
Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy

Assistance Publique-Hôpitaux de Paris, Service de Gynécologie-Obstétrique, Hôpital Bicêtre, Université Paris Saclay, 94270 Le Kremlin-Bicetre, France
*
Author to whom correspondence should be addressed.
Metabolites 2023, 13(5), 633; https://doi.org/10.3390/metabo13050633
Submission received: 29 March 2023
/
Revised: 26 April 2023
/
Accepted: 3 May 2023
/
Published: 6 May 2023
(This article belongs to the Special Issue Fetal–Maternal–Neonatal Metabolomics)
Abstract
:Thyroid hormones and iodine are required to increase basal metabolic rate and to regulate protein synthesis, long bone growth and neuronal maturation. They are also essential for protein, fat and carbohydrate metabolism regulation. Imbalances in thyroid and iodine metabolism can negatively affect these vital functions. Pregnant women are at risk of hypo or hyperthyroidism, in relation to or regardless of their medical history, with potential dramatic outcomes. Fetal development highly relies on thyroid and iodine metabolism and can be compromised if they malfunction. As the interface between the fetus and the mother, the placenta plays a crucial role in thyroid and iodine metabolism during pregnancy. This narrative review aims to provide an update on current knowledge of thyroid and iodine metabolism in normal and pathological pregnancies. After a brief description of general thyroid and iodine metabolism, their main modifications during normal pregnancies and the placental molecular actors are described. We then discuss the most frequent pathologies to illustrate the upmost importance of iodine and thyroid for both the mother and the fetus.
1. Introduction
Iodine is an essential mineral nutrient, mainly involved in thyroid metabolism. It is a key component of thyroid hormones (TH), which are essential for kidney, liver and brain metabolism. A healthy adult individual contains 15–20 mg of iodine, 70–80% of which is located in the thyroid gland [1,2]. Iodine deficiency (ID) is frequent worldwide, affecting approximately 40% of the world’s population [1]. Pregnancy tends to worsen ID, and around two third of European pregnant women are iodine deficient [3]. Maternal thyroid metabolism can be heavily impaired during pregnancy because of ID, auto-immune disease or pregnancy-related changes. As TH are mandatory for fetal development, the placenta plays a crucial role during the pregnancy [2,4]. Indeed, it provides the fetus with maternal TH as long as the fetal thyroid is not fully functional [5]. It also supplies the fetal thyroid with iodine in order to synthesize fetal TH from the end of the first trimester [5,6]. Here, we provide a narrative review of the data concerning normal and pathological iodine and thyroid metabolism during pregnancy.
2. General Iodine and Thyroid Metabolism
2.1. Iodine Absorption and Excretion
Iodine’s primary source is dietary intake. Animals and plants of marine origin usually present the highest amount of iodine among types of food because they concentrate iodine from seawater. Unfortunately, dietary iodine intake is rather low all around the world. Indeed, most of the consumed sources only provide 3–80 µg per serving, whereas WHO recommends a daily dose of 150 µg of iodine [7,8]. The main iodine sources in many western countries are bread and dairy products, and the low iodine amount of those aliments explains the high prevalence of ID in those areas [9]. The level of iodine in bread is linked to its addition in flour, while that of dairy products comes from the products themselves. In Asian countries—especially in Japan—consumption of seawater food diminishes the prevalence of ID [10]. In order to maximize dietary iodine intake, cooking salt is iodized worldwide [11,12]. This constitutes a major source of additional iodine [13,14]. However, its contribution alone is not completely sufficient to ensure adequate intake [15].
In addition to dietary intake, iodine is efficiently recycled by the human body. Bloodstream iodine that is not imported into the thyroid or that comes from TH degradation in peripheral tissues is accumulated in the salivary glands and stomach [16]. Both can excrete iodine, increasing the pool that will be absorbed by the duodenum, jejunum, and ileum.
One of the main proteins involved in iodine metabolism is the NIS symporter (Sodium/Iodine Symporter, SLC515), a transmembrane glycoprotein weighting 90–100 kDa [17]. NIS is one of the most effective iodine transporters as it has a twice-higher affinity for iodine than the other ones. With its 13 transmembrane domains, it can transport two sodium cations (Na+) for each iodine anion (also known as iodide, I−). The transport direction depends on the sodium gradient, which is created by Na+/K+ ATPase. The energy obtained by exporting sodium—which follows the electrochemical gradient of Na+/K+ ATPase—is used to import iodine against its own electrochemical gradient.
In the salivary glands, NIS is located on the ductal cells’ basal membrane. It imports iodine from the bloodstream into the salivary gland cells. Iodine can then diffuse by osmosis to the apical surface, where it is taken in charge by other transporters such as CFTR, Anoctamin 1 (Tmem16A) and/or Pendrin (SLC26A4) in order to release it into the oral cavity thus increasing the diet pool [18,19]. The intracellular iodine pool can influence its own transport. For instance, Pendrin can be activated by a high concentration of intracellular iodine [20]. A similar phenomenon takes place in the stomach, where the accumulation of NIS on the basal membrane of mucin-secreting and parietal cells suggests iodine transfer from the bloodstream to the stomach cells [21]. The export from those cells to the gastric fluid is realized by CFTR and Anoctamin 1 [22].
Dietary and recycled iodine accumulates in the gut lumen. They are mainly absorbed in the duodenum, jejunum, and ileum [23]. NIS once again plays a major role in iodine import, this time located on the apical side of enterocytes, allowing import into the cells in a manner similar to the one in the salivary glands (working in conjunction with the Na/K ATPases). How iodine is exported from the gut cells to the bloodstream remains unclear, but chloride and iodine symporters located on the basal surface of enterocytes, such as Chloride Channel 2 (ClC-2), may have a role in this process [16,24].
When dietary iodine intake is adequate, more than 80% of iodine is passively excreted in the urine via the glomerulus [25]. When it is inadequate, the kidney is the ultimate location to reabsorb iodin. This reabsorption probably involves the usual transporters (NIS, CFTR, Pendrin, Anoctamin 1), but the iodine metabolism in the kidney has not been fully elucidated [26,27,28,29].
Urinary iodine concentration results in a balance between dietary intake, thyroidal metabolism, and renal function. It is the most convenient method to assess the iodine status. The best urinary iodine excretion evaluation method applied to a population consists of a 24 h urinary collection [30,31]. Unfortunately, it has low individual value. A population with correct intakes should present a urinary iodine concentration between 100–199 µg/L [32]. ID is suspected when UIC is <100 µg/L in non-pregnant women (severe if IUC <20 µg/L, moderate if IUC = 20 − 49 µg/L and mild if IUC = 50 − 99 µg/L). The risk of iodine-induced hyperthyroidism exists if the IUC is >300 µg/L [33].
2.2. Thyroid Metabolism
Iodine (I) is the oxidized form of iodide (I−) and the main component of TH. Those TH are phenolic rings bound with an ether link. They can be iodinated in three positions, forming 3,5,3′-tri-iodo-L-thyronine (also known as triiodothyronine, T3) or four positions, forming 3,5,3′,5′-tetra-iodo-L-thyronine (also known as thyroxine, T4) [17,34].
I− is imported from the blood circulation into the thyrocytes (thyroid follicular epithelial cells) by NIS–located on their basolateral side–and released into the follicular lumen by Pendrin, and eventually Chloride Channel 5 (ClC5) and/or Anoctamin. The fraction of iodine imported into the thyroid varies according to the iodine intake [2]. Less than 20% of the absorbed iodine is taken up by the thyroid when the dietary intake is adequate, whereas more than 80% can be imported in cases of chronic ID.
Into the follicular lumen, accumulated I− is oxidized into diatomic iodine (I2) by Thyroid peroxidase (also called thyroperoxidase or iodide peroxidase, TPO) [17,34]. TPO is located on the apical side of the thyroid epithelial cell and faces the follicular lumen. The energy needed for this oxidation is provided by hydrogen peroxide (H2O2), generated mainly by a dual oxidase 2 and additionally by a dual oxidase 1 [35,36]. Dual oxidase 1 and 2 both belong to the NADPH oxidase family.
Iodine is afterward covalently linked on three or four positions of tyrosyl residues on Thyroglobulin (TG) [34]. TG is synthetized in the thyrocytes by the rough endoplasmic reticulum ribosomes and exported by exocytosis into the follicular lumen [34]. Its concentration in the follicular lumen is approximately 100–400 mg/mL. Iodine binding on TG creates iodinated TH intermediates, 3-mono-iodotyrosine (MIT) and 3,5-di-iodotyrosine (DIT). In order to form T3 and T4, MIT and DIT are respectively coupled on the TG polypeptide backbone by TPO. Follicular TG is re-internalized into thyrocytes by endocytosis and proteolyzed. This frees thyroid hormones from the polypeptide backbone. T3 and T4 are then secreted into the blood stream by transporters such as (Mono-Carboxylate Transporter 8 (MCT8) at the basolateral membrane of thyrocytes [34].
T4 is considered to be a prohormone, as it is three to four times less potent than T3 and can be converted into T3 by deiodinases [37]. The ratio of T4 to T3 released into the bloodstream is approximately 14:1, and the major part of T3 in the human body is thus generated in peripheral tissues by the deiodination of T4.
In the blood stream, TH are in the vast majority (more than 99%) transported by proteins such as albumin, Thyroxine-Binding Globulin (TBG) or transthyretin [38]. Only a fraction of TH are unbound; they are called free T3 (fT3) or free T4 (fT4) and are biologically active.
Thyroid-stimulating hormone (TSH) is secreted by the pituitary gland and plays a major role in TH synthesis. It binds the TSH receptor (TSHR) on the thyrocyte basolateral membrane [39]. This activates a G-protein signal cascade which results in the intracellular rising of cyclic adenosine monophosphate. The ultimate consequence is the increase in TH synthesis. The TSH/TSHR link has many other effects. It up-regulates the NIS activity, thus raising the iodine trapping and stimulating the iodination of TG in the follicular lumen and the conjugation of tyrosine residues. It also increases endocytosis of the iodinated thyroglobulin protein across the apical membrane back into the follicular cell, proteolysis of iodinated thyroglobulin to form free T3 and T4, and secretion of T3 and T4 across the thyrocytes basolateral membrane.
Three transcription factors are of the upmost importance for NIS gene transcription in the thyrocytes: NkX2.1, PAX8 and FOXE1. Nkx2.1 binds a proximal NIS promoter, up-regulating NIS activity [40,41]. PAX8 has the highest impact on NIS transcription, binding a distant region of the proximal promoter called the NIS Upstream Enhancer (NUE) [42,43,44]. Each promoter has one PAX8 binding site. In rodents, Nkx2.1 and FOXE1 are also able to bind the NUE, but this has not been proven yet in man [42,45]. Many other factors are able to enhance the NUE, such as a cAMP response element (CRE-like), the GLI-similar 3 (GLIS3), the Nuclear Factor-kappa B (NF-kB), the Upstream Transcription Factor 1 (USF1). GLIS3 is essential in order to enhance the NIS transcription mediated by TSH, and the NF-KB p65 subunit seems to interact with PAX8 to increase NIS transcription [46,47,48].
3. Physiologic Metabolism during Pregnancy
Thyroid and iodine metabolism are modified during pregnancy to ensure proper maternal and fetal thyroid function.
The human placenta is hemochorial with villous trophoblast; maternal and fetal circulations are fully separated by the placental barrier. Exchanges between these two circulations take place in the syncytiotrophoblast (SCT), where SCT cells are in contact with maternal circulation on their apical side and in contact with fetal capillaries on their basal side [49]. Different patterns are involved for substances to cross the placenta: passive transport (passive diffusion and facilitated diffusion), active transport with transporters, and endocytosis/exocytosis. Passive diffusion is the main placental transport mean for non-ionized liposoluble with low molecular weight (<500 Da) molecules. However, most of the molecules implied in iodine and thyroid metabolism require different means of transport because of their specificities. Placenta is permeable to iodine, anti-thyroid drugs (ATD), TSH Receptor Autoantibodies (TRAb), and Thyroid Peroxidase Antibodies (TPOAb).
3.1. Iodine
Iodine requirement is rapidly increased by 50% during pregnancy in order to maintain maternal and fetal homeostasis [50]. As the fetus is unable to produce its own TH during the first trimester, it initially fully depends on maternal TH import. Maternal T4 production is thus rapidly increased by approximately 50% to meet both mother and fetal needs, which implies higher iodine needs. The fetal thyroid becomes fully functional after 17–19 WG but still requires adequate fetal iodine supply in order to operate [4,5]. Finally, median UIC is increased during pregnancy because of a physiological increased renal clearance. Different studies revealed that the median UIC was higher in pregnant women when compared to non-pregnant ones (for instance, 172 µg/L versus 132 µg/L for Stilwell et al.) [51,52,53,54]. Moreover, the percentage of pregnant women with UIC < 50 µg/L is inferior to the percentage of non-pregnant women (7.3% versus 16.8% for Stilwell et al.). The median UIC is believed to be constant throughout pregnancy, but this still has to be proven.
The increased needs and the frequent iodine deficiency have led many scientific societies to recommend a systematic iodine supplementation with 150–250 µg of iodine/day for women who are pregnant, planning pregnancy, or breastfeeding. [55,56,57]. A median IUC of 150–249 µg/L is believed to indicate adequate iodine intake in pregnant women [58,59]. Unfortunately, ID during pregnancy persists and can cause adverse maternal and fetal outcomes, as will be discussed further.
