: A Randomized Trial. Parkinsonism Relat. Disord. 2020, 75, 50–54. [Google Scholar] [CrossRef]
- Hadi, F.; Agah, E.; Tavanbakhsh, S.; Mirsepassi, Z.; Mousavi, S.V.; Talachi, N.; Tafakhori, A.; Aghamollaii, V. Safety and Efficacy of Melatonin, Clonazepam, and Trazodone in Patients with Parkinson’s Disease and Sleep Disorders: A Randomized, Double-Blind Trial. Neurol. Sci. 2022, 43, 6141–6148. [Google Scholar] [CrossRef]
- Delgado-Lara, D.L.; González-Enríquez, G.V.; Torres-Mendoza, B.M.; González-Usigli, H.; Cárdenas-Bedoya, J.; Macías-Islas, M.A.; de la Rosa, A.C.; Jiménez-Delgado, A.; Pacheco-Moisés, F.; Cruz-Serrano, J.A.; et al. Effect of Melatonin Administration on the PER1 and BMAL1 Clock Genes in Patients with Parkinson’s Disease. Biomed. Pharmacother. 2020, 129, 110485. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Delgado, A.; Ortiz, G.G.; Delgado-Lara, D.L.; González-Usigli, H.A.; González-Ortiz, L.J.; Cid-Hernández, M.; Cruz-Serrano, J.A.; Pacheco-Moisés, F.P. Effect of Melatonin Administration on Mitochondrial Activity and Oxidative Stress Markers in Patients with Parkinson’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 5577541. [Google Scholar] [CrossRef] [PubMed]
- Wongprayoon, P.; Govitrapong, P. Melatonin as a Mitochondrial Protector in Neurodegenerative Diseases. Cell. Mol. Life Sci. 2017, 74, 3999–4014. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, B.S. The Neuroprotective Role of Melatonin in Neurological Disorders. J. Neurosci. Res. 2018, 96, 1136–1149. [Google Scholar] [CrossRef]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT 1 and MT 2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef]
- Videnovic, A.; Willis, G.L. Circadian System—A Novel Diagnostic and Therapeutic Target in Parkinson’s Disease? Mov. Disord. 2016, 31, 260–269. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Reiter, R.J.; Alipoor, R.; Dadgostar, E.; Kouchaki, E.; Asemi, Z. Melatonin and Parkinson Disease: Current Status and Future Perspectives for Molecular Mechanisms. Cell. Mol. Neurobiol. 2020, 40, 15–23. [Google Scholar] [CrossRef]
- Ma, H.; Yan, J.; Sun, W.; Jiang, M.; Zhang, Y. Melatonin Treatment for Sleep Disorders in Parkinson’s Disease: A Meta-Analysis and Systematic Review. Front. Aging Neurosci. 2022, 14, 784314. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Wang, X.; Liu, H.; Wang, T.; Lin, Z.; Xiong, N. Neuroprotective Effect of Melatonin on Sleep Disorders Associated with Parkinson’s Disease. Antioxidants 2023, 12, 396. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional Cloning of the Mouse Obese Gene and Its Human Homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Palhinha, L.; Liechocki, S.; Hottz, E.D.; da Pereira, J.A.S.; de Almeida, C.J.; Moraes-Vieira, P.M.M.; Bozza, P.T.; Maya-Monteiro, C.M. Leptin Induces Proadipogenic and Proinflammatory Signaling in Adipocytes. Front. Endocrinol. 2019, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhong, L.; Zhu, C.; Zhao, H.; Zhao, F.; Cui, R.; Gao, S.; Li, B. Role of Leptin in Mood Disorder and Neurodegenerative Disease. Front. Neurosci. 2019, 13, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chua, S., Jr. Leptin Function and Regulation. Compr. Physiol. 2017, 8, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Signore, A.P.; Zhang, F.; Weng, Z.; Gao, Y.; Chen, J. Leptin Neuroprotection in the CNS: Mechanisms and Therapeutic Potentials. J. Neurochem. 2008, 106, 1977–1990. [Google Scholar] [CrossRef] [PubMed]
- Lorefält, B.; Toss, G.; Granérus, A.-K. Weight Loss, Body Fat Mass, and Leptin in Parkinson’s Disease. Mov. Disord. 2009, 24, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, M.; Rasul Chaudhry, S.; Yasin, H.; Zhao, Y.; Stadlbauer, A.; Buchfelder, M.; Kinfe, T. Emerging Roles of Leptin in Parkinson’s Disease: Chronic Inflammation, Neuroprotection and More? Brain. Behav. Immun. 2023, 107, 53–61. [Google Scholar] [CrossRef]
- Rocha, N.P.; Scalzo, P.L.; Barbosa, I.G.; de Sousa, M.S.; Morato, I.B.; Vieira, É.L.M.; Christo, P.P.; Reis, H.J.; Teixeira, A.L. Circulating Levels of Adipokines in Parkinson’s Disease. J. Neurol. Sci. 2014, 339, 64–68. [Google Scholar] [CrossRef]
- Acquarone, E.; Monacelli, F.; Borghi, R.; Nencioni, A.; Odetti, P. Resistin: A Reappraisal. Mech. Ageing Dev. 2019, 178, 46–63. [Google Scholar] [CrossRef]
- Lu, D.-Y.; Chen, J.-H.; Tan, T.-W.; Huang, C.-Y.; Yeh, W.-L.; Hsu, H.-C. Resistin Protects against 6-Hydroxydopamine-Induced Cell Death in Dopaminergic-like MES23.5 Cells. J. Cell. Physiol. 2013, 228, 563–571. [Google Scholar] [CrossRef]
- Filková, M.; Haluzík, M.; Gay, S.; Šenolt, L. The Role of Resistin as a Regulator of Inflammation: Implications for Various Human Pathologies. Clin. Immunol. 2009, 133, 157–170. [Google Scholar] [CrossRef]
- Codoñer-Franch, P.; Alonso-Iglesias, E. Resistin: Insulin Resistance to Malignancy. Clin. Chim. Acta 2015, 438, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.S.; Yan, S.; Lü, J.; Liang, Z.; Yao, Q.; Chen, C. Resistin Increases Monolayer Permeability of Human Coronary Artery Endothelial Cells. PLoS ONE 2013, 8, e84576. [Google Scholar] [CrossRef] [PubMed]
- Sardi, F.; Fassina, L.; Venturini, L.; Inguscio, M.; Guerriero, F.; Rolfo, E.; Ricevuti, G. Alzheimer’s Disease, Autoimmunity and Inflammation. The Good, the Bad and the Ugly. Autoimmun. Rev. 2011, 11, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Silswal, N.; Singh, A.K.; Aruna, B.; Mukhopadhyay, S.; Ghosh, S.; Ehtesham, N.Z. Human Resistin Stimulates the Pro-Inflammatory Cytokines TNF-α and IL-12 in Macrophages by NF-ΚB-Dependent Pathway. Biochem. Biophys. Res. Commun. 2005, 334, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Xiaoying, L.; Li, T.; Yu, S.; Jiusheng, J.; Jilin, Z.; Jiayi, W.; Dongxin, L.; Wengang, F.; Xinyue, Z.; Hao, Y.; et al. Resistin-Inhibited Neural Stem Cell-Derived Astrocyte Differentiation Contributes to Permeability Destruction of the Blood–Brain Barrier. Neurochem. Res. 2019, 44, 905–916. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of Adiponectin by Adipose Tissue-Derived Cytokines: In Vivo and In Vitro Investigations in Humans. Am. J. Physiol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef]