Maternal iodine crosses the placenta from the maternal circulation into the fetal circulation. NIS and Pendrin are deeply involved in this transport. Even though they were initially described in the thyroid, there is evidence of their presence in placental tissue [60,61,62]. The precise mechanisms involved in fetal iodine import remain unknown. Immunohistochemical analysis of the primary culture of villous cytotrophoblastic cells and of villous syncytiotrophoblast showed that Pendrin and NIS are mainly expressed respectively at the brush border membrane of SCT—facing the maternal blood—and in the cytotrophoblast (CT) [63]. However, these locations would imply an inverted iodine transport direction when compared to thyrocytes. On the contrary, a study on BeWo choriocarcinoma cells used as a model of iodine transport by placenta suggests more classic mechanisms, iodine being imported into the trophoblast through NIS and being exported into the fetal circulation through Pendrin [64].
NIS and Pendrin placental mRNA expression obtained using RT-PCR vary throughout pregnancy [63]. NIS mRNA expression is similar during the first and the third trimesters, whereas Pendrin mRNA expression is higher during the third trimester. This is consistent with the increased fetal iodine needs at term. Pendrin’s utmost importance is highlighted by Pendred syndrome. This autosomal recessive disease is caused by SLC26A4 mutation on chromosome 7q31 and makes Pendrin unfunctional [63]. It is characterized by severe hypothyroidism, goiter and variable deafness.
Regulation of placental and thyroidal iodine transport is HCG-dependent. HCG (human chorionic gonadotrophin) is a specific to humans 37 kDa glycoprotein composed of two glycosylated subunits, non-covalently linked [65]. Alpha sub-unit contains 92 amino acids and is common with the alpha sub-units of other pituitary gonadotropin hormones (LH, FSH, and TSH) [66]. It contains two N-linked oligosaccharide sidechains. Conversely, the beta sub-unit is hormone specific. It is composed of 145 amino acid residues with two N-linked and four O-linked oligosaccharides [66]. Even though the HCG beta sub-unit confers the hormone’s biological specificity, it shares an overall three-dimensional structure with the TSH beta sub-unit. Indeed, both possess 12 half-cysteine residues, one N-linked oligosaccharide and three disulfide bonds at highly conservated positions [67]. In addition, glycoproteins hormone receptors of HCG and TSH are highly homogenous. They have a single polypeptide chain forming seven hydrophobic alpha-helices that constitute transmembrane segments [68].
The similarities between the beta sub-units and the receptors explain why HCG is able to cross-react with the TSH receptor [69]. In Jar and BeWo cells exposed to HCG, NIS expression and iodine import are increased [70,71]. On the contrary, they are decreased when HCG is withdrawn.
Many different pregnancy-related hormones can affect iodine placental import. When exposed in vitro to HCG, oxytocin, and prolactin, primary cultures of term placental trophoblast cells increased Iodine 125 (I125) uptake by 60, 45, and 32%, respectively [72]. Accordingly to this increased I125 uptake, NIS mRNA was higher in exposed cells when compared to controls. Surprisingly, no increased expression of Pendrin was observed, which leads us to believe that other unknown mechanisms must be involved.
3.2. Maternal and Fetal Thyroid
3.2.1. Maternal Thyroid Metabolism
Maternal thyroid activity is highly impacted by pregnancy for various reasons. As described earlier, HCG has a TSH-like effect on thyroid tissue [69]. This leads to a rise in fT4 synthesis and, conversely, to a decrease in TSH hypophyseal secretion by a negative feed-back [76]. The fT4 synthesis rise is temporary, lasting mostly during the first trimester. It ensures adequate fT4 fetal supply until the fetal thyroid is fully functional. Indeed, the fT4 synthesis diminishes when the HCG serum concentration falls at the end of the first trimester [76,77]. TSH is usually normalized in 2 to 3 weeks after the end of the negative feed-back phenomenon, but fT4 concentration constantly declines throughout the pregnancy [78,79].
Also, plasma volume expansion in pregnant women tends to increase the T3 and T4 pool size. In iodine-deficient areas, the physiological rise of iodine renal clearance can be responsible for a decrease in TH synthesis.
3.2.2. Fetal Thyroid
The fetal thyroid gland develops from a thickening of the pharyngeal floor from the median oral endoderm and from two caudal extensions of the fourth branchial pouches at approximately day 24 of human gestation [81]. The thyroid is initially located at the foramen caecum, which is the junction of the anterior two-thirds and posterior one-third of the tongue. At four WG, the embryonic structures are fused, and the thyroid gland consists of two lateral lobes with superior and inferior poles connected by a median isthmus [82]. When the isthmus is absent, the two lobes are distinct from each other. Between five and seven WG, the thyroid migrates to its almost definitive position between the second and the fourth cervical vertebrae [83]. This descent through the neck tissues mainly happens during the first trimester and follows the thyroglossal duct. In 50% of the cases, the distal part of the thyroglossal duct differentiates into the additional pyramidal lobe of the thyroid gland [82]. The rest of the duct is afterward obliterated before the end of the first trimester in most cases. After the first trimester, the thyroid migration drastically slows down but still continues throughout the pregnancy. Its final site between the 5th cervical and the 1st thoracic vertebrae is probably reached only in adults [83].
Alongside its migration, thyroid tissues are functionally differentiated [84,85]. The first stage of this differentiation takes place between seven to nine WG. At that time, the thyroid is only composed of unpolarized thyrocyte precursors developed from the medial endoderm, which are unable to synthesize TH. Between 10 and 11 WG, small follicles are formed by polarizing thyrocytes, which allow the fetal thyroid to accumulate iodine and synthesize TH. In parallel, calcitonin-secreting cells (also known as parafollicular or C cells) are developed from the fourth branchial pouches [40,86]. By 12 WG, the thyroid organogenesis is complete.
The NIS gene and the NIS gene transcription factors (nkx2.1, FOXE1 and PAX8) are highly involved in fetal thyroid development [85]. Trueba et al. studied human embryos and fetuses from seven to 33 WG with quantitative PCR to analyze mRNA expression [87]. They proved that FOXE1, PAX8 and NKX2.1 were constantly expressed from seven to 33 WG. NIS mRNA was the last to be detected at eight WG, but their quantity greatly rose between nine and 10 WG, underlining NIS’s key role in fetal thyroid function.
Fetal TH synthesis begins around 10 WG, and fetal T4 can be detected in the fetal circulation by 11 WG [5,6]. It is fully dependent on the maternal iodine transport through the placenta. However, it is not entirely functional before 17–19 WG [5]. Until then, maternal TH are mandatory for fetal physiological development, and the fetus relies entirely on them in the first trimester [4].
After the beginning of the second trimester, TH from both maternal and fetal thyroid glands is present in the fetal circulation. The fetal concentration of total and free circulating T3, T4 and TGB increase throughout pregnancy and reach the mean adult values at approximately 36 WG. This reflects the increased maturation of the fetal pituitary gland and liver [6]. However, fetal TSH is also increased even when TH are high, suggesting that the fetal pituitary gland sensitivity to negative feed-back is limited.
3.3. Thyroid Metabolites Placental Transport
3.3.1. TH Placental Transport
Placenta is the interface between the mother and fetal circulations. It is poorly permeable to T4 and T3, into and out of the fetal circulation [5]. However, fetuses with athyreosis display at least one-third to half of the mother’s fT4 serum concentration. This indicates a potential consequent mother-to-fetus TH passage under high gradient concentration. Transplacental T4 circulation seems to be asymmetrical, according to an in vitro study [88]. The maternal-to-fetal passage is limited, but the fetal-to-maternal is way faster. Placenta could thus play a protective role in order to avoid too high T4 concentration in fetal circulation, but more studies are needed.
The exact mechanisms of TH import across the placenta and into the fetal circulation still have to be studied. TH bound to TGB, transthyretin, or serum albumin in the maternal serum are transported to the placenta [89]. Afterward, cellular membrane transporters are in charge of carrying TH into and across the placenta.
Placental transport patterns vary throughout the pregnancy. From five to six WG to the end of the first trimester, the exocoelomic cavity is a privileged area for maternal-fetal exchange. TH are carried across the placenta into the exocoelomic cavity and then pass into the fetal circulation through the yolk sac and then the fetal intestines [90,91]. The yolk sac is an extension of the fetal intestinal circulation and is able to absorb and deliver different proteins—such as TH—to the fetus. Early T3 and T4 concentrations in fetal tissues are more than 100-fold times lower than in the maternal serum [5]. However, the concentrations of fT4 in the coelomic and amniotic fluid are equivalent to approximately a third of the maternal one, reaching values high enough to induce biological effects.
From the second trimester, the placenta becomes the main area of materno-fetal exchange. TH are transported directly into the fetal blood via the placenta after crossing cytotrophoblast and/or syncytiotrophoblast [5].
TH transporters are involved in order for TH to cross the placenta. Their presence in human choriocarcinoma cell lines has been known for over 30 years [92]. More than 15 TH transporters have been identified, but only a fraction of those are involved in placental traffic [93,94]. mRNA sequencing of human placentas showed that L-type Amino-acid Transporters (LAT1 and LAT2), Mono-Carboxylate Transporters (MCT8 and MCT10), and Organic-Anion Transporting Peptides (OATP1A2 and OATP4A1) are the ones playing an active role in transporting TH to the fetus [95]. They are located on the plasma membranes of cytotrophoblasts and syncytiotrophoblast, and each one of them can interact with TH. Studies are still ongoing in order to determine how they link and transport TH. For instance, the T3 carboxyl group could be bound by H-bonds to a positively charged arginine and a histidine at the substrate-binding center of MCT8, but the exact transport mechanism remains unclear [96,97].
3.3.2. TSH
Placenta is usually considered to be impermeable to TSH. Cord blood TSH measurements in infants born with congenital absence of the anterior pituitary gland show very low TSH levels [110]. This suggests that maternal TSH cannot cross the placenta. However, an in vitro study of dually perfused human term placenta proved that TSH actually crosses both the placenta and the fetal membranes, but so sparingly that it cannot influence fetal thyroid metabolism [111]. TSH is present in amniotic fluid from 18 WG, and its fetal origin is confirmed by the high correlation between amniotic fluid and blood cord concentration [112]. Fetal TSH level slowly rises to a peak value of 15 mU/L at 25 WG [113]. Whether it remains stable or slightly decreases during the last trimester remains debated [114].
3.3.3. TRH
TRH is a hypothalamic-releasing factor produced by paraventricular neurons in the hypothalamus. After being produced and activated in the anterior pituitary, it binds TRH-receptors, inducing the synthesis and secretion of the TSH beta-subunit by the hypophysis.
Placenta is highly permeable to maternal TRH. It is present in the fetal brain way before the fetal hypothalamus is developed [115]. The fetal hypothalamus and hypophysis are functional from 20 WG, but TRH can be detected in fetal blood as soon as 12 WG. Maternal TRH could influence fetal TSH synthesis, but this is uncertain. TRH deprivation in rodent studies is not responsible for altered TSH synthesis, but it seems to affect fetal TSH after 37 WG [114,116].
Interestingly, TRH accelerates fetal lung maturation in rodents and increases the amount of surfactant secreted using extra-thyroidal pathway stimulation [117]. However, the widespread use of antenatal corticosteroids has supplanted that of TRH in this indication. Interestingly, TRH accelerates fetal lung maturation in rodents and increases the amount of surfactant secreted using extra-thyroidal pathway stimulation [117]. However, the widespread use of antenatal corticosteroids has supplanted that of TRH in this indication.
3.3.4. TRAb/TPOAb
Thyroid antibodies (TRAb and TPOAb) are both class G immunoglobulins (Ig G). They have a tetrameric T-like shaped structure containing two heavy and two light chains. Each IgG weight of approximately 150 kDa consists of an antigen-binding site (Fab) and a constant region (Fc) [118]. Fc binds Fc receptors on a wide variety of specific targets.
Studies revealed that Fab and Fc do not cross the placenta in a similar way. When radiomarked and injected into pregnant women, Fc and complete IgG transport are greatly more efficient than Fab transport alone [119]. This lead to the discovery of placental FcRN [120,121]. FcRN is a membranal protein of 40–45 kDa, non-covalently linked to a b2-microglobulin. It is highly sensitive to pH as its ligand affinity is a hundred times higher when exposed to neutral than acid pH (7.4 versus 6).
Placental FcRn is poorly expressed in CT. It is preferentially located in SCT and in the fetal capillary endothelial cells [122,123]. IgG placental transfer requires both FcRn and endocytosis [124,125,126]. FcRn binds IgG on the placental maternal side, and the FcRn-IgG complex is imported into and carried across SCT by endocytosis. The acid environment in the vesicle reinforces the FcRn-IgG link and protects it from lysosomal degradation. At the SCT basal side, the FcRn-IgG complex is freed from the vesicle and dissociated by the physiological neutral pH. IgG is transferred across the SCT basal membrane directly into the fetal capillaries of the placental villi and indirectly after stromal transit.
IgG transplacental transport begins at approximately 13 WG. Fetal IgG serum concentration increases throughout the pregnancy, starting at 5–10% of maternal blood concentration at 17–22 WG, reaching 50% of maternal blood concentration by 32 WG and finally exceeding maternal blood concentration (120–130%) at birth [127,128].
TRAb and TPOAb require active transport in order to cross the placenta because of their high molecular weight. This placental transport is mandatory for fetal metabolism and implies the FnCR [129,130]. However, the specific thyroid antibody pathways involved in placental import have not been identified yet.
3.4. Deiodinases
Deionidase are a family of selenoproteins. Each one of them has specific tissue regulation and distribution, and both deionidase 2 (D-II) and 3 (D-III) have been identified in the human placenta [131]. D-II is located in CT cells and inconstantly in SCT during the first trimester. D-II binds preferably T4 over rT3 and converts it into active T3 [132]. D-III is conversely located in SCT and is able to catalyze inner ring deiodination of T3 and T4, inactivating T3 to T2 or T4 to rT3 [133]. It has a higher affinity for T3 than D-I.