- Cnop, M.; Havel, P.J.; Utzschneider, K.M.; Carr, D.B.; Sinha, M.K.; Boyko, E.J.; Retzlaff, B.M.; Knopp, R.H.; Brunzell, J.D.; Kahn, S.E. Relationship of Adiponectin to Body Fat Distribution, Insulin Sensitivity and Plasma Lipoproteins: Evidence for Independent Roles of Age and Sex. Diabetologia 2003, 46, 459–469. [Google Scholar] [CrossRef]
- Kadowaki, T. Adiponectin and Adiponectin Receptors in Insulin Resistance, Diabetes, and the Metabolic Syndrome. J. Clin. Investig. 2006, 116, 1784–1792. [Google Scholar] [CrossRef]
- Gavrila, A.; Chan, J.L.; Yiannakouris, N.; Kontogianni, M.; Miller, L.C.; Orlova, C.; Mantzoros, C.S. Serum Adiponectin Levels Are Inversely Associated with Overall and Central Fat Distribution but Are Not Directly Regulated by Acute Fasting or Leptin Administration in Humans: Cross-Sectional and Interventional Studies. J. Clin. Endocrinol. Metab. 2003, 88, 4823–4831. [Google Scholar] [CrossRef]
- Halleux, C.M.; Takahashi, M.; Delporte, M.L.; Detry, R.; Funahashi, T.; Matsuzawa, Y.; Brichard, S.M. Secretion of Adiponectin and Regulation of ApM1 Gene Expression in Human Visceral Adipose Tissue. Biochem. Biophys. Res. Commun. 2001, 288, 1102–1107. [Google Scholar] [CrossRef]
- Keller, P.; Møller, K.; Krabbe, K.S.; Pedersen, B.K. Circulating Adiponectin Levels during Human Endotoxaemia. Clin. Exp. Immunol. 2003, 134, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Adiponectin: Action, Regulation and Association to Insulin Sensitivity. Obes. Rev. 2005, 6, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Polito, R.; Di Meo, I.; Barbieri, M.; Daniele, A.; Paolisso, G.; Rizzo, M.R. Adiponectin Role in Neurodegenerative Diseases: Focus on Nutrition Review. Int. J. Mol. Sci. 2020, 21, 9255. [Google Scholar] [CrossRef] [PubMed]
- Haugen, F.; Drevon, C.A. Activation of Nuclear Factor-ΚB by High Molecular Weight and Globular Adiponectin. Endocrinology 2007, 148, 5478–5486. [Google Scholar] [CrossRef]
- Beitz, J.M. Parkinson s Disease a Review. Front. Biosci. 2014, S6, S415. [Google Scholar] [CrossRef]
- Bloemer, J.; Pinky, P.D.; Govindarajulu, M.; Hong, H.; Judd, R.; Amin, R.H.; Moore, T.; Dhanasekaran, M.; Reed, M.N.; Suppiramaniam, V. Role of Adiponectin in Central Nervous System Disorders. Neural Plast. 2018, 2018, 4593530. [Google Scholar] [CrossRef]
- Santaella, A.; Kuiperij, H.B.; van Rumund, A.; Esselink, R.A.J.; van Gool, A.J.; Bloem, B.R.; Verbeek, M.M. Inflammation Biomarker Discovery in Parkinson’s Disease and Atypical Parkinsonisms. BMC Neurol. 2020, 20, 26. [Google Scholar] [CrossRef]
- Kim, J.-Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-Associated Improvements in Metabolic Profile through Expansion of Adipose Tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef]
- Chinta, S.J.; Lieu, C.A.; DeMaria, M.; Laberge, R.-M.; Campisi, J.; Andersen, J.K. Environmental Stress, Ageing and Glial Cell Senescence: A Novel Mechanistic Link to Parkinson’s Disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, Progression, and Mortality. Neurology 1967, 17, 427. [Google Scholar] [CrossRef]
- Mańka, S.; Baj, Z.; Majewska, E. The Influence of Melatonin on Apoptosis of Human Neutrophils. Postep. Hig. Med. Dosw. 2019, 73, 81–91. [Google Scholar] [CrossRef]
- Cipolla-Neto, J.; Amaral, F.G.D. Melatonin as a Hormone: New Physiological and Clinical Insights. Endocr. Rev. 2018, 39, 990–1028. [Google Scholar] [CrossRef] [PubMed]
- Vural, E.M.S.; van Munster, B.C.; de Rooij, S.E. Optimal Dosages for Melatonin Supplementation Therapy in Older Adults: A Systematic Review of Current Literature. Drugs Aging 2014, 31, 441–451. [Google Scholar] [CrossRef] [PubMed]
- del Valle Bessone, C.; Fajreldines, H.D.; de Barboza, G.E.D.; Tolosa de Talamoni, N.G.; Allemandi, D.A.; Carpentieri, A.R.; Quinteros, D.A. Protective Role of Melatonin on Retinal Ganglionar Cell: In Vitro an in Vivo Evidences. Life Sci. 2019, 218, 233–240. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical Perspectives in Neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed]
- Grivas, T.B.; Savvidou, O.D. Melatonin the “Light of Night” in Human Biology and Adolescent Idiopathic Scoliosis. Scoliosis 2007, 2, 6. [Google Scholar] [CrossRef]
- Scholtens, R.M.; van Munster, B.C.; van Kempen, M.F.; de Rooij, S.E.J.A. Physiological Melatonin Levels in Healthy Older People: A Systematic Review. J. Psychosom. Res. 2016, 86, 20–27. [Google Scholar] [CrossRef]
- Xie, S.; Fan, W.; He, H.; Huang, F. Role of Melatonin in the Regulation of Pain. J. Pain Res. 2020, 13, 331–343. [Google Scholar] [CrossRef]
- Tchekalarova, J.; Tzoneva, R. Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin. Int. J. Mol. Sci. 2023, 24, 3022. [Google Scholar] [CrossRef]
- Breen, D.P.; Nombela, C.; Vuono, R.; Jones, P.S.; Fisher, K.; Burn, D.J.; Brooks, D.J.; Reddy, A.B.; Rowe, J.B.; Barker, R.A. Hypothalamic Volume Loss Is Associated with Reduced Melatonin Output in Parkinson’s Disease. Mov. Disord. 2016, 31, 1062–1066. [Google Scholar] [CrossRef]
- Gunata, M.; Parlakpinar, H.; Acet, H.A. Melatonin: A Review of Its Potential Functions and Effects on Neurological Diseases. Rev. Neurol. 2020, 176, 148–165. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Du, Y.; Yuan, S.; Shen, J.; Lin, X.; Zheng, Z. Serum Melatonin Is an Alternative Index of Parkinson’s Disease Severity. Brain Res. 2014, 1547, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Golombek, D. Circadian Dysregulation in Parkinson’s Disease. Neurobiol. Sleep Circadian Rhythm. 2017, 2, 53–58. [Google Scholar] [CrossRef]