Placental D-III is much more active and highly expressed than D-II (approximately 200 times at the beginning of the pregnancy and up to 400 times at term). Both their activity and expression decrease throughout pregnancy and seem to be regulated by fetal circulating T4 [134,135]. This could explain why fetal T3 levels are relatively low and fetal rT3 are high. Polak suggested that this mechanism could cause low in-utero thermogenesis [114].
Deionidase is also expressed in fetal tissues. For instance, D-II is expressed in the fetal brain. It is believed to protect the fetal brain from hypothyroidism [136].
4. Maternal Pathologies during Pregnancy
4.1. Gestational Transient Hyperthyroidism (GTH)
HCG cross-stimulation of the TSH receptor is responsible for gestational transient hyperthyroidism in 2.4–11% of pregnant women [137,138]. It is usually more common between eight to 11 WG than 12–14 WG, as the HCG decreases at the end of the first trimester. The GTH is, by definition, transient, resolving with the decline of HCG. TSH concentrations are normalized in less than 3 weeks [138]. The more severe conditions are caused by hydatidiform moles/choriocarcinomas or multiple pregnancies because a large amount of HCG is secreted in those cases [139].
GTH is often revealed by pregnancy symptoms being more intense and severe than usual. In addition to asthenia and nausea, it may provoke tremors, tachycardia, vomiting, and weight loss (up-to hyperemesis gravidarum).
GTH is seldom responsible for thyrotoxicosis. In those rare cases, ATD might be necessary in order to treat the symptoms and to rapidly lower the maternal TH blood concentration [140]. If the ATD do not properly control the hyperadrenergic symptoms, beta-blockers can be used in addition [141]. These medications should be used with caution because they do cross the placental and can be responsible for neonatal hypoglycemia and/or bradycardia [142].
4.2. Gestational Transient Hyperthyroidism (GTH)
Graves’ disease is an auto-immune disease caused by a certain type of TSH Receptor Antibodies (TRAb), which binds and stimulates TSHR [143]. Inhibiting TRAb also exist but only represent less than 10% of the total TRAb and do not play any role in Graves’ disease. It affects approximately 0.2% of pregnant women and is rarely discovered during pregnancy [144]. Graves’ disease is often revealed by rather unspecific symptoms that are shared with GTH. The TRAb increase during the first trimester and a co-existing GTH can both worsen the symptoms at the beginning of the pregnancy [145]. Post-partum is also a period at risk for thyrotoxicosis because of potential immune system rebound [146].
Uncontrolled thyrotoxicosis in cases of Graves’ disease is associated with miscarriage, gestational hypertension and pre-eclampsia, thyroid storm and maternal congestive heart failure [147,148].
As for non-pregnant women, ATD are the main treatment for GH [141]. The objective is to maintain total T4 under 150% of the non-pregnant upper limit value and fT4 under the upper limit value. The doses should be lowered when T3 and T4 values are corrected and, ultimately, when TSH becomes detectable. A thyroidectomy is an option during the second trimester if a woman has an uncontrollable GH, a highly symptomatic goiter, or cannot tolerate ATD [148].
4.3. Distinguishing GHT from Graves’ Disease
It is crucial to be able to distinguish Graves’ disease from GTH. Women affected by Graves’ disease usually present familial and personal history of auto-immune diseases. The presence of three other elements is highly in favor of Graves’ disease: maternal goiter, ophthalmopathy and TRAb presence. In addition, some maternal biological and sonographic markers may be helpful [149]. Usually, in Graves’ disease, the fT3/fT4 ratio is >2.7, the total T3/total T4 ratio is >20, TSH is <0.01, and HCG are <70,000 mUI/mL [149,150]. fT3/fT4 ratio is the best biological parameter. Thyroid ultrasound can measure the mean Systolic Peak Velocity of the Superior Thyroid Artery (STA-PSV) and the Superior Thyroid Artery Diastolic internal diameter (STA-D). STA-PSV < 40 cm/s and STA-D > 2.0 mm are associated with Graves’ disease, whereas STA-PSV > 40 cm/s and STA-D are associated with gestational transient hyperthyroidism [151,152].
4.4. Thyroid Storm
Thyroid storm is a scarce condition that affects less than 1% of pregnant women with hyperthyroidism [147,153]. It can be caused by an inciting event such as surgery, infection, pre-eclampsia, or labor. Atrial fibrillation, congestive heart failure and liver failure can develop, with mortality rates up to 25% [154]. It is therefore recommended to preventively treat all hyperthyroid pregnant women, especially before undergoing surgery [155].
4.5. Maternal Hypothyroidism
The prevalence of maternal hypothyroidism during pregnancy ranges between 1–2% [130]. Worldwide, the most common cause of maternal hypothyroidism is ID. Indeed, the thyroid usually uses its iodine stores to compensate for any insufficient supply. An iodine-deficient woman becoming pregnant can easily experience hypothyroidism as their iodine needs suddenly and rapidly increase. In areas of iodine insufficiency, the main etiology is autoimmune disease (especially Hashimoto’s thyroiditis caused by TPOAb) [156]. The other causes of hypothyroidism during pregnancy are thyroidectomy, ablative iodine therapies and over-treatment of a GTH [157].
Maternal hypothyroidism can lead to adverse pregnancy outcomes, such as miscarriage and gestational hypertensive disorders, including preeclampsia, abruption placenta and post-partum hemorrhage [158,159,160,161]. The risk of adverse outcomes in chronic autoimmune thyroiditis is higher than in ID hypothyroidism, and TPOAb-positive mothers have an increased risk of placental malperfusion irrespective of their TSH serum concentrations [162]. Isolated maternal hypothyroxinemia (serum low free thyroxine and normal thyroid stimulating hormone levels) is also associated with maternal and neonatal adverse outcomes such as premature rupture of membranes and low Apgar scores at birth [163].
5. Fetal Consequences of Pathological Thyroid Metabolism
5.1. Fetal Anomalies Caused by Hyper or Hypothyroidism
TH perfect balance in the fetal compartment is essential to ensure normal embryogenesis, brain development and fetal growth.
5.1.1. Congenital Heart Defects
Rodents with fetal hyperthyroidism induced by D-III knock-out are more likely to present congenital heart defects than those with normal thyroid metabolism. Three hundred and 64 misregulated genes were identified in D-III knock-out hearts with RNA sequencing, such as TBX5, MEF2C, TGFB1, TP53, and FOXO4 [164]. Dong et al. confirmed the relationship between hyperthyroidism and congenital heart defect in humans [165]. They proved that high concentrations of fT4 measured between 12 and 18 WG are associated with an increased risk of congenital heart defects. In their work, each 1% of free-to-total thyroxine proportion was associated with a 1.41-fold higher risk of congenital heart defect.
5.1.2. Impairments to Brain Development
Neurological impairment is a negative outcome of fetal hypothyroidism. Neuronal proliferation, neuronal migration, axonal growth, dendritic synaptogenesis, neural differentiation and migration and myelination require adequate levels of TH [4]. For instance, neural differentiation of neural stem cells depends on sequential molecular cascade [166]. Pax6, a sequence-specific DNA binding transcription factor, binds the Tbr2 enhancer region and induces its expression [167]. Tb2r is a T-box transcription factor, and neural stem cells differentiate into intermediate progenitor cells under its influence [168]. Tbr2 up-regulates Tbr1 expression, which induces intermediate progenitor cells’ differentiation into postmitotic projection neurons. This molecular cascade could be even more complex, implying Neurog2, Insm1 and Neurod1. Maternal TH deficiency in rodents down-regulates Pax6, thus diminishing Tbr2 expression. This results in the persistent decrease of intermediate progenitor cells and reduces neuron-specific nuclear protein positivity in the first and third layers of the neocortex, which reflects a depletion in mature neurons. The main consequence of this pathological phenomenon is reduced cortical thickness, which indicates a non-compensatory impairment in neurogenesis (Mohan 2012) [169]. Congenital hypothyroidism in mice highly also alters the subventricular zone—which is the largest neural stem cell harbor area in the adult mammalian brain—and is responsible for underperformances on short-term memory tests [170]. Finally, Shimokawa and al. proved that congenitally hypothyroid rodents show retardation of cerebellar morphogenesis (poor dendritic arborization of Purkinje cells and retarded migration of granule cells) [171]. These are responsible for severe impairment of motor coordination and balance. They also proved that congenitally hypothyroid rodents show intense anxiety and depression, which could be explained by deterioration in the axonal transport of dopamine in the nigrostriatal pathway.
As the fetus is fully dependent on maternal TH during the first trimester, pathological variations of the maternal serum TH concentration can also result in neurodevelopmental disorders. For instance, high maternal T3 and both high or low maternal fT4 are associated with an increased risk of weakened neuropsychological development and attention-deficit hyperactivity disorder in infancy [172,173].
5.1.3. Birthweight
In addition to their role in heart and brain development, TH are of the utmost importance in birth weight regulation. Indeed, maternal subclinical hypothyroidism during pregnancy is associated with a higher risk of low birth weight, and isolated hypothyroxinemia is associated with a higher risk of high birth weight [174]. The relationship between maternal TH serum concentration and birthweight could be mediated by triglycerides, as triglycerides are correlated with maternal fT4 and neonatal birthweight [175,176]. Maternal carnitine could also be involved in this process because its serum concentration during the second trimester–alongside fT4–has a negative influence on birthweight [177]. Precise mechanisms involved in this process still have to be elucidated.
5.2. Fetal Hyperthyroidism
Congenital hyperthyroidism is less frequent than congenital hypothyroidism, but its consequences can also be disastrous. The most common cause is maternal Graves’ disease. As described earlier, TRAb cross the placenta and stimulate fetal TSHR, leading to an increase in fetal TH secretion. As the fetal TSHR is able to respond to TSH stimulation from 17–19WG, fetal hyperthyroidism develops during the second trimester, especially in women with high levels of TRAb [178]. It can lead to fetal and then post-natal thyrotoxicosis. The maternal TRAb can take as long as 4 months to disappear from the child’s blood circulation. 1% of children born of women with Graves’ disease present hyperthyroidism [179].
Two other causes of fetal hyperthyroidism are TSHR mutations and McCune-Albright syndrome. They both result from signal transduction modifications in the fetal thyroid.
Familial nonautoimmune hyperthyroidism (FNAH) and Sporadic nonautoimmune hyperthyroidism (SNAH) are two types of activating TSHR germline mutations. The mutated TSHR, in a pathway involving GTP-binding proteins, adenylate cyclase, cAMP, cAMP-dependent protein kinase, phospholipase C, diacylglycerol and calcium, increases the TH synthesis and secretion with no feedback loop [180].
FNAH is an autosomal dominant disorder that has never led to a prenatal hyperthyroidism diagnosis. The first hyperthyroidism symptoms indeed appear from 1 to 23 years old [180,181].
SNAH is caused by de novo mutations. Seventeen patients have been reported in the literature with 12 identified mutations. Interestingly, two cases presented prenatal features (fetal tachycardia in one case and a goiter associated with craniosynostosis in the other one), which raised suspicions [182,183].
Among various other manifestations, McCune-Albright syndrome can cause hyperthyroidism. This syndrome results from activating mutations at the R201 position in the GNAS gene, which encodes the alpha subunit of G proteins [184]. Cases with neonatal hyperthyroidism have been reported–disturbing the same signaling pathway as the TSHR mutations described above–but fetal hyperthyroidism was not suspected [185,186,187]. However, McCune-Albright syndrome has been described prenatally in a fetus with ovarian cysts [188]. It should thus be evoked in fetal hyperthyroidism when there is no argument for maternal Graves’ disease.
Classic neonatal and fetal hyperthyroidism include fetal tachycardia and goiter. Hepatomegaly, splenomegaly, gynecomastia and cardiac insufficiency can also occur [189]. Fetal hyperthyroidism must be carefully looked for in fetuses from mothers with Graves’ disease, McCune-Albright and FNAH/SNAH. Prenatal sonographic features such as goiter with central vascularization, tachycardia, hepatomegaly, splenomegaly and cardiac insufficiency can easily be diagnosed (Figure 1) [190]. Fetal thyroid size curves are available in order to diagnose fetal goiter [191,192]. Fetal therapy for hyperthyroidism consists of administering ATD to the mother because they can cross the placenta [189].
5.3. Fetal Hypothyroidism
Congenital hypothyroidism affects one in 4000 newborns. The most frequent causes of neonatal hypothyroidism are thyroid dysgenesis, which consists of an impairment of the thyroidal gland development, and thyroid dyshormonogenesis, which consists of thyroid hormone synthesis disorder [193]. Dyshormonogenesis is usually caused by genetic disorders (mainly TPO mutations but also thyroglobulin mutations, NIS mutations, Pendred syndrome, Allan–Herndon–Dudley syndrome, etc.), ATD overload (especially when over-correcting GHT), iodine overload by iodine-based medicine consumption, maternal, or TSH blocking antibodies [194]. TPOAb can cross the placenta, but unlike TRAb, they cannot cause fetal dysthyroidism.
Hypothyroidism may manifest in the fetus only in the event of dyshormonogenesis or ATD overload. Fetal hypothyroidism is responsible for delayed bone age, surprisingly increased fetal movements and eventually bulky goiter with peripheral vascularization (Figure 2) [190,195]. Fetal blood sampling by cordocentesis (percutaneous umbilical blood sampling) is often needed to confirm hypothyroidism and dosing fetal TSH, T3, and T4 [189]. After the confirmation of fetal hyperthyroidism, prenatal treatment can be undertaken. It consists of repeated intra-amniotic injections of thyroxine.
Severe ID is the main cause of post-natal endemic “cretinism.” Two types of this condition have been described. The neurological one is characterized by intellectual disability with an intellectual quotient around 30, mutism, and spastic diplegia. The myxoedematous one is characterized by intellectual disability—however less severe than the neurological one—and delayed bone age [196]. Both are caused by hypothyroidism, which affects fetal neurological development.