- Kataoka, H.; Saeki, K.; Kurumatani, N.; Sugie, K.; Obayashi, K. Melatonin Secretion in Patients with Parkinson’s Disease Receiving Different-Dose Levodopa Therapy. Sleep Med. 2020, 75, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Bolitho, S.J.; Naismith, S.L.; Rajaratnam, S.M.W.; Grunstein, R.R.; Hodges, J.R.; Terpening, Z.; Rogers, N.; Lewis, S.J.G. Disturbances in Melatonin Secretion and Circadian Sleep–Wake Regulation in Parkinson Disease. Sleep Med. 2014, 15, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.; Irving, A.J.; Harvey, J. Leptin Induces a Novel Form of NMDA Receptor-Dependent Long-Term Depression. J. Neurochem. 2005, 95, 396–405. [Google Scholar] [CrossRef]
- Ozdilek, B.; Kenangil, G. Serum Leptin Concentrations in Turkish Parkinson’s Disease Population. Parkinsons. Dis. 2014, 2014, 576020. [Google Scholar] [CrossRef]
- Fiszer, U.; Michałowska, M.; Baranowska, B.; Wolińska-Witort, E.; Jeske, W.; Jethon, M.; Piaścik-Gromada, M.; Marcinowska-Suchowierska, E. Leptin and Ghrelin Concentrations and Weight Loss in Parkinson’s Disease. Acta Neurol. Scand. 2010, 121, 230–236. [Google Scholar] [CrossRef]
- Evidente, V.G.H.; Caviness, J.N.; Adler, C.H.; Gwinn-Hardy, K.A.; Pratley, R.E. Serum Leptin Concentrations and Satiety in Parkinson’s Disease Patients with and without Weight Loss. Mov. Disord. 2001, 16, 924–927. [Google Scholar] [CrossRef]
- Markaki, E.; Ellul, J.; Kefalopoulou, Z.; Trachani, E.; Theodoropoulou, A.; Kyriazopoulou, V.; Constantoyannis, C. The Role of Ghrelin, Neuropeptide Y and Leptin Peptides in Weight Gain after Deep Brain Stimulation for Parkinson’s Disease. Stereotact. Funct. Neurosurg. 2012, 90, 104–112. [Google Scholar] [CrossRef]
- Adams, F.; Boschmann, M.; Lobsien, E.; Kupsch, A.; Lipp, A.; Franke, G.; Leisse, M.C.; Janke, J.; Gottschalk, S.; Spranger, J.; et al. Influences of Levodopa on Adipose Tissue and Skeletal Muscle Metabolism in Patients with Idiopathic Parkinson’s Disease. Eur. J. Clin. Pharmacol. 2008, 64, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, C.G.; Trenkwalder, C. Body Weight in Patients with Parkinson’s Disease. Mov. Disord. 2006, 21, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Park, K.-H.; Cho, Y.M.; Chung, S.S.; Cho, H.J.; Cho, S.Y.; Kim, S.J.; Kim, S.Y.; Lee, H.K.; Park, K.S. Resistin Is Secreted from Macrophages in Atheromas and Promotes Atherosclerosis. Cardiovasc. Res. 2006, 69, 76–85. [Google Scholar] [CrossRef] [PubMed]
- McTernan, P.G.; Kusminski, C.M.; Kumar, S. Resistin. Curr. Opin. Lipidol. 2006, 17, 170–175. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Mcternan, P.G.; Kumar, S. Role of Resistin in Obesity, Insulin Resistance and Type II Diabetes. Clin. Sci. 2005, 109, 243–256. [Google Scholar] [CrossRef]
- De Rui, M.; Inelmen, E.M.; Trevisan, C.; Pigozzo, S.; Manzato, E.; Sergi, G. Parkinson’s Disease and the Non-Motor Symptoms: Hyposmia, Weight Loss, Osteosarcopenia. Aging Clin. Exp. Res. 2020, 32, 1211–1218. [Google Scholar] [CrossRef]
- Jenner, P. Molecular Mechanisms of L-DOPA-Induced Dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef]
- Kataoka, H.; Sugie, K. Serum Adiponectin Levels between Patients with Parkinson’s Disease and Those with PSP. Neurol. Sci. 2020, 41, 1125–1131. [Google Scholar] [CrossRef]
- Sekiyama, K.; Waragai, M.; Akatsu, H.; Sugama, S.; Takenouchi, T.; Takamatsu, Y.; Fujita, M.; Sekigawa, A.; Rockenstein, E.; Inoue, S.; et al. Disease Modifying Effect of Adiponectin in Model of α-synucleinopathies. Ann. Clin. Transl. Neurol. 2014, 1, 479–489. [Google Scholar] [CrossRef]
- Sharma, J.C.; Bachmann, C.G.; Linazasoro, G. Classifying Risk Factors for Dyskinesia in Parkinson’s Disease. Park. Relat. Disord. 2010, 16, 490–497. [Google Scholar] [CrossRef]
- Sharma, J.C.; Macnamara, L.; Hasoon, M.; Vassallo, M.; Ross, I. Cascade of Levodopa Dose and Weight-Related Dyskinesia in Parkinson’s Disease (LD–WD-PD Cascade). Park. Relat. Disord. 2006, 12, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Cassani, E.; Cancello, R.; Cavanna, F.; Maestrini, S.; Di Blasio, A.M.; Liuzzi, A.; Pezzoli, G.; Barichella, M. Serum Adiponectin Levels in Advanced-Stage Parkinson’s Disease Patients. Park. Dis. 2011, 2011, 624764. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Parkinson’s Disease | Control | p Value | Power of a Test | ||
|---|---|---|---|---|---|---|
| No Dyskinesia | With Dyskinesia | |||||
| n (Female/Male) | 20 (9/11) | 24 (10/14) | 20 (8/12) | - | - | |
| Age [years] | Mean | 68.250 | 68.417 | 66.950 | 0.6220 | 0.1069 |
| SEM | 1.326 | 1.302 | 0.540 | |||
| Median | 69.000 | 68.500 | 67.500 | |||
| IQR | 7.250 | 6.500 | 4.000 | |||
| Body mass [kg] | Mean | 81.250 | 74.021 | 78.500 | 0.7065 | 1.0000 |
| SEM | 4.852 | 3.147 | 2.162 | |||
| Median | 78.500 | 74.000 | 77.500 | |||
| IQR | 21.500 | 14.500 | 15.250 | |||
| Height [cm] | Mean | 170.011 | 166.958 | 169.700 | 0.2443 | 0.2374 |
| SEM | 1.761 | 1.372 | 1.093 | |||
| Median | 169.000 | 167.500 | 169.000 | |||
| IQR | 10.000 | 6.500 | 6.250 | |||
| BMI [kg/m2] | Mean | 27.829 | 26.419 | 27.218 | 0.4982 | 0.9604 |
| SEM | 1.214 | 1.006 | 0.608 | |||
| Median | 26.037 | 25.510 | 27.184 | |||
| IQR | 4.183 | 4.412 | 3.899 | |||
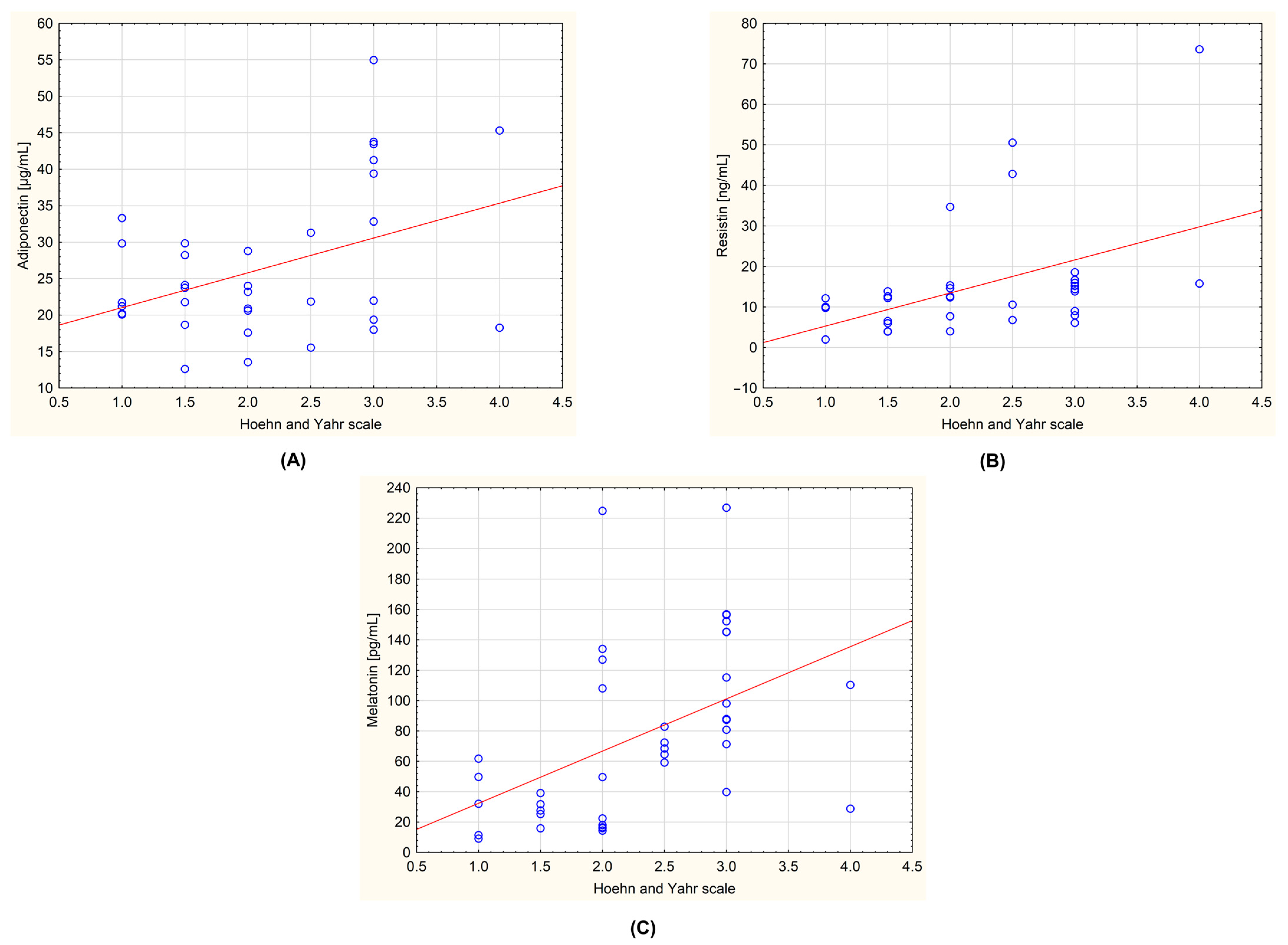
| Hoehn–Yahr scale | Mean | 1.500 | 2.813 | 0.000 | <0.0001 | 0.9999 |
| SEM | 0.089 | 0.108 | 0.000 | |||
| Median | 1.500 | 3.000 | 0.000 | |||
| IQR | 1.000 | 0.500 | 0.000 | |||
| Years since diagnosis [years] | Mean | 1.300 | 8.167 | 0.000 | <0.0001 | 1.0000 |
| SEM | 0.105 | 0.354 | 0.000 | |||
| Median | 1.000 | 8.000 | 0.000 | |||
| IQR | 1.000 | 3.250 | 0.000 | |||
| Drugs, Dose | PD No Dyskinesia [n = 20] | PD with Dyskinesia [n = 24] |
|---|---|---|
| no treatment | 0 | 0 |
| madopar HBS | 14 | 12 |
| madopar 250 + HBS | 0 | 7 |
| madopar HBS + madopar 125 | 0 | 3 |
| madopar HBS + madopar 125 + madopar 625 | 1 | 1 |
| madopar 625 + HBS | 4 | 0 |
| madopar 125 + madopar 125 | 1 | 0 |
| madopar x4 | 0 | 1 |
| Parameter | Parkinson’s Disease | Control | p Value | Power of a Test | ||
|---|---|---|---|---|---|---|
| No Dyskinesia | With Dyskinesia | |||||
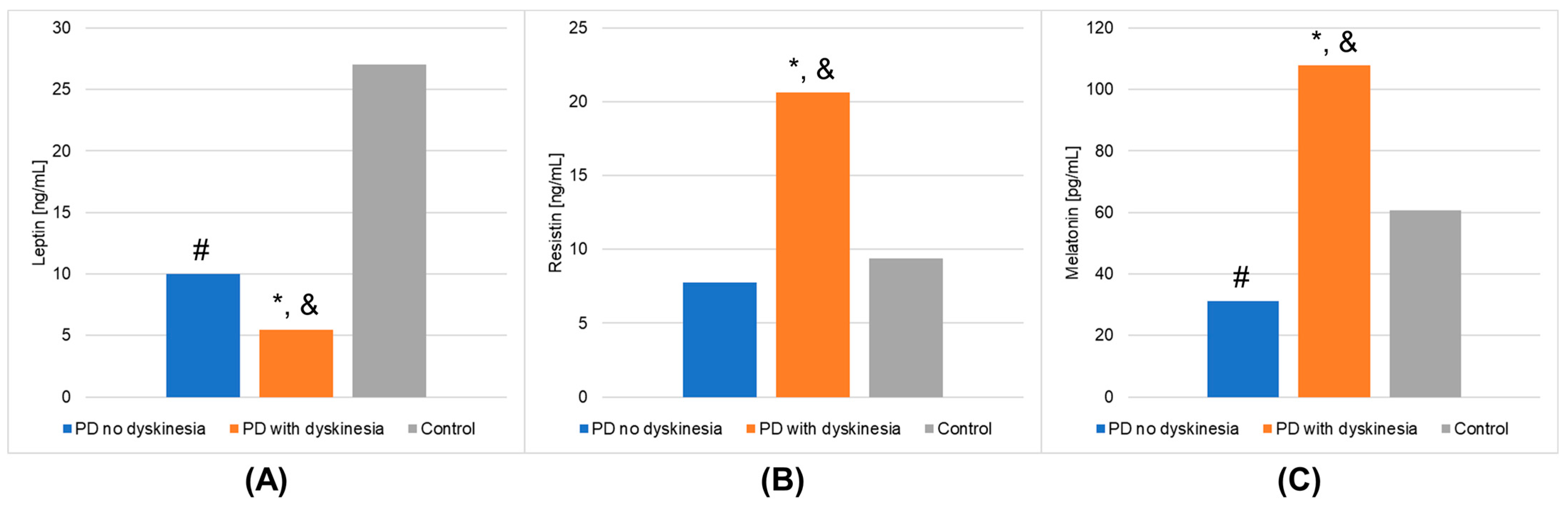