Allan–Herndon–Dudley syndrome is another example of the utmost importance of maternal TH for fetal brain development and, thus, of placental TH transporters. It is a singular syndrome caused by mutations in the Solute Carrier Family 16, Member 2 (SLC12A2) gene on the X chromosome [197]. This gene encodes for the MCT8. The mutations include truncations, in-frame deletions, nonsense and missense mutations. They lead to central hypothyroidism and peripheric hyperthyroidism. Allan–Herndon–Dudley syndrome manifests in boys as congenital hypotonia, which is detectable in the early weeks/months of life. It also causes severe intellectual deficits and possibly a distinctive dysmorphism. fT3 serum concentration is abnormally high, whereas fT4 can be low or normal, and TSH is slightly elevated or normal.
6. Conclusions
Iodine and thyroid role in pregnancy is central. Any impairment in their metabolism can affect both the mother and the fetus and eventually put at risk the newborn development. Iodine, thyroid hormone and thyroid metabolites in the fetal compartment are precisely regulated, and the placenta is highly involved in this regulation.
This review underlines the utmost importance of iodine and thyroid metabolism during pregnancy and the need for careful screening and treatment of mothers, fetuses and new-born to ensure optimal health.
Author Contributions
Conceptualization, D.L. and C.M.; writing—original draft preparation, C.M.; writing—review and editing, D.L., G.D. and C.M.; supervision, D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ma, J.; Hatch-Mcchesney, A.; Lieberman, H.R. Iodine and Iodine Deficiency: A Comprehensive Review of a Re-Emerging Issue. Nutrients 2022, 14, 3474. [Google Scholar] [CrossRef]
- Zimmermann, M.B. The effects of iodine deficiency in pregnancy and infancy. Paediatr. Perinat. Epidemiol. 2012, 26, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Ittermann, T.; Albrecht, D.; Arohonka, P.; Bilek, R.; De Castro, J.J.; Dahl, L.; Filipsson Nystrom, H.; Gaberscek, S.; Garcia-Fuentes, E.; Gheorghiu, M.L.; et al. Standardized Map of Iodine Status in Europe. Thyroid 2020, 30, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- De Escobar, G.M.; Obregón, M.J.; Del Rey, F.E. Role of thyroid hormone during early brain development. Eur. J. Endocrinol. 2004, 151 (Suppl. S3), U25–U37. [Google Scholar] [CrossRef] [PubMed]
- Calvo, R.M.; Jauniaux, E.; Gulbis, B.; Asunción, M.; Gervy, C.; Contempré, B.; De Escobar, G.M. Fetal Tissues Are Exposed to Biologically Relevant Free Thyroxine Concentrations during Early Phases of Development. J. Clin. Endocrinol. Metab. 2002, 87, 1768–1777. [Google Scholar] [CrossRef]
- Thorpe-Beeston, J.G.; Nicolaides, K.H.; Felton, C.V.; Butler, J.; McGregor, A.M. Maturation of the secretion of thyroid hormone and thyroid-stimulating hormone in the fetus. N. Engl. J. Med. 1991, 324, 532–536. [Google Scholar] [CrossRef]
- Haldimann, M.; Alt, A.; Blanc, A.; Blondeau, K. Iodine content of food groups. J. Food Compos. Anal. 2005, 18, 461–471. [Google Scholar] [CrossRef]
- De Miranda Milagres, R.C.R.; De Souza, E.C.G.; Do Carmo Gouveia Peluzio, M.; Do Carmo Castro Franceschini, S.; Duarte, M.S.L. Food Iodine Content Table compiled from international databases. Rev. Nutr. 2020, 33, e190222. [Google Scholar] [CrossRef]
- Pearce, E.N.; Pino, S.; He, X.; Bazrafshan, H.R.; Lee, S.L.; Braverman, L.E. Sources of dietary iodine: Bread, cows’ milk, and infant formula in the Boston area. J. Clin. Endocrinol. Metab. 2004, 89, 3421–3424. [Google Scholar] [CrossRef]
- Katagiri, R.; Asakura, K.; Uechi, K.; Masayasu, S.; Sasaki, S. Iodine Excretion in 24-hour Urine Collection and Its Dietary Determinants in Healthy Japanese Adults. J. Epidemiol. 2016, 26, 613. [Google Scholar] [CrossRef]
- Tadesse, S.; Hymete, A.; Lieberman, M.; Gebreyesus, S.H.; Ashenef, A. Iodine status, household salt iodine content, knowledge and practice assessment among pregnant women in Butajira, South Central Ethiopia. PLoS ONE 2022, 17, e0277208. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Du, Y.; Meng, F.; Liu, L.; Li, M.; Liu, P.; Sun, D. How to Decide the Iodine Content in Salt for a Country-China as an Example. Nutrients 2022, 14, 4606. [Google Scholar] [CrossRef] [PubMed]
- Clar, C.; Wu, T.; Liu, G.; Li, P. Iodized salt for iodine deficiency disorders. A systematic review. Endocrinol. Metab. Clin. North Am. 2002, 31, 681–698. [Google Scholar] [CrossRef] [PubMed]
- Esche, J.; Thamm, M.; Remer, T. Contribution of iodized salt to total iodine and total salt intake in Germany. Eur. J. Nutr. 2020, 59, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Rosen, S.R.; Ovadia, Y.S.; Anteby, E.Y.; Fytlovich, S.; Aharoni, D.; Zamir, D.; Gefel, D.; Shenhav, S. Low intake of iodized salt and iodine containing supplements among pregnant women with apparently insufficient iodine status-time to change policy? Isr. J. Health Policy Res. 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- De La Vieja, A.; Santisteban, P. Role of iodide metabolism in physiology and cancer. Endocr. Relat. Cancer 2018, 25, R225–R245. [Google Scholar] [CrossRef]
- Portulano, C.; Paroder-Belenitsky, M.; Carrasco, N. The Na+/I− Symporter (NIS): Mechanism and Medical Impact. Endocr. Rev. 2014, 35, 106–149. [Google Scholar] [CrossRef]
- Shcheynikov, N.; Yang, D.; Wang, Y.; Zeng, W.; Karniski, L.P.; So, I.; Wall, S.M.; Muallem, S. The Slc26a4 transporter functions as an electroneutral Cl−/I−/HCO3− exchanger: Role of Slc26a4 and Slc26a6 in I− and HCO3− secretion and in regulation of CFTR in the parotid duct. J. Physiol. 2008, 586, 3813–3824. [Google Scholar] [CrossRef]
- Perez-Cornejo, P.; Gokhale, A.; Duran, C.; Cui, Y.; Xiao, Q.; Hartzell, H.C.; Faundez, V. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc. Natl. Acad. Sci. USA 2012, 109, 10376–10381. [Google Scholar] [CrossRef]
- Yoshida, A.; Hisatome, I.; Taniguchi, S.; Sasaki, N.; Yamamoto, Y.; Miake, J.; Fukui, H.; Shimizu, H.; Okamura, T.; Okura, T.; et al. Mechanism of iodide/chloride exchange by pendrin. Endocrinology 2004, 145, 4301–4308. [Google Scholar] [CrossRef]
- Altorjay, Á.; Dohán, O.; Szilágyi, A.; Paroder, M.; Wapnir, I.L.; Carrasco, N. Expression of the Na+/l-symporter (NIS) is markedly decreased or absent in gastric cancer and intestinal metaplastic mucosa of Barrett esophagus. BMC Cancer 2007, 7, 5. [Google Scholar] [CrossRef]
- Mazzone, A.; Bernard, C.E.; Strege, P.R.; Beyder, A.; Galietta, L.J.V.; Pasricha, P.J.; Rae, J.L.; Parkman, H.P.; Linden, D.R.; Szurszewski, J.H.; et al. Altered expression of ano1 variants in human diabetic gastroparesis. J. Biol. Chem. 2011, 286, 13393–13403. [Google Scholar] [CrossRef] [PubMed]
- Pablo Nicola, J.; Basquin, C.; Portulano, C.; Reyna-Neyra, A.; Paroder, M.; Carrasco, N. The Na/I symporter mediates active iodide uptake in the intestine. Am. J. Physiol. Cell Physiol. 2009, 296, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Fahlke, C. Ion permeation and selectivity in ClC-type chloride channels. Am. J. Physiol. Renal Physiol. 2001, 280, F748–F757. [Google Scholar] [CrossRef]
- Zimmermann, M.B.; Andersson, M. Assessment of iodine nutrition in populations: Past, present, and future. Nutr. Rev. 2012, 70, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Spitzweg, C.; Dutton, C.M.; Castro, M.R.; Bergert, E.R.; Goellner, J.R.; Heufelder, A.E.; Morris, J.C. Expression of the sodium iodide symporter in human kidney. Kidney Int. 2001, 59, 1013–1023. [Google Scholar] [CrossRef]
- Wapnir, I.L.; Van De Rijn, M.; Nowels, K.; Amenta, P.S.; Walton, K.; Montgomery, K.; Greco, R.S.; Dohán, O.; Carrasco, N. Immunohistochemical Profile of the Sodium/Iodide Symporter in Thyroid, Breast, and Other Carcinomas Using High Density Tissue Microarrays and Conventional Sections. J. Clin. Endocrinol. Metab. 2003, 88, 1880–1888. [Google Scholar] [CrossRef]
- Xu, J.; Barone, S.; Li, H.; Holiday, S.; Zahedi, K.; Soleimani, M. Slc26a11, a chloride transporter, localizes with the vacuolar H+ -ATPase of A-intercalated cells of the kidney. Kidney Int. 2011, 80, 926–937. [Google Scholar] [CrossRef]
- Svenningsen, P.; Nielsen, M.R.; Marcussen, N.; Walter, S.; Jensen, B.L. TMEM16A is a Ca2+-activated Cl− channel expressed in the renal collecting duct. Acta Physiol. 2014, 212, 166–174. [Google Scholar] [CrossRef]
- Pearce, E.N.; Caldwell, K.L. Urinary iodine, thyroid function, and thyroglobulin as biomarkers of iodine status. Am. J. Clin. Nutr. 2016, 104 (Suppl. S3), 898S–901S. [Google Scholar] [CrossRef]
- Remer, T.; Fonteyn, N.; Alexy, U.; Berkemeyer, S. Longitudinal examination of 24-h urinary iodine excretion in schoolchildren as a sensitive, hydration status-independent research tool for studying iodine status. Am. J. Clin. Nutr. 2006, 83, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Karumbunathan, V.; Zimmermann, M.B. Global iodine status in 2011 and trends over the past decade. J. Nutr. 2012, 142, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Zhong, G.; Ma, X. Urinary iodine concentration (UIC) could be a promising biomarker for predicting goiter among school-age children: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0174095. [Google Scholar] [CrossRef]
- Citterio, C.E.; Targovnik, H.M.; Arvan, P. The role of thyroglobulin in thyroid hormonogenesis. Nat. Rev. Endocrinol. 2019, 15, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Ris-Stalpers, C. Physiology and pathophysiology of the DUOXes. Antioxid. Redox Signal. 2006, 8, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Aycan, Z.; Cangul, H.; Muzza, M.; Bas, V.N.; Fugazzola, L.; Chatterjee, V.K.; Persani, L.; Schoenmakers, N. Digenic DUOX1 and DUOX2 Mutations in Cases with Congenital Hypothyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 3085. [Google Scholar] [CrossRef] [PubMed]
- Pilo, A.; Iervasi, G.; Vitek, F.; Ferdeghini, M.; Cazzuola, F.; Bianchi, R. Thyroidal and peripheral production of 3,5,3′-triiodothyronine in humans by multicompartmental analysis. Am. J. Physiol. 1990, 258, E715–E726. [Google Scholar] [CrossRef]
- Janssen, S.T.; Janssen, O.E. Directional thyroid hormone distribution via the blood stream to target sites. Mol. Cell. Endocrinol. 2017, 458, 16–21. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol. Rev. 2014, 94, 355. [Google Scholar] [CrossRef]
- De Felice, M.; Di Lauro, R. Thyroid development and its disorders: Genetics and molecular mechanisms. Endocr. Rev. 2004, 25, 722–746. [Google Scholar] [CrossRef]
- Ohmori, M.; Endo, T.; Harii, N.; Onaya, T. A Novel Thyroid Transcription Factor Is Essential for Thyrotropin-Induced Up-Regulation of Na+/I− Symporter Gene Expression. Mol. Endocrinol. 1998, 12, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Zannini, M.; Levy, O.; Carrasco, N.; di Lauro, R. The Paired-Domain Transcription Factor Pax8 Binds to the Upstream Enhancer of the Rat Sodium/Iodide Symporter Gene and Participates in Both Thyroid-Specific and Cyclic-AMP-Dependent Transcription. Mol. Cell. Biol. 1999, 19, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.L.; Espinoza, C.R.; Loos, U. Characterization of a Thyroid-Specific and Cyclic Adenosine Monophosphate-Responsive Enhancer Far Upstream from the Human Sodium Iodide Symporter Gene. Thyroid 2004, 12, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Taki, K.; Kogai, T.; Kanamoto, Y.; Hershman, J.M.; Brent, G.A. A Thyroid-Specific Far-Upstream Enhancer in the Human Sodium/Iodide Symporter Gene Requires Pax-8 Binding and Cyclic Adenosine 3′,5′-Monophosphate Response Element-Like Sequence Binding Proteins for Full Activity and Is Differentially Regulated in Normal and Thyroid Cancer Cells. Mol. Endocrinol. 2002, 16, 2266–2282. [Google Scholar] [CrossRef]
- Fernández, L.P.; López-Márquez, A.; Martínez, Á.M.; Gómez-López, G.; Santisteban, P. New Insights into FoxE1 Functions: Identification of Direct FoxE1 Targets in Thyroid Cells. PLoS ONE 2013, 8, e62849. [Google Scholar] [CrossRef]
- Kang, H.S.; Kumar, D.; Liao, G.; Lichti-Kaiser, K.; Gerrish, K.; Liao, X.H.; Refetoff, S.; Jothi, R.; Jetten, A.M. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J. Clin. Investig. 2022, 127, 4326–4337. [Google Scholar] [CrossRef]