| Adiponectin [µg/mL] | Mean | 22.833 | 29.778 | 29.082 | 0.0626 | 0.9998 |
| SEM | 1.486 | 2.824 | 1.981 | |||
| Median | 21.753 | 23.595 | 30.513 | |||
| IQR | 8.638 | 20.103 | 7.331 | |||
| Leptin [ng/mL] | Mean | 9.983 | 5.431 | 27.030 | <0.0001 | 1.0000 |
| SEM | 1.414 | 0.933 | 2.860 | |||
| Median | 8.725 | 4.070 | 26.664 | |||
| IQR | 7.995 | 4.076 | 19.910 | |||
| Resistin [ng/mL] | Mean | 7.768 | 20.645 | 9.392 | 0.0013 | 0.9082 |
| SEM | 1.145 | 4.025 | 0.993 | |||
| Median | 7.140 | 14.640 | 9.143 | |||
| IQR | 8.188 | 6.159 | 4.724 | |||
| Melatonin [pg/mL] | Mean | 31.205 | 107.742 | 60.580 | <0.0001 | 0.9997 |
| SEM | 6.256 | 10.719 | 4.844 | |||
| Median | 23.841 | 93.025 | 60.292 | |||
| IQR | 17.858 | 74.497 | 31.985 | |||
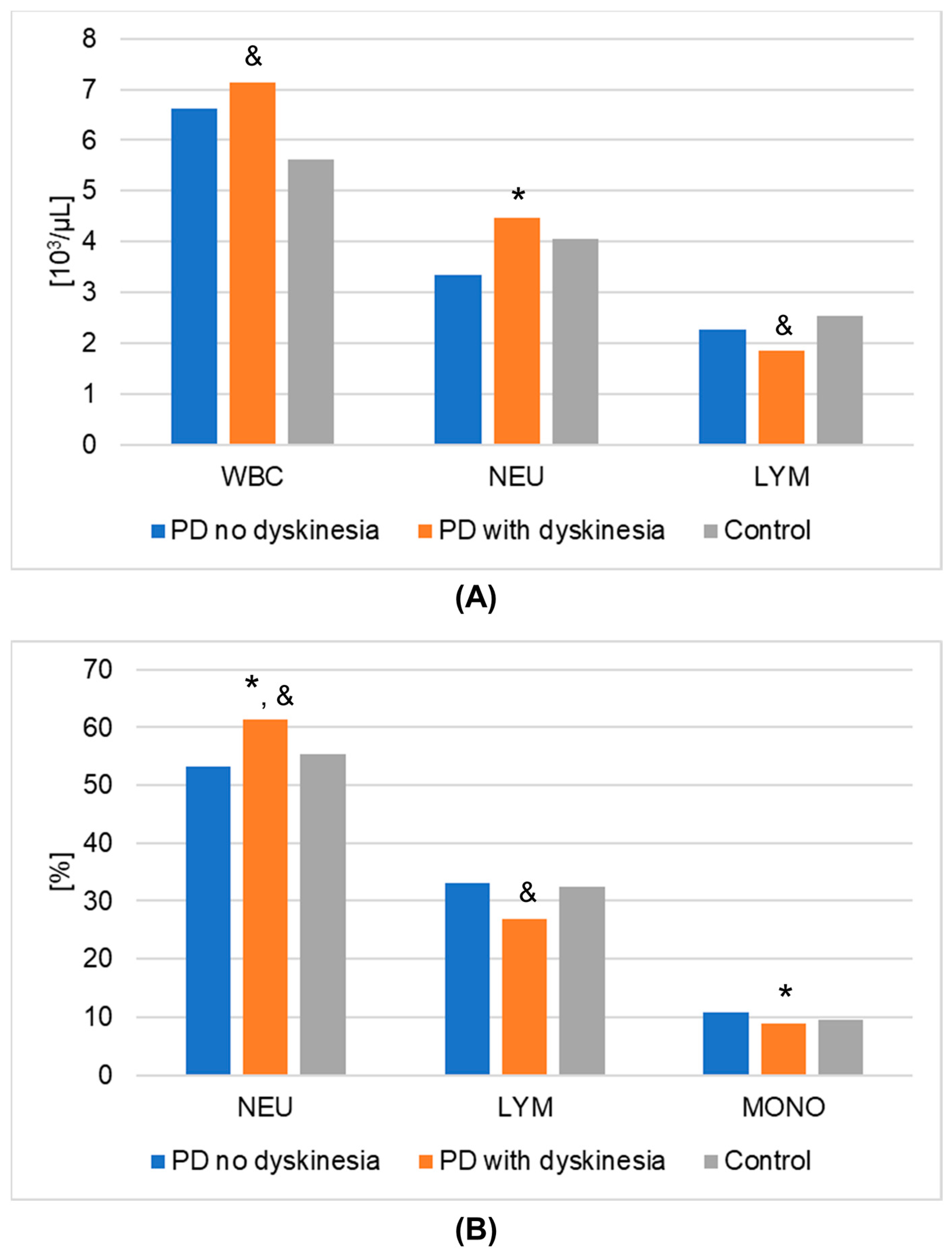
| WBC [103/µL] | Mean | 6.635 | 7.150 | 5.625 | 0.0245 | 0.5693 |
| SEM | 0.511 | 0.388 | 0.211 | |||
| Median | 6.570 | 6.725 | 5.350 | |||
| IQR | 2.195 | 2.025 | 1.475 | |||
| RBC [106/μL] | Mean | 4.641 | 4.622 | 4.382 | 0.1117 | 0.3545 |
| SEM | 0.097 | 0.093 | 0.090 | |||
| Median | 4.715 | 4.620 | 4.370 | |||
| IQR | 0.480 | 0.762 | 0.283 | |||
| Hb [g/dL] | Mean | 14.225 | 14.154 | 13.865 | 0.6297 | 0.1053 |
| SEM | 0.232 | 0.295 | 0.273 | |||
| Median | 14.500 | 14.000 | 14.100 | |||
| IQR | 1.425 | 2.025 | 1.600 | |||
| HCT [%] | Mean | 42.090 | 40.421 | 40.560 | 0.1383 | 0.9929 |
| SEM | 0.686 | 1.747 | 0.396 | |||
| Median | 42.850 | 41.150 | 40.200 | |||
| IQR | 4.475 | 4.800 | 1.375 | |||
| MCV [fL] | Mean | 90.915 | 89.504 | 90.135 | 0.9075 | 0.9579 |
| SEM | 0.817 | 2.013 | 1.000 | |||
| Median | 91.000 | 91.900 | 91.300 | |||
| IQR | 3.750 | 5.125 | 4.400 | |||
| MCH [pg] | Mean | 30.700 | 30.588 | 31.050 | 0.3596 | 0.2495 |
| SEM | 0.273 | 0.291 | 0.341 | |||
| Median | 30.700 | 30.700 | 31.200 | |||
| IQR | 1.150 | 1.675 | 1.825 | |||
| MCHC [g/dL] | Mean | 33.765 | 33.471 | 33.767 | 0.1925 | 0.1517 |
| SEM | 0.162 | 0.174 | 0.316 | |||
| Median | 33.700 | 33.400 | 34.055 | |||
| IQR | 0.850 | 0.625 | 1.666 | |||
| RDW [%] | Mean | 13.710 | 14.079 | 14.115 | 0.2986 | 0.2080 |
| SEM | 0.172 | 0.184 | 0.235 | |||
| Median | 13.600 | 13.900 | 14.150 | |||
| IQR | 0.875 | 1.150 | 0.850 | |||
| PLT [103/μL] | Mean | 234.450 | 217.667 | 254.250 | 0.0654 | 0.4336 |
| SEM | 13.984 | 9.871 | 8.587 | |||
| Median | 216.500 | 211.000 | 246.000 | |||
| IQR | 83.500 | 81.750 | 59.750 | |||
| MPV [fL] | Mean | 10.705 | 10.925 | 9.315 | <0.0001 | 0.9889 |
| SEM | 0.201 | 0.221 | 0.212 | |||
| Median | 10.900 | 10.850 | 9.050 | |||
| IQR | 1.250 | 1.375 | 1.150 | |||
| % NEU [%] | Mean | 53.320 | 61.458 | 55.293 | 0.0059 | 0.7305 |
| SEM | 2.446 | 1.669 | 1.242 | |||
| Median | 55.700 | 62.350 | 55.410 | |||
| IQR | 18.425 | 10.800 | 9.389 | |||
| % LYM [%] | Mean | 33.075 | 26.925 | 32.541 | 0.0168 | 0.6166 |
| SEM | 2.157 | 1.555 | 1.186 | |||
| Median | 33.350 | 24.750 | 31.589 | |||
| IQR | 18.800 | 11.450 | 6.511 | |||
| % MONO [%] | Mean | 10.730 | 8.888 | 9.468 | 0.0447 | 0.9950 |