- Giuliani, C.; Bucci, I.; Napolitano, G. The role of the transcription factor Nuclear Factor-kappa B in thyroid autoimmunity and cancer. Front. Endocrinol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Nicola, J.P.; Nazar, M.; Mascanfroni, I.D.; Pellizas, C.G.; Masini-Repiso, A.M. NF-κB p65 Subunit Mediates Lipopolysaccharide-Induced Na+/I− Symporter Gene Expression by Involving Functional Interaction with the Paired Domain Transcription Factor Pax8. Mol. Endocrinol. 2010, 24, 1846–1862. [Google Scholar] [CrossRef]
- Elefant, É. Placental immunoglobulin transfer. Bull. Acad. Natl. Med. 2012, 196, 1601–1612. [Google Scholar] [CrossRef]
- Zimmermann, M.B. Iodine deficiency in pregnancy and the effects of maternal iodine supplementation on the offspring: A review. Am. J. Clin. Nutr. 2009, 89, 668S–672S. [Google Scholar] [CrossRef]
- Caldwell, K.L.; Jones, R.; Hollowell, J.G. Urinary iodine concentration: United States National Health And Nutrition Examination Survey 2001–2002. Thyroid 2005, 15, 692–699. [Google Scholar] [CrossRef]
- Brander, L.; Als, C.; Buess, H.; Haldimann, F.; Harder, M.; Hänggi, W.; Herrmann, U.; Lauber, K.; Niederer, U.; Zürcher, T.; et al. Urinary iodine concentration during pregnancy in an area of unstable dietary iodine intake in Switzerland. J. Endocrinol. Investig. 2003, 26, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Stilwell, G.; Reynolds, P.J.; Parameswaran, V.; Blizzard, L.; Greenaway, T.M.; Burgess, J.R. The Influence of Gestational Stage on Urinary Iodine Excretion in Pregnancy. J. Clin. Endocrinol. Metab. 2008, 93, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Cai, Y.; Ji, C.; Zhao, C.; Tian, C.; Pang, B.; Shi, M.; Li, X.; Liu, Y.; Sun, D. Evaluation of iodine nutritional status during pregnancy by estimated 24-h urinary iodine excretion: Population variation range and individual accuracy. Public Health Nutr. 2022, 25, 237–247. [Google Scholar] [CrossRef] [PubMed]
- De Groot, L.; Abalovich, M.; Alexander, E.K.; Amino, N.; Barbour, L.; Cobin, R.H.; Eastman, C.J.; Lazarus, J.H.; Luton, D.; Mandel, S.J.; et al. Management of Thyroid Dysfunction during Pregnancy and Postpartum: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2012, 97, 2543–2565. [Google Scholar] [CrossRef]
- Alexander, E.K.; Pearce, E.N.; Brent, G.A.; Brown, R.S.; Chen, H.; Dosiou, C.; Grobman, W.A.; Laurberg, P.; Lazarus, J.H.; Mandel, S.J.; et al. 2017 Guidelines of the American Thyroid Association for the Diagnosis and Management of Thyroid Disease during Pregnancy and the Postpartum. Thyroid 2017, 27, 315–389. [Google Scholar] [CrossRef]
- Lazarus, J.; Brown, R.S.; Daumerie, C.; Hubalewska-Dydejczyk, A.; Negro, R.; Vaidya, B. 2014 European thyroid association guidelines for the management of subclinical hypothyroidism in pregnancy and in children. Eur. Thyroid J. 2014, 3, 76–94. [Google Scholar] [CrossRef]
- Andersson, M.; De Benoist, B.; Delange, F.; Zupan, J. Prevention and control of iodine deficiency in pregnant and lactating women and in children less than 2-years-old: Conclusions and recommendations of the Technical Consultation. Public Health Nutr. 2007, 10, 1606–1611. [Google Scholar] [CrossRef]
- Miles, E.A.; Vahlberg, T.; Calder, P.C.; Houttu, N.; Pajunen, L.; Koivuniemi, E.; Mokkala, K.; Laitinen, K. Iodine status in pregnant women and infants in Finland. Eur. J. Nutr. 2022, 61, 2919–2927. [Google Scholar] [CrossRef]
- Bidart, J.-M.; Lacroix, L.; Evain-Brion, D.; Caillou, B.; Lazar, V.; Frydman, R.; Bellet, D.; Filetti, S.; Schlumberger, M. Expression of Na+/I− symporter and Pendred syndrome genes in trophoblast cells. J. Clin. Endocrinol. Metab. 2000, 85, 4367–4372. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Manley, S.W.; Morris, J.C.; Powell, K.A.; Bergert, E.R.; Mortimer, R.H. Sodium iodide symporter (NIS) gene expression in human placenta. Placenta 2001, 22, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Degrelle, S.A.; Guibourdenche, J.; Galland, F.; Bidart, J.M.; Fournier, T.; Evain-Brion, D. Iodide transporters expression in early human invasive trophoblast. Placenta 2013, 34, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Bidart, J.-M.; Mian, C.; Lazar, V.; Russo, D.; Filetti, S.; Caillou, B.; Schlumberger, M. Expression of Pendrin and the Pendred Syndrome (PDS) Gene in Human Thyroid Tissues*. J. Clin. Endocrinol. Metab. 2000, 85, 2028–2033. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.W.; Li, H.; Mortimer, R.H. The BeWo choriocarcinoma cell line as a model of iodide transport by placenta. Placenta 2005, 26, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Nwabuobi, C.; Arlier, S.; Schatz, F.; Guzeloglu-Kayisli, O.; Lockwood, C.J.; Kayisli, U.A. hCG: Biological Functions and Clinical Applications. Int. J. Mol. Sci. 2017, 18, 2037. [Google Scholar] [CrossRef]
- Morgan, F.J.; Birken, S.; Canfield, R.E. The amino acid sequence of human chorionic gonadotropin. The alpha subunit and beta subunit. J. Biol. Chem. 1975, 250, 5247–5258. [Google Scholar] [CrossRef]
- Kraiem, Z.; Sadeh, O.; Blithe, D.L.; Nisula, B.C. Human chorionic gonadotropin stimulates thyroid hormone secretion, iodide uptake, organification, and adenosine 3′,5′-monophosphate formation in cultured human thyrocytes. J. Clin. Endocrinol. Metab. 1994, 79, 595–599. [Google Scholar] [CrossRef]
- Yoshimura, M.; Hershman, J.M. Thyrotropic action of human chorionic gonadotropin. Thyroid 1995, 5, 425–434. [Google Scholar] [CrossRef]
- Pekonen, F.; Alfthan, H.; Stenman, U.H.; Ylikorkala, O. Human chorionic gonadotropin (hCG) and thyroid function in early human pregnancy: Circadian variation and evidence for intrinsic thyrotropic activity of hCG. J. Clin. Endocrinol. Metab. 1988, 66, 853–856. [Google Scholar] [CrossRef]
- Arturi, F.; Presta, I.; Scarpelli, D.; Bidart, J.M.; Schlumberger, M.; Filetti, S.; Russo, D. Stimulation of iodide uptake by human chorionic gonadotropin in FRTL-5 cells: Effects on sodium/iodide symporter gene and protein expression. Eur. J. Endocrinol. 2002, 147, 655–661. [Google Scholar] [CrossRef]
- Li, H.; Richard, K.; McKinnon, B.; Mortimer, R.H. Effect of Iodide on Human Choriogonadotropin, Sodium-Iodide Symporter Expression, and Iodide Uptake in BeWo Choriocarcinoma Cells. J. Clin. Endocrinol. Metab. 2007, 92, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.; O’Herlihy, C.; Smyth, P.P.A. Regulation of Iodide Uptake in Placental Primary Cultures. Eur. Thyroid J. 2013, 2, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Schröder-Van Der Elst, J.P.; Van Der Heide, D.; Kastelijn, J.; Rousset, B.; Obregón, M.J. The expression of the sodium/iodide symporter is up-regulated in the thyroid of fetuses of iodine-deficient rats. Endocrinology 2001, 142, 3736–3741. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.; O’Herlihy, C.; Smyth, P.P.A. The placenta as a compensatory iodine storage organ. Thyroid 2011, 21, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Neven, K.Y.; Marien, C.B.D.; Janssen, B.G.; Roels, H.A.; Waegeneers, N.; Nawrot, T.S.; Ruttens, A. Variability of iodine concentrations in the human placenta. Sci. Rep. 2020, 10, 161. [Google Scholar] [CrossRef]
- Brent, G.A. Maternal thyroid function: Interpretation of thyroid function tests in pregnancy. Clin. Obstet. Gynecol. 1997, 40, 3–15. [Google Scholar] [CrossRef]
- Lazarus, J.H. Thyroid function in pregnancy. Br. Med. Bull. 2011, 97, 137–148. [Google Scholar] [CrossRef]
- Lee, R.H.; Spencer, C.A.; Mestman, J.H.; Miller, E.A.; Petrovic, I.; Braverman, L.E.; Goodwin, T.M. Free T4 immunoassays are flawed during pregnancy. Am. J. Obstet. Gynecol. 2009, 200, 260.e1–260.e6. [Google Scholar] [CrossRef]
- Knøsgaard, L.; Andersen, S.; Hansen, A.B.; Vestergaard, P.; Andersen, S.L. Classification of maternal thyroid function in early pregnancy using repeated blood samples. Eur. Thyroid J. 2022, 11, e210055. [Google Scholar] [CrossRef]
- Ain, K.B.; Mori, Y.; Refetoff, S. Reduced clearance rate of thyroxine-binding globulin (TBG) with increased sialylation: A mechanism for estrogen-induced elevation of serum TBG concentration. J. Clin. Endocrinol. Metab. 1987, 65, 689–696. [Google Scholar] [CrossRef]
- Pradhan, R.; Agarwal, A.; Lombardi, C.P.; Raffaelli, M. Applied Embryology of the Thyroid and Parathyroid Glands. Surg. Thyroid Parathyr. Gland. 2021, 1, 15–25.e4. [Google Scholar] [CrossRef]
- Arrangoiz, R.; Cordera, F.; Caba, D.; Muñoz, M.; Moreno, E.; de León, E.L.; Arrangoiz, R.; Cordera, F.; Caba, D.; Muñoz, M.; et al. Comprehensive Review of Thyroid Embryology, Anatomy, Histology, and Physiology for Surgeons. Int. J. Otolaryngol. Head Neck Surg. 2018, 7, 160–188. [Google Scholar] [CrossRef]
- Ozguner, G.; Sulak, O. Size and location of thyroid gland in the fetal period. Surg. Radiol. Anat. 2014, 36, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Dom, G.; Dmitriev, P.; Lambot, M.A.; Van Vliet, G.; Glinoer, D.; Libert, F.; Lefort, A.; Dumont, J.E.; Maenhaut, C. Transcriptomic Signature of Human Embryonic Thyroid Reveals Transition From Differentiation to Functional Maturation. Front. Cell Dev. Biol. 2021, 9, 1481. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Fagman, H. Development of the thyroid gland. Development 2017, 144, 2123–2140. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Mishra, S.; Kaul, J.M. Development of Parafollicular Cells and their Relationship with Developing Thyroid Follicles in Human Foetuses. J. Clin. Diagn. Res. 2017, 11, AC01. [Google Scholar] [CrossRef]
- Trueba, S.S.; Augé, J.; Mattei, G.; Etchevers, H.; Martinovic, J.; Czernichow, P.; Vekemans, M.; Polak, M.; Attié-Bitach, T. PAX8, TITF1, and FOXE1 gene expression patterns during human development: New insights into human thyroid development and thyroid dysgenesis-associated malformations. J. Clin. Endocrinol. Metab. 2005, 90, 455–462. [Google Scholar] [CrossRef]
- Chen, Z.; Peeters, R.P.; Leeuwenburgh, S.; Broekhuizen, M.; Neuman, R.I.; Hitzerd, E.; Tan, L.; Jongejan, R.M.S.; de Rijke, Y.B.; Reiss, I.K.M.; et al. Asymmetrical Transport of Thyroxine Across Human Term Placenta. Thyroid, 2023; ahead of print. [Google Scholar] [CrossRef]
- Landers, K.; Richard, K. Traversing barriers—How thyroid hormones pass placental, blood-brain and blood-cerebrospinal fluid barriers. Mol. Cell. Endocrinol. 2017, 458, 22–28. [Google Scholar] [CrossRef]
- Costa, A.; Arisio, R.; Benedetto, C.; Bertino, E.; Fabris, C.; Giraudi, G.; Marozio, L.; Maulà, V.; Pagliano, M.; Testori, O.; et al. Thyroid hormones in tissues from human embryos and fetuses. J. Endocrinol. Investig. 1991, 14, 559–568. [Google Scholar] [CrossRef]
- Contempré, B.; Jauniaux, E.; Calvo, R.; Jurkovic, D.; Campbell, S.; De Escobar, G.M. Detection of thyroid hormones in human embryonic cavities during the first trimester of pregnancy. J. Clin. Endocrinol. Metab. 1993, 77, 1719–1722. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Manley, S.W.; Mortimer, R.H. Membrane transport of thyroid hormone in the human choriocarcinoma cell line, JAR. Mol. Cell. Endocrinol. 1992, 87, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Zuñiga, L.F.F.; Muñoz, Y.S.; Pustovrh, M.C. Thyroid hormones: Metabolism and transportation in the fetoplacental unit. Mol. Reprod. Dev. 2022, 89, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Meima, M.E.; Peeters, R.P.; Visser, W.E. Thyroid Hormone Transporters in Pregnancy and Fetal Development. Int. J. Mol. Sci. 2022, 23, 15113. [Google Scholar] [CrossRef] [PubMed]
- Roost, M.S.; Van Iperen, L.; Ariyurek, Y.; Buermans, H.P.; Arindrarto, W.; Devalla, H.D.; Passier, R.; Mummery, C.L.; Carlotti, F.; De Koning, E.J.P.; et al. KeyGenes, a Tool to Probe Tissue Differentiation Using a Human Fetal Transcriptional Atlas. Stem Cell Rep. 2015, 4, 1112–1124. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Jose, A.; Poojari, V.G.; Shetty, S.; Prabhu RV, K.; Rao, M. Role and Clinical Significance of Monocarboxylate Transporter 8 (MCT8) During Pregnancy. Reprod. Sci. 2023. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, S.; De Souza, E.C.L.; Meima, M.E.; Peeters, R.P.; Edward Visser, W.; Visser, T.J. Outward-Open Model of Thyroid Hormone Transporter Monocarboxylate Transporter 8 Provides Novel Structural and Functional Insights. Endocrinology 2017, 158, 3292–3306. [Google Scholar] [CrossRef]
- Friesema, E.C.H.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem. 2003, 278, 40128–40135. [Google Scholar] [CrossRef]
- Friesema, E.C.H.; Docter, R.; Moerings, E.P.C.M.; Verrey, F.; Krenning, E.P.; Hennemann, G.; Visser, T.J. Thyroid Hormone Transport by the Heterodimeric Human System L Amino Acid Transporter. Endocrinology 2001, 142, 4339–4348. [Google Scholar] [CrossRef]
- Zevenbergen, C.; Meima, M.E.; De Souza, E.C.L.; Peeters, R.P.; Kinne, A.; Krause, G.; Edward Visser, W.; Visser, T.J. Transport of Iodothyronines by Human L-Type Amino Acid Transporters. Endocrinology 2015, 156, 4345–4355. [Google Scholar] [CrossRef]
- Kinne, A.; Wittner, M.; Wirth, E.K.; Hinz, K.M.; Schülein, R.; Köhrle, J.; Krause, G. Involvement of the L-Type Amino Acid Transporter Lat2 in the Transport of 3,3’-Diiodothyronine across the Plasma Membrane. Eur. Thyroid J. 2015, 4, 42–50. [Google Scholar] [CrossRef]
- Hinz, K.M.; Neef, D.; Rutz, C.; Furkert, J.; Köhrle, J.; Schülein, R.; Krause, G. Molecular features of the L-type amino acid transporter 2 determine different import and export profiles for thyroid hormones and amino acids. Mol. Cell. Endocrinol. 2017, 443, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Wirth, E.K.; Roth, S.; Blechschmidt, C.; Hölter, S.M.; Becker, L.; Racz, I.; Zimmer, A.; Klopstock, T.; Gailus-Durner, V.; Fuchs, H.; et al. Neuronal 3′,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J. Neurosci. 2009, 29, 9439–9449. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Adachi, H.; Nishio, T.; Unno, M.; Tokui, T.; Okabe, M.; Onogawa, T.; Suzuki, T.; Asano, N.; Tanemoto, M.; et al. Identification of Thyroid Hormone Transporters in Humans: Different Molecules Are Involved in a Tissue-Specific Manner. Endocrinology 2001, 142, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.H.; Jansen, J.; Jachtenberg, J.W.; Visser, W.E.; Kester, M.H.A.; Visser, T.J. Effective Cellular Uptake and Efflux of Thyroid Hormone by Human Monocarboxylate Transporter 10. Mol. Endocrinol. 2008, 22, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.H.; Grueters, P.A.; Biebermann, H.; Krude, H.; Von Moers, A.; Reeser, M.; Barrett, T.G.; Mancilla, E.E.; Svensson, J.; Kester, M.H.A.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Chan, S.Y.; Martín-Santos, A.; Loubière, L.S.; González, A.M.; Stieger, B.; Logan, A.; Mccabe, C.J.; Franklyn, J.A.; Kilby, M.D. The expression of thyroid hormone transporters in the human fetal cerebral cortex during early development and in N-Tera-2 neurodifferentiation. J. Physiol. 2011, 589, 2827–2845. [Google Scholar] [CrossRef]
- Heuer, H.; Maier, M.K.; Iden, S.; Mittag, J.; Friesema, E.C.H.; Visser, T.J.; Bauer, K. The Monocarboxylate Transporter 8 Linked to Human Psychomotor Retardation Is Highly Expressed in Thyroid Hormone-Sensitive Neuron Populations. Endocrinology 2005, 146, 1701–1706. [Google Scholar] [CrossRef]
- Friesema, E.C.H.; Visser, T.J.; Borgers, A.J.; Kalsbeek, A.; Swaab, D.F.; Fliers, E.; Alkemade, A. Thyroid hormone transporters and deiodinases in the developing human hypothalamus. Eur. J. Endocrinol. 2012, 167, 379–386. [Google Scholar] [CrossRef]
- Heinrichs, C.; De Zegher, F.; Vansnick, F.; Vokaer, A.; Christophe, C.; Frankenne, F. Fetal hypopituitarism: Perinatal endocrine and morphological studies in two cases. Acta Paediatr. 1994, 83, 448–451. [Google Scholar] [CrossRef]
- Bajoria, R.; Fisk, N.M. Permeability of Human Placenta and Fetal Membranes to Thyrotropin-Stimulating Hormone in Vitro. Pediatr. Res. 1998, 43, 621–628. [Google Scholar] [CrossRef]
- Yoshida, K.; Sakurada, T.; Takahashi, T.; Furuhashi, N.; Kaise, K.; Yoshinaga, K. Measurement of tsh in human amniotic fluid: Diagnosis of fetal thyroid abnormality in utero. Clin. Endocrinol. 1986, 25, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Guibourdenche, J.; Noël, M.; Chevenne, D.; Vuillard, E.; Voluménie, J.L.; Polak, M.; Boissinot, C.; Porquet, D.; Luton, D. Biochemical investigation of foetal and neonatal thyroid function using the ACS-180SE analyser: Clinical application. Ann. Clin. Biochem. 2001, 38, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Polak, M. Human fetal thyroid function. Endocr. Dev. 2014, 26, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Sousa, N. Maternal hormonal milieu influence on fetal brain development. Brain Behav. 2018, 8, e00920. [Google Scholar] [CrossRef]
- Theodoropoulos, T.; Braverman, L.E.; Vagenakis, A.G. Thyrotropin-Releasing Hormone is not Required for Thyrotropin Secretion in the Perinatal Rat. J. Clin. Investig. 1979, 63, 588–594. [Google Scholar] [CrossRef]
- Ansari, M.A.; Demello, D.E.; Polk, D.H.; Devaskar, U.P. Thyrotropin-releasing hormone accelerates fetal mouse lung ultrastructural maturation via stimulation of extra thyroidal pathway. Pediatr. Res. 1997, 42, 709–714. [Google Scholar] [CrossRef]
- Yang, C.; He, B.; Zhang, H.; Wang, X.; Zhang, Q.; Dai, W. IgG Fc Affinity Ligands and Their Applications in Antibody-Involved Drug Delivery: A Brief Review. Pharmaceutics 2023, 15, 187. [Google Scholar] [CrossRef]
- Gitlin, D.; Kumate, J.; Urrusti, J.; Morales, C. The selectivity of the human placenta in the transfer of plasma proteins from mother to fetus. J. Clin. Investig. 1964, 43, 1938–1951. [Google Scholar] [CrossRef]
- Volkov, M.; Brinkhaus, M.; van Schie, K.A.; Bondt, A.; Kissel, T.; van der Kooi, E.J.; Bentlage, A.E.H.; Koeleman, C.A.M.; de Taeye, S.W.; Derksen, N.I.; et al. IgG Fab Glycans Hinder FcRn-Mediated Placental Transport. J. Immunol. 2023, 210, 158–167. [Google Scholar] [CrossRef]
- Firan, M.; Bawdon, R.; Radu, C.; Ober, R.J.; Eaken, D.; Antohe, F.; Ghetie, V.; Ward, E.S. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int. Immunol. 2001, 13, 993–1002. [Google Scholar] [CrossRef]
- Szlauer, R.; Ellinger, I.; Haider, S.; Saleh, L.; Busch, B.L.; Knöfler, M.; Fuchs, R. Functional expression of the human neonatal Fc-receptor, hFcRn, in isolated cultured human syncytiotrophoblasts. Placenta 2009, 30, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, B.K. Human placental Fc gamma-binding proteins in the maternofetal transfer of IgG. APMIS. Suppl. 1996, 64, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Bonagura, V.R.; Morrison, S.L.; Bjorkman, P.J. Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry 1995, 34, 14649–14657. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Chen, M.Y.; Gastinel, L.N.; Bjorkman, P.J. Investigation of the interaction between the class I MHC-related Fc receptor and its immunoglobulin G ligand. Immunity 1994, 1, 303–315. [Google Scholar] [CrossRef]
- Ober, R.J.; Martinez, C.; Lai, X.; Zhou, J.; Ward, E.S. Exocytosis of IgG as mediated by the receptor, FcRn: An analysis at the single-molecule level. Proc. Natl. Acad. Sci. USA 2004, 101, 11076–11081. [Google Scholar] [CrossRef]
- Malek, A.; Sager, R.; Kuhn, P.; Nicolaides, K.H.; Schneider, H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am. J. Reprod. Immunol. 1996, 36, 248–255. [Google Scholar] [CrossRef]
- Lozano, N.A.; Lozano, A.; Marini, V.; Saranz, R.J.; Blumberg, R.S.; Baker, K.; Agresta, M.F.; Ponzio, M.F. Expression of FcRn receptor in placental tissue and its relationship with IgG levels in term and pre-term newborns HHS Public Access. Am. J. Reprod. Immunol. 2018, 80, 12972. [Google Scholar] [CrossRef]
- Ciobanu, A.M.; Dumitru, A.E.; Gica, N.; Botezatu, R.; Peltecu, G.; Panaitescu, A.M. Benefits and Risks of IgG Transplacental Transfer. Diagnostics 2020, 10, 583. [Google Scholar] [CrossRef]
- Seror, J.; Amand, G.; Guibourdenche, J.; Ceccaldi, P.F.; Luton, D. Anti-TPO antibodies diffusion through the placental barrier during pregnancy. PLoS ONE 2014, 9, e0084647. [Google Scholar] [CrossRef]
- Köhrle, J. The deiodinase family: Selenoenzymes regulating thyroid hormone availability and action. Cell. Mol. Life Sci. 2000, 57, 1853–1863. [Google Scholar] [CrossRef]
- Salvatore, D.; Bartha, T.; Harney, J.W.; Larsen, P.R. Molecular biological and biochemical characterization of the human type 2 selenodeiodinase. Endocrinology 1996, 137, 3308–3315. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. Local activation and inactivation of thyroid hormones: The deiodinase family. Mol. Cell. Endocrinol. 1999, 151, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kachilele, S.; Hobbs, E.; Bulmer, J.N.; Boelaert, K.; McCabe, C.J.; Driver, P.M.; Bradwell, A.R.; Kester, M.; Visser, T.J.; et al. Placental iodothyronine deiodinase expression in normal and growth-restricted human pregnancies. J. Clin. Endocrinol. Metab. 2003, 88, 4488–4495. [Google Scholar] [CrossRef] [PubMed]
- Koopdonk-Kool, J.M.; de Vijlder, J.J.; Veenboer, G.J.; Ris-Stalpers, C.; Kok, J.H.; Vulsma, T.; Boer, K.; Visser, T.J. Type II and type III deiodinase activity in human placenta as a function of gestational age. J. Clin. Endocrinol. Metab. 1996, 81, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Vulsma, T.; Gons, M.H.; de Vijlder, J.J.M. Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis. N. Engl. J. Med. 1989, 321, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Glinoer, D.; de Nayer, P.; Bourdoux, P.; Lemone, M.; Robyn, C.; van Steirteghem, A.; Kinthaert, J.; Lejeune, B. Regulation of Maternal Thyroid during Pregnancy. J. Clin. Endocrinol. Metab. 1990, 71, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.P.; Khoo, D.H.C.; Eng, P.H.K.; Tan, H.K.; Yo, S.L.; Jacob, E. Prevalence of gestational thyrotoxicosis in Asian women evaluated in the 8th to 14th weeks of pregnancy: Correlations with total and free beta human chorionic gonadotrophin. Clin. Endocrinol. 2001, 55, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Virmani, S.; Srinivas, S.B.; Bhat, R.; Rao, R.; Kudva, R. Transient Thyrotoxicosis in Molar Pregnancy. J. Clin. Diagn. Res. 2017, 11, QD01. [Google Scholar] [CrossRef]
- Glinoer, D. The regulation of thyroid function in pregnancy: Pathways of endocrine adaptation from physiology to pathology. Endocr. Rev. 1997, 18, 404–433. [Google Scholar] [CrossRef]
- Nguyen, C.T.; Sasso, E.B.; Barton, L.; Mestman, J.H. Graves’ hyperthyroidism in pregnancy: A clinical review. Clin. Diabetes Endocrinol. 2018, 4, 1–9. [Google Scholar] [CrossRef]
- De Bruin, R.; Van Dalen, S.L.; Franx, S.J.; Simons, S.H.P.; Flint, R.B.; Van Den Bosch, G.E. Risk for neonatal hypoglycaemia and bradycardia after beta-blocker use during pregnancy or lactation: A systematic review and meta-analysis protocol. BMJ Open 2022, 12, e055292. [Google Scholar] [CrossRef] [PubMed]
- Kotwal, A.; Stan, M. Thyrotropin Receptor Antibodies—An Overview. Ophthal. Plast. Reconstr. Surg. 2018, 34, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S.; Laurberg, P. Hyperthyroidism in pregnancy. Lancet Diabetes Endocrinol. 2013, 1, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Amino, N.; Tanizawa, O.; Mori, H.; Iwatani, Y.; Yamada, T.; Kurachi, K.; Kumahara, Y.; Miyai, K. Aggravation of thyrotoxicosis in early pregnancy and after delivery in Graves’ disease. J. Clin. Endocrinol. Metab. 1982, 55, 108–112. [Google Scholar] [CrossRef]
- Tagami, T.; Hagiwara, H.; Kimura, T.; Usui, T.; Shimatsu, A.; Naruse, M. The incidence of gestational hyperthyroidism and postpartum thyroiditis in treated patients with Graves’ disease. Thyroid 2007, 17, 767–772. [Google Scholar] [CrossRef]
- Davis, L.E.; Lucas, M.J.; Hankins, G.D.V.; Roark, M.L.; Cunningham, F.G. Thyrotoxicosis complicating pregnancy. Am. J. Obstet. Gynecol. 1989, 160, 63–70. [Google Scholar] [CrossRef]
- Laurberg, P.; Bournaud, C.; Karmisholt, J.; Orgiazzi, J. Management of Graves’ hyperthyroidism in pregnancy: Focus on both maternal and foetal thyroid function, and caution against surgical thyroidectomy in pregnancy. Eur. J. Endocrinol. 2009, 160, 1–8. [Google Scholar] [CrossRef]
- Guo, N.; Xue, M.; Liang, Z. Advances in the differential diagnosis of transient hyperthyroidism in pregnancy and Graves’ disease. Arch. Gynecol. Obstet. 2022. [Google Scholar] [CrossRef]
- Yoshihara, A.; Noh, J.Y.; Mukasa, K.; Suzuki, M.; Ohye, H.; Matsumoto, M.; Kunii, Y.; Watanabe, N.; Suzuki, N.; Kameda, T.; et al. Serum human chorionic gonadotropin levels and thyroid hormone levels in gestational transient thyrotoxicosis: Is the serum hCG level useful for differentiating between active Graves’ disease and GTT? Endocr. J. 2015, 62, 557–560. [Google Scholar] [CrossRef]