| SEM | 0.647 | 0.615 | 0.431 | |||
| Median | 9.900 | 8.200 | 9.352 | |||
| IQR | 2.850 | 3.225 | 2.967 | |||
| % EOS [%] | Mean | 2.740 | 2.196 | 2.362 | 0.6014 | 0.3214 |
| SEM | 0.355 | 0.260 | 0.275 | |||
| Median | 2.400 | 2.050 | 2.276 | |||
| IQR | 2.450 | 1.100 | 1.444 | |||
| % BASO [%] | Mean | 0.495 | 0.425 | 0.489 | 0.5830 | 0.0548 |
| SEM | 0.080 | 0.069 | 0.057 | |||
| Median | 0.300 | 0.350 | 0.541 | |||
| IQR | 0.450 | 0.325 | 0.426 | |||
| NEU [103/μL] | Mean | 3.348 | 4.477 | 4.058 | 0.0173 | 0.8609 |
| SEM | 0.257 | 0.271 | 0.288 | |||
| Median | 3.200 | 4.485 | 4.271 | |||
| IQR | 1.945 | 1.470 | 2.481 | |||
| LYM [103/μL] | Mean | 2.261 | 1.846 | 2.531 | 0.0064 | 0.7222 |
| SEM | 0.166 | 0.137 | 0.148 | |||
| Median | 2.105 | 1.770 | 2.668 | |||
| IQR | 1.253 | 0.865 | 0.981 | |||
| MONO [103/μL] | Mean | 0.698 | 0.691 | 0.715 | 0.2321 | 0.0505 |
| SEM | 0.043 | 0.080 | 0.051 | |||
| Median | 0.685 | 0.565 | 0.701 | |||
| IQR | 0.230 | 0.248 | 0.315 | |||
| EOS [103/μL] | Mean | 0.187 | 0.256 | 0.208 | 0.3068 | 0.0540 |
| SEM | 0.025 | 0.099 | 0.022 | |||
| Median | 0.210 | 0.140 | 0.214 | |||
| IQR | 0.168 | 0.068 | 0.127 | |||
| BASO [103/μL] | Mean | 0.029 | 0.030 | 0.026 | 0.8760 | 0.0500 |
| SEM | 0.003 | 0.003 | 0.001 | |||
| Median | 0.030 | 0.030 | 0.024 | |||
| IQR | 0.015 | 0.020 | 0.012 | |||
| Parameter | p Value | ||
|---|---|---|---|
| No Dyskinesia vs. with Dyskinesia | No Dyskinesia vs. Control | With Dyskinesia vs. Control | |
| Hoehn–Yahr scale | <0.0001 | <0.0001 | <0.0001 |
| Years since diagnosis | <0.0001 | <0.0001 | <0.0001 |
| Leptin | 0.0360 | <0.0001 | <0.0001 |
| Resistin | 0.0030 | 0.8922 | 0.0064 |
| Melatonin | <0.0001 | 0.0025 | 0.0010 |
| WBC | 0.6085 | 0.1895 | 0.0192 |
| MPV | 1.0000 | 0.0002 | <0.0001 |
| % NEU | 0.0280 | 0.8577 | 0.0151 |
| % LYM | 0.0777 | 0.9951 | 0.0192 |
| % MONO | 0.0455 | 0.6250 | 0.6476 |
| NEU | 0.0120 | 0.2033 | 1.0000 |
| LYM | 0.1262 | 0.4398 | 0.0050 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanowski, J.; Kozerawski, K.; Falęcka, W.; Dudek, D.; Lisewska, B.; Lisewski, P.; Nuszkiewicz, J.; Wesołowski, R.; Wojtasik, J.; Mila-Kierzenkowska, C.; et al. Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies. Metabolites 2023, 13, 668. https://doi.org/10.3390/metabo13050668
Milanowski J, Kozerawski K, Falęcka W, Dudek D, Lisewska B, Lisewski P, Nuszkiewicz J, Wesołowski R, Wojtasik J, Mila-Kierzenkowska C, et al. Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies. Metabolites. 2023; 13(5):668. https://doi.org/10.3390/metabo13050668
Chicago/Turabian StyleMilanowski, Jan, Kamil Kozerawski, Weronika Falęcka, Dominik Dudek, Beata Lisewska, Paweł Lisewski, Jarosław Nuszkiewicz, Roland Wesołowski, Jakub Wojtasik, Celestyna Mila-Kierzenkowska, and et al. 2023. "Changes in the Secretion of Melatonin and Selected Adipokines during the Progression of Parkinson’s Disease—Preliminary Studies" Metabolites 13, no. 5: 668. https://doi.org/10.3390/metabo13050668