- Xue, M.; Shi, Q.L.; Tan, K.N.; Wu, Y.; Zhou, R. The role of color doppler ultrasonography, thyroid function and auto antibody for the screening of Graves’ disease in pregnancy. Zhonghua Nei Ke Za Zhi 2016, 55, 470–473. [Google Scholar] [CrossRef]
- Hiraiwa, T.; Tsujimoto, N.; Tanimoto, K.; Terasaki, J.; Amino, N.; Hanafusa, T. Use of color Doppler ultrasonography to measure thyroid blood flow and differentiate graves’ disease from painless thyroiditis. Eur. Thyroid J. 2013, 2, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Metsman, J.H. Hyperthyroidism in pregnancy. Best Pract. Res. Clin. Endocrinol. Metab. 2004, 18, 267–288. [Google Scholar] [CrossRef]
- Patel, K.N.; Yip, L.; Lubitz, C.C.; Grubbs, E.G.; Miller, B.S.; Shen, W.; Angelos, P.; Chen, H.; Doherty, G.M.; Fahey, T.J.; et al. The American Association of Endocrine Surgeons Guidelines for the Definitive Surgical Management of Thyroid Disease in Adults. Ann. Surg. 2020, 271, E21–E93. [Google Scholar] [CrossRef] [PubMed]
- de Mul, N.; Damstra, J.; Nieveen van Dijkum, E.J.M.; Fischli, S.; Kalkman, C.J.; Schellekens, W.J.M.; Immink, R.V. Risk of perioperative thyroid storm in hyperthyroid patients: A systematic review. Br. J. Anaesth. 2021, 127, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Abalovich, M.; Gutierrez, S.; Alcaraz, G.; Maccallini, G.; Garcia, A.; Levalle, O. Overt and subclinical hypothyroidism complicating pregnancy. Thyroid 2002, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.M.; Middleton, P.; Cossich, M.C.; Crowther, C.A.; Bain, E. Interventions for clinical and subclinical hypothyroidism pre-pregnancy and during pregnancy. Cochrane Database Syst. Rev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Dhillon-Smith, R.K.; Tobias, A.; Smith, P.P.; Middleton, L.J.; Sunner, K.K.; Baker, K.; Farrell-Carver, S.; Bender-Atik, R.; Agrawal, R.; Bhatia, K.; et al. The Prevalence of Thyroid Dysfunction and Autoimmunity in Women with History of Miscarriage or Subfertility. J. Clin. Endocrinol. Metab. 2020, 105, 2667–2677. [Google Scholar] [CrossRef]
- Toloza, F.J.K.; Derakhshan, A.; Männistö, T.; Bliddal, S.; Popova, P.V.; Carty, D.M.; Chen, L.; Taylor, P.; Mosso, L.; Oken, E.; et al. Association between maternal thyroid function and risk of gestational hypertension and pre-eclampsia: A systematic review and individual-participant data meta-analysis. Lancet Diabetes Endocrinol. 2022, 10, 243–252. [Google Scholar] [CrossRef]
- Geng, X.; Chen, Y.; Li, S.; Wang, W.; Wu, W.; Sun, C.; Li, N.; Wang, L. Systematic review and meta-analysis on the influence of thyroid dysfunction in early pregnancy on pregnancy outcomes under ultrasound guidance. Ann. Palliat. Med. 2022, 11, 1001–1016. [Google Scholar] [CrossRef]
- Thangaratinam, S.; Tan, A.; Knox, E.; Kilby, M.D.; Franklyn, J.; Coomarasamy, A. Association between thyroid autoantibodies and miscarriage and preterm birth: Meta-analysis of evidence. BMJ 2011, 342, d2616. [Google Scholar] [CrossRef]
- Spinillo, A.; De Maggio, I.; Ruspini, B.; Bellingeri, C.; Cavagnoli, C.; Giannico, S.; Boschetti, A.; Magri, F.; Lovati, E.; Beneventi, F. Placental pathologic features in thyroid autoimmunity. Placenta 2021, 112, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ye, E.; Sun, M.; Lin, H.; Yu, L.; Lin, Z.; Peng, M.; Lin, D.; Lu, X. Association between third trimester maternal isolated hypothyroxinemia and adverse pregnancy outcomes. Endocr. J. 2023, EJ22-0528. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.E.; Pinz, I.; Preda, M.; Norton, C.R.; Gridley, T.; Hernandez, A. DIO3 protects against thyrotoxicosis-derived cranio-encephalic and cardiac congenital abnormalities. JCI Insight 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Peng, T.; Li, M.Q.; Xie, F.; Wu, J.N. Association between Maternal Thyroxine and Risk of Fetal Congenital Heart Defects: A Hospital-Based Cohort Study. Int. J. Endocrinol. 2022, 2022, 3859388. [Google Scholar] [CrossRef]
- Ochi, S.; Manabe, S.; Kikkawa, T.; Osumi, N. Thirty Years’ History since the Discovery of Pax6: From Central Nervous System Development to Neurodevelopmental Disorders. Int. J. Mol. Sci. 2022, 23, 6115. [Google Scholar] [CrossRef]
- Xu, Y.; Xi, J.; Wang, G.; Guo, Z.; Sun, Q.; Lu, C.; Ma, L.; Wu, Y.; Jia, W.; Zhu, S.; et al. PAUPAR and PAX6 sequentially regulate human embryonic stem cell cortical differentiation. Nucleic Acids Res. 2021, 49, 1935–1950. [Google Scholar] [CrossRef]
- Hevner, R.F. Intermediate progenitors and Tbr2 in cortical development. J. Anat. 2019, 235, 616–625. [Google Scholar] [CrossRef]
- Mohan, V.; Sinha, R.A.; Pathak, A.; Rastogi, L.; Kumar, P.; Pal, A.; Godbole, M.M. Maternal thyroid hormone deficiency affects the fetal neocorticogenesis by reducing the proliferating pool, rate of neurogenesis and indirect neurogenesis. Exp. Neurol. 2012, 237, 477–488. [Google Scholar] [CrossRef]
- Vancamp, P.; Le Blay, K.; Butruille, L.; Sébillot, A.; Boelen, A.; Demeneix, B.A.; Remaud, S. Developmental thyroid disruption permanently affects the neuroglial output in the murine subventricular zone. Stem Cell Rep. 2022, 17, 459. [Google Scholar] [CrossRef]
- Shimokawa, N.; Yousefi, B.; Morioka, S.; Yamaguchi, S.; Ohsawa, A.; Hayashi, H.; Azuma, A.; Mizuno, H.; Kasagi, M.; Masuda, H.; et al. Altered cerebellum development and dopamine distribution in a rat genetic model with congenital hypothyroidism. J. Neuroendocrinol. 2014, 26, 164–175. [Google Scholar] [CrossRef]
- Engel, S.M.; Villanger, G.D.; Herring, A.; Nethery, R.C.; Drover, S.S.M.; Zoeller, R.T.; Meltzer, H.M.; Zeiner, P.; Knudsen, G.P.; Reichborn-Kjennerud, T.; et al. Gestational thyroid hormone concentrations and risk of attention-deficit hyperactivity disorder in the Norwegian Mother, Father and Child Cohort Study. Paediatr. Perinat. Epidemiol. 2023, 37, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, Z.C.; Zhang, T.; Fan, P.; Ma, R.; Zhang, J.; Ouyang, F. Maternal Thyroid Dysfunction and Neuropsychological Development in Children. J. Clin. Endocrinol. Metab. 2023, 108, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, A.; Peeters, R.P.; Taylor, P.N.; Bliddal, S.; Carty, D.M.; Meems, M.; Vaidya, B.; Chen, L.; Knight, B.A.; Ghafoor, F.; et al. Association of maternal thyroid function with birth weight: A systematic review and individual-participant data meta-analysis HHS Public Access. Lancet Diabetes Endocrinol. 2020, 8, 501–510. [Google Scholar] [CrossRef]
- Wu, W.; Zhou, Y.; Liu, Y.; Liu, C.; Ren, J.; Liu, X.; Korevaar, T.I.M.; Fan, J. Triglycerides Mediate the Relationship between Maternal Free Thyroxine and Birth Weight: A Prospective Cohort Study from China. Thyroid 2023. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhong, H. Correlation between Hypothyroidism During Pregnancy and Glucose and Lipid Metabolism in Pregnant Women and Its Influence on Pregnancy Outcome and Fetal Growth and Development. Front. Surg. 2022, 9, 863286. [Google Scholar] [CrossRef]
- Yang, M.; Sun, M.; Jiang, C.; Wu, Q.; Jiang, Y.; Xu, J.; Luo, Q. Thyroid hormones and carnitine in the second trimester negatively affect neonate birth weight: A prospective cohort study. Front. Endocrinol. 2023, 14, 1080969. [Google Scholar] [CrossRef]
- Zakarija, M.; McKenzie, J.M. Pregnancy-associated changes in the thyroid-stimulating antibody of Graves’ disease and the relationship to neonatal hyperthyroidism. J. Clin. Endocrinol. Metab. 1983, 57, 1036–1040. [Google Scholar] [CrossRef]
- Polak, M. Hyperthyroidism in early infancy: Pathogenesis, clinical features and diagnosis with a focus on neonatal hyperthyroidism. Thyroid 1998, 8, 1171–1177. [Google Scholar] [CrossRef]
- Hébrant, A.; Van Staveren, W.C.G.; Maenhaut, C.; Dumont, J.E.; Leclère, J. Genetic hyperthyroidism: Hyperthyroidism due to activating TSHR mutations. Eur. J. Endocrinol. 2011, 164, 1–9. [Google Scholar] [CrossRef]
- Paschke, R.; Niedziela, M.; Vaidya, B.; Persani, L.; Rapoport, B.; Leclere, J.; Paschke, R. 2012 European Thyroid Association Guidelines for the Management of Familial and Persistent Sporadic Non-Autoimmune Hyperthyroidism Caused by Thyroid-Stimulating Hormone Receptor Germline Mutations. Eur. Thyroid J. 2012, 1, 142–147. [Google Scholar] [CrossRef]
- Cho, W.K.; Ahn, M.B.; Jang, W.; Chae, H.; Kim, M.; Suh, B.K. Nonautoimmune congenital hyperthyroidism due to p.Asp633Glu mutation in the TSHR gene. Ann. Pediatr. Endocrinol. Metab. 2018, 23, 235. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, H.P.; Wonerow, P.; Von Petrykowski, W.; Henschen, M.; Scherbaum, W.A.; Paschke, R. Sporadic congenital hyperthyroidism due to a spontaneous germline mutation in the thyrotropin receptor gene. J. Clin. Endocrinol. Metab. 1997, 82, 3879–3884. [Google Scholar] [CrossRef]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, R.; Dias, P.; Gouveia, R.; Sousa, A.B.; Oliveira, G. Neonatal McCune-Albright syndrome with systemic involvement: A case report. J. Med. Case Rep. 2015, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Shenker, A.; Weinstein, L.S.; Moran, A.; Pescovitz, O.H.; Charest, N.J.; Boney, C.M.; Van Wyk, J.J.; Merino, M.J.; Feuillan, P.P.; Spiegel, A.M. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein GS. J. Pediatr. 1993, 123, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Nakayama, M.; Baba, T.; Uehara, Y.; Niikawa, N.; Ito, M.; Tsuji, Y. A case of neonatal McCune-Albright syndrome with Cushing syndrome and hyperthyroidism. Acta Paediatr. Scand. 1991, 80, 984–987. [Google Scholar] [CrossRef]
- Gaspari, L.; Paris, F.; Nicolino, M.; Hameury, F.; Bonnaure, H.; Pienkowski, C.; Servant, N.; Kalfa, N.; Sultan, C. Fetal ovarian cysts: An early manifestation of McCune-Albright syndrome? Prenat. Diagn. 2012, 32, 859–863. [Google Scholar] [CrossRef]
- Polak, M.; Legac, I.; Vuillard, E.; Guibourdenche, J.; Castanet, M.; Luton, D. Congenital Hyperthyroidism: The Fetus as a Patient. Horm. Res. Paediatr. 2006, 65, 235–242. [Google Scholar] [CrossRef]
- Huel, C.; Guibourdenche, J.; Vuillard, E.; Ouahba, J.; Piketty, M.; Oury, J.F.; Luton, D. Use of ultrasound to distinguish between fetal hyperthyroidism and hypothyroidism on discovery of a goiter. Ultrasound Obstet. Gynecol. 2009, 33, 412–420. [Google Scholar] [CrossRef]
- Ranzini, A.C.; Ananth, C.V.; Smulian, J.C.; Kung, M.; Limbachia, A.; Vintzileos, A.M. Ultrasonography of the fetal thyroid: Nomograms based on biparietal diameter and gestational age. J. Ultrasound Med. 2001, 20, 613–617. [Google Scholar] [CrossRef]
- Luton, D.; Le Gac, I.; Vuillard, E.; Castanet, M.; Guibourdenche, J.; Noel, M.; Toubert, M.E.; Léger, J.; Boissinot, C.; Schlageter, M.H.; et al. Management of Graves’ Disease during Pregnancy: The Key Role of Fetal Thyroid Gland Monitoring. J. Clin. Endocrinol. Metab. 2005, 90, 6093–6098. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.V.; LaFranchi, S.H. Congenital hypothyroidism. Orphanet J. Rare Dis. 2010, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.; Bellisario, R.L.; Botero, D.; Fournier, L.; Abrams, C.A.; Cowger, M.L.; David, R.; Fort, P.; Richman, R.A. Incidence of transient congenital hypothyroidism due to maternal thyrotropin receptor-blocking antibodies in over one million babies. J. Clin. Endocrinol. Metab. 1996, 81, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Gutch, M.; Philip, R.; Philip, R.; Toms, A.; Saran, S.; Gupta, K. Skeletal manifestations of juvenile hypothyroidism and the impact of treatment on skeletal system. Indian J. Endocrinol. Metab. 2013, 17, 181. [Google Scholar] [CrossRef] [PubMed]
- Vanderpas, J. Nutritional epidemiology and thyroid hormone metabolism. Annu. Rev. Nutr. 2006, 26, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Centre de Référence des Déficiences Intellectuelles de Causes Rares Filière DéfiScience Maladies Rares du Développement Cérébral et Déficience Intellectuelle Protocole National de Diagnostic et de Soins (PNDS) Syndrome de Allan Herndon-Dudley (SAHD) (MCT8 Thyroid Hormone Transporter) Argumentaire. Centre de Référence des Déficiences Intelectuelles de Causes Rares, Encéphalopathies, Centre de Compétence des Leucodystrophies et Leuco Encéphalopathies Rares, HAS. Protocole National de Diagnostic et de Soins: Syndrome de Allan Herndon-Dudley. 2019. Available online: https://www.has-sante.fr/upload/docs/application/pdf/2020-05/sahd_mct8_argumentaire.pdf (accessed on 12 March 2023).
Figure 1.
Sonographic view of fetal thyroid goiter from a sagittal plane (personal data from Bicetre Hospital Fetal Medicine Unit). The arrow points toward the fetal neck, which is distorted by a hyperthyroidic goiter.
Figure 1.
Sonographic view of fetal thyroid goiter from a sagittal plane (personal data from Bicetre Hospital Fetal Medicine Unit). The arrow points toward the fetal neck, which is distorted by a hyperthyroidic goiter.

Figure 2.
Sonographic views of fetal thyroids in an axial plane at 23 WG (personal data from Bicetre Hospital Fetal Medicine Unit). Image (A) represents a normal fetal thyroid with a transverse diameter of 13.57 mm and a perimeter of 36.73 mm, whereas image (B) represents a large hypothyroidic fetal goiter with a transverse diameter of 26.0 mm and a perimeter of 71.9 mm.
Figure 2.
Sonographic views of fetal thyroids in an axial plane at 23 WG (personal data from Bicetre Hospital Fetal Medicine Unit). Image (A) represents a normal fetal thyroid with a transverse diameter of 13.57 mm and a perimeter of 36.73 mm, whereas image (B) represents a large hypothyroidic fetal goiter with a transverse diameter of 26.0 mm and a perimeter of 71.9 mm.


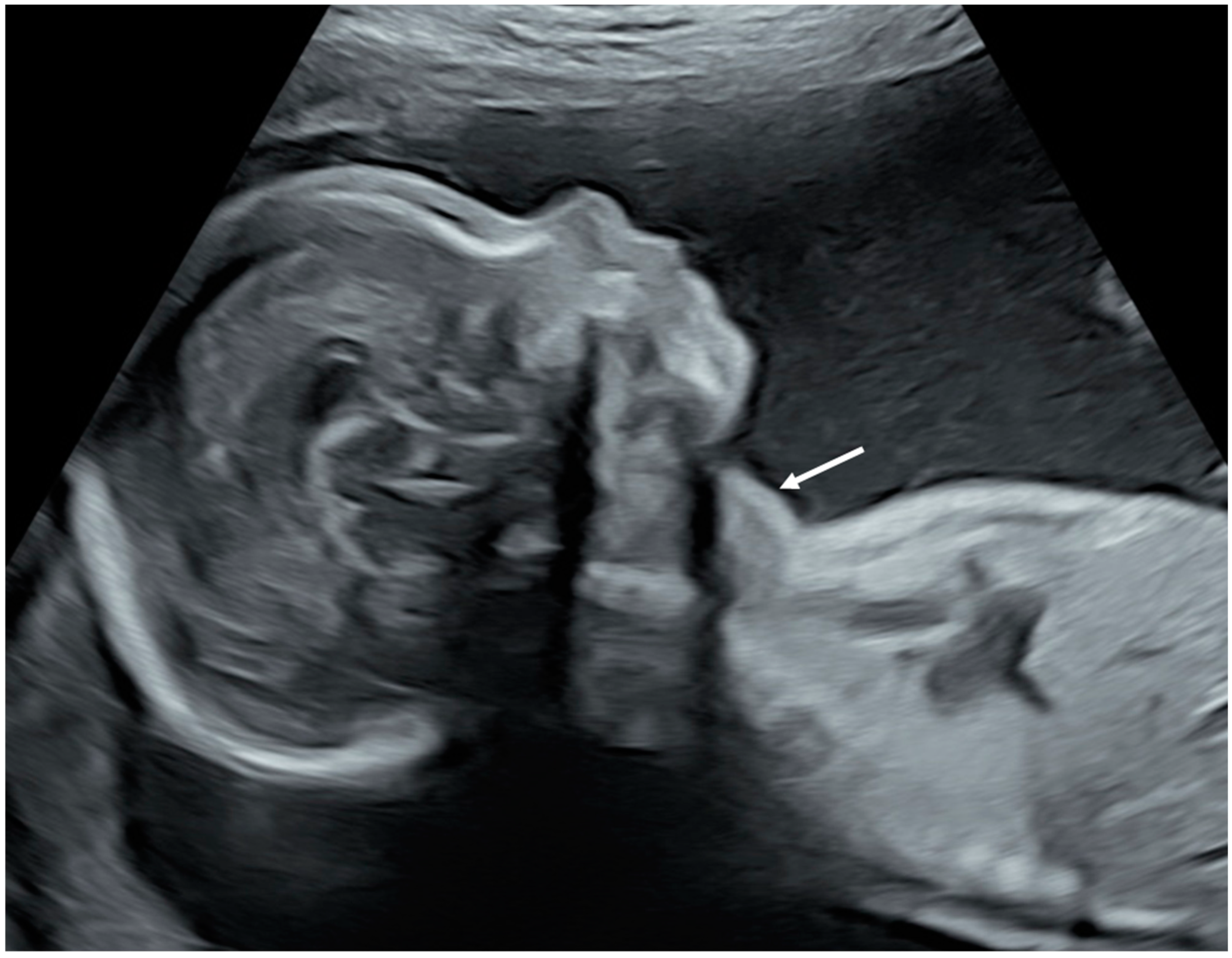{kind=link}
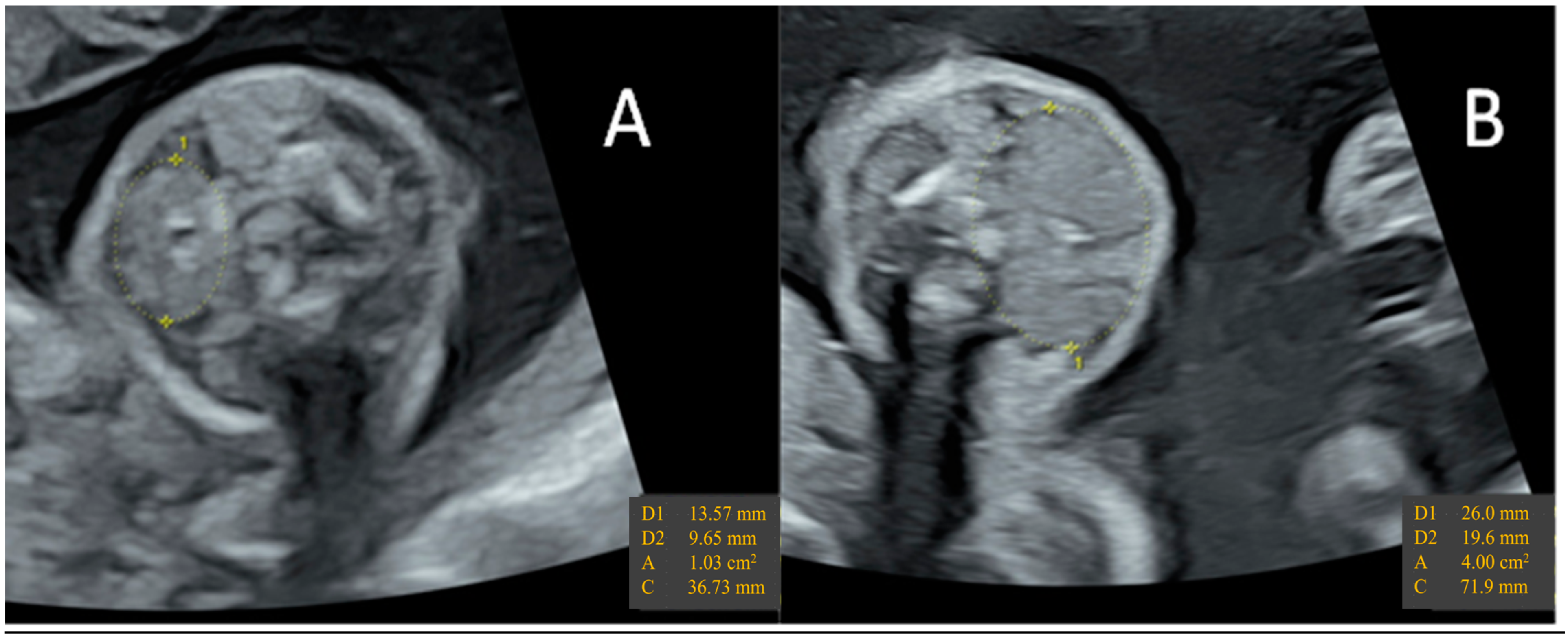{kind=link}
Table 1.
TH transporters’ locations in the placenta and their variations throughout pregnancy [94,98,99,100,101,102,103,104,105,106,107,108,109].
| Placental Location | Evolution throughout Pregnancy | Substrates Uptake | Substrates Efflux | |||
|---|---|---|---|---|---|---|
| T1 | T2 | T3 | ||||
| LAT1 | SCT apical surface |  | T2 > rT3 > T3 > T4 | T2 | ||
| LAT2 | SCT |  | T2 > T3 | * | ||
| MCT8 | CT + SCT | * | T3, T4 > rT3 > T2 | T3, T4 | ||
| MCT10 | CT + SCT at T1 SCT at T3 | * | T3 > T4 | T3 | ||
| OATP1A2 | SCT basal surface CT and CT EV |  | T3 > rT3, T4 | * | ||
| OAPT4A1 | SCT apical surface |  | T3 > T4, rT3 | * | ||
T2: Diiodothyronine; T3: Triiodothyronine; rT3: Reverse Triiodothyronine; T4: Thyroxine; LAT1: L-type Amino-acid Transporter 1; LAT2: L-type Amino-acid Transporter 2; MCT8: Mono-Carboxylate Transporters 8; MCT10: Mono-Carboxylate Transporters 8; OATP1A2: Organic-Anion Transporting Peptide 1A2; OATP4A1: Organic-Anion Transporting Peptide 4A1; CT: Cytotrophoblast; EV CT: Extra-villous Cytotrophoblast; SCT: Syncytiotrophoblast; T1: first trimester of pregnancy; T2: second trimester of pregnancy; T3: third trimester of pregnancy; *: no data available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mégier, C.; Dumery, G.; Luton, D. Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy. Metabolites 2023, 13, 633. https://doi.org/10.3390/metabo13050633
AMA Style
Mégier C, Dumery G, Luton D. Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy. Metabolites. 2023; 13(5):633. https://doi.org/10.3390/metabo13050633
Chicago/Turabian StyleMégier, Charles, Grégoire Dumery, and Dominique Luton. 2023. "Iodine and Thyroid Maternal and Fetal Metabolism during Pregnancy" Metabolites 13, no. 5: 633. https://doi.org/10.3390/metabo13050633
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.