
Influence of Cholesterol on the Regulation of Osteoblast Function

Abstract
:1. Introduction
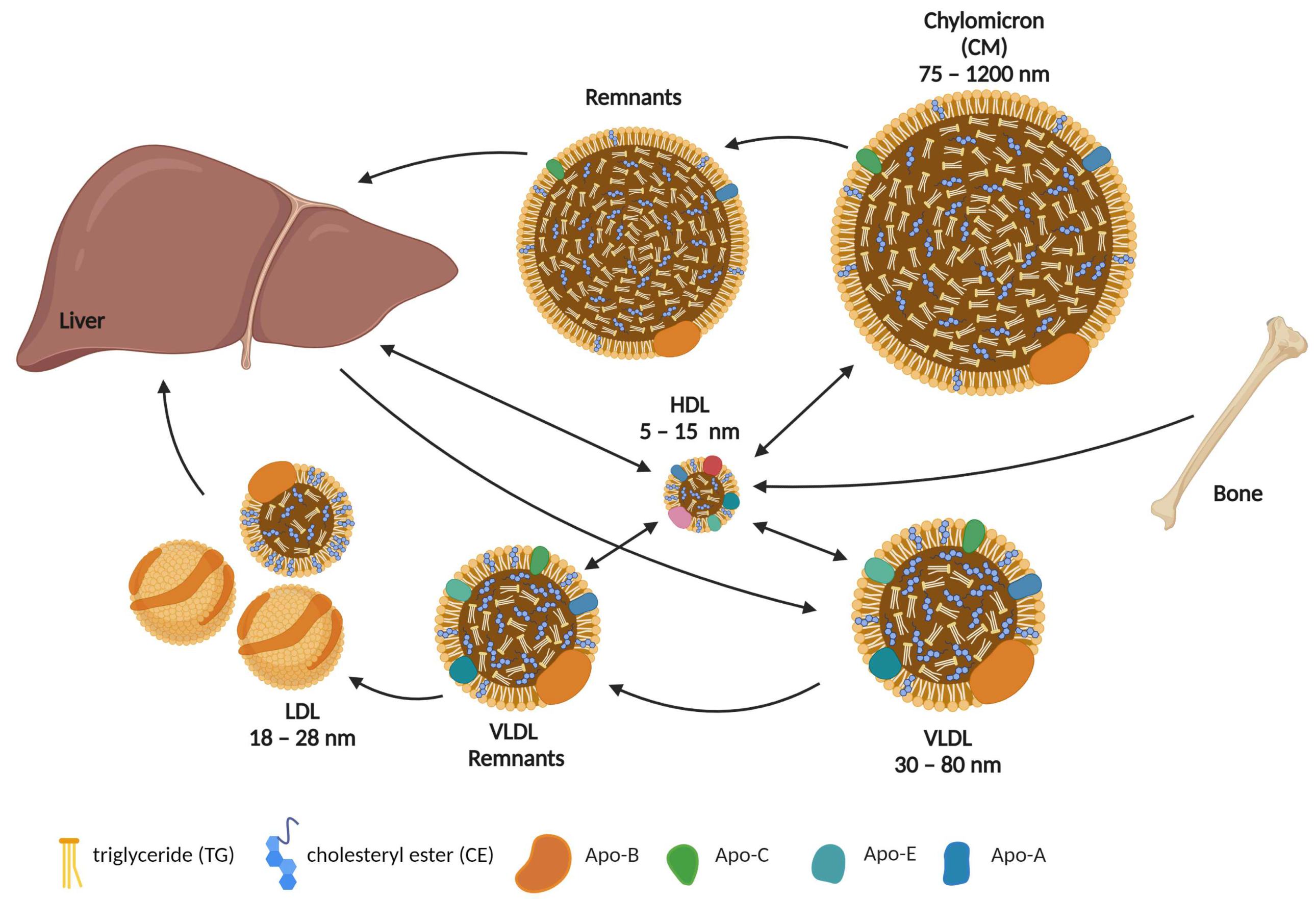
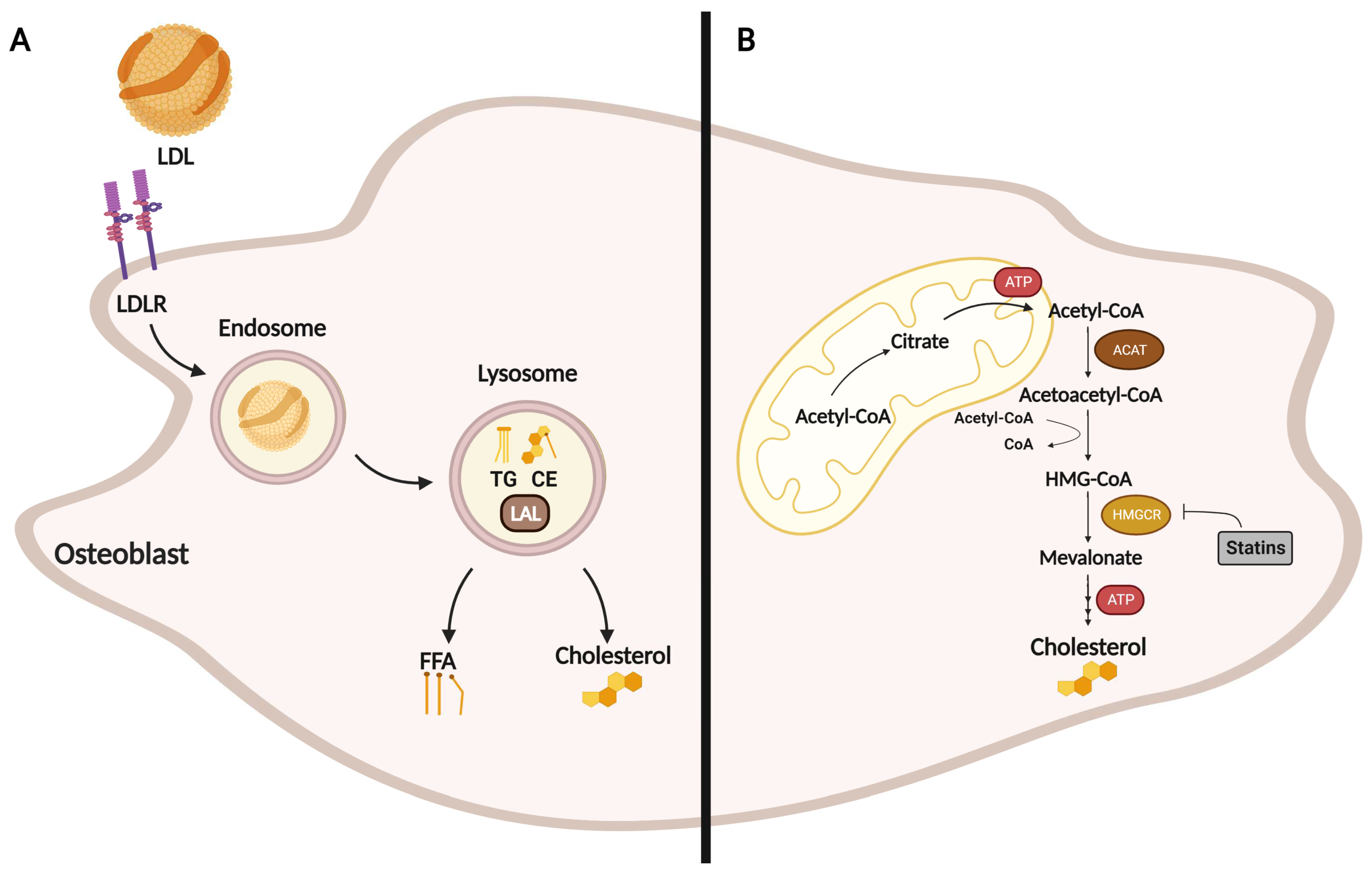
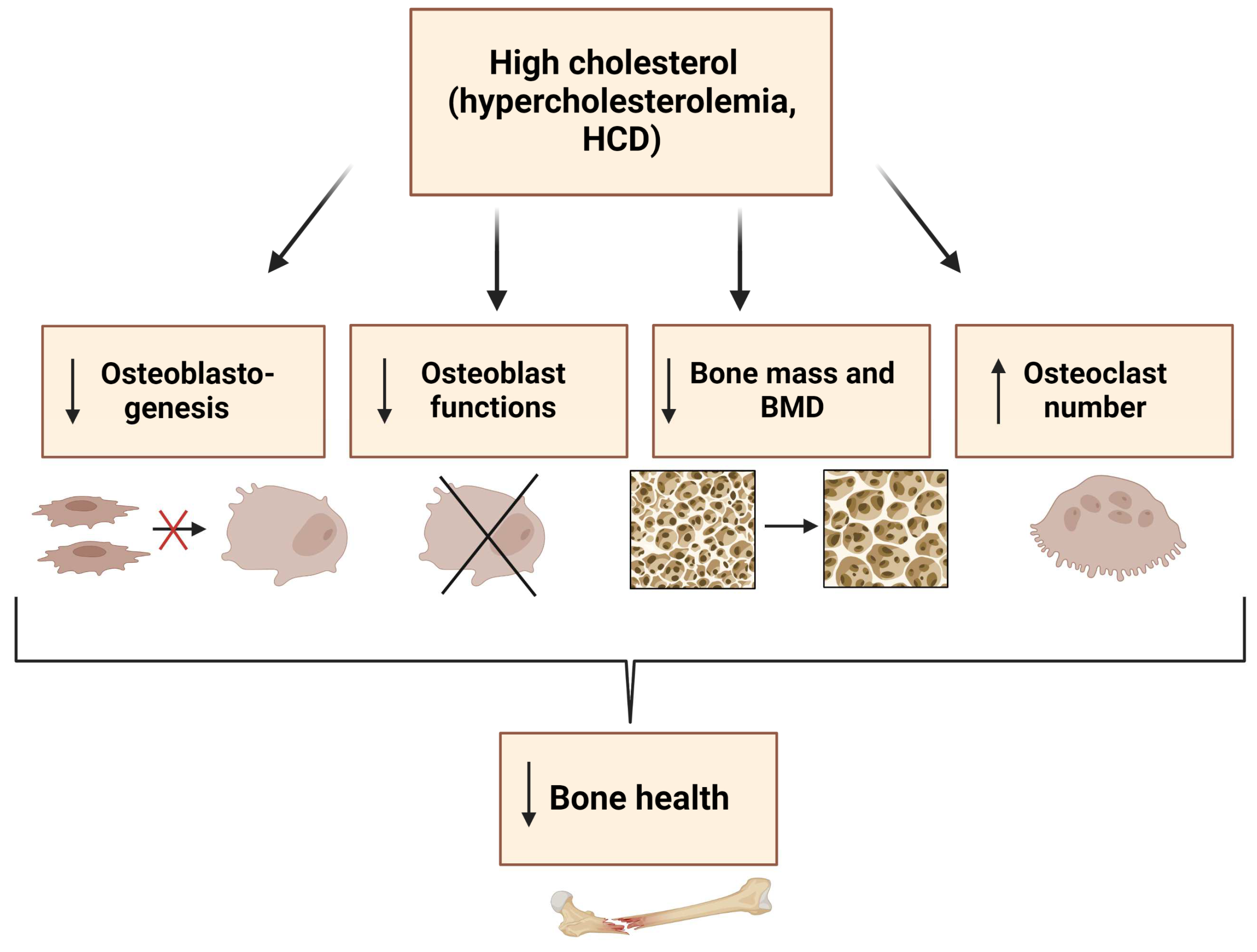
2. Role of Cholesterol in Osteoblast Formation, Function, and Metabolism
3. Effect of Statins on Osteoblasts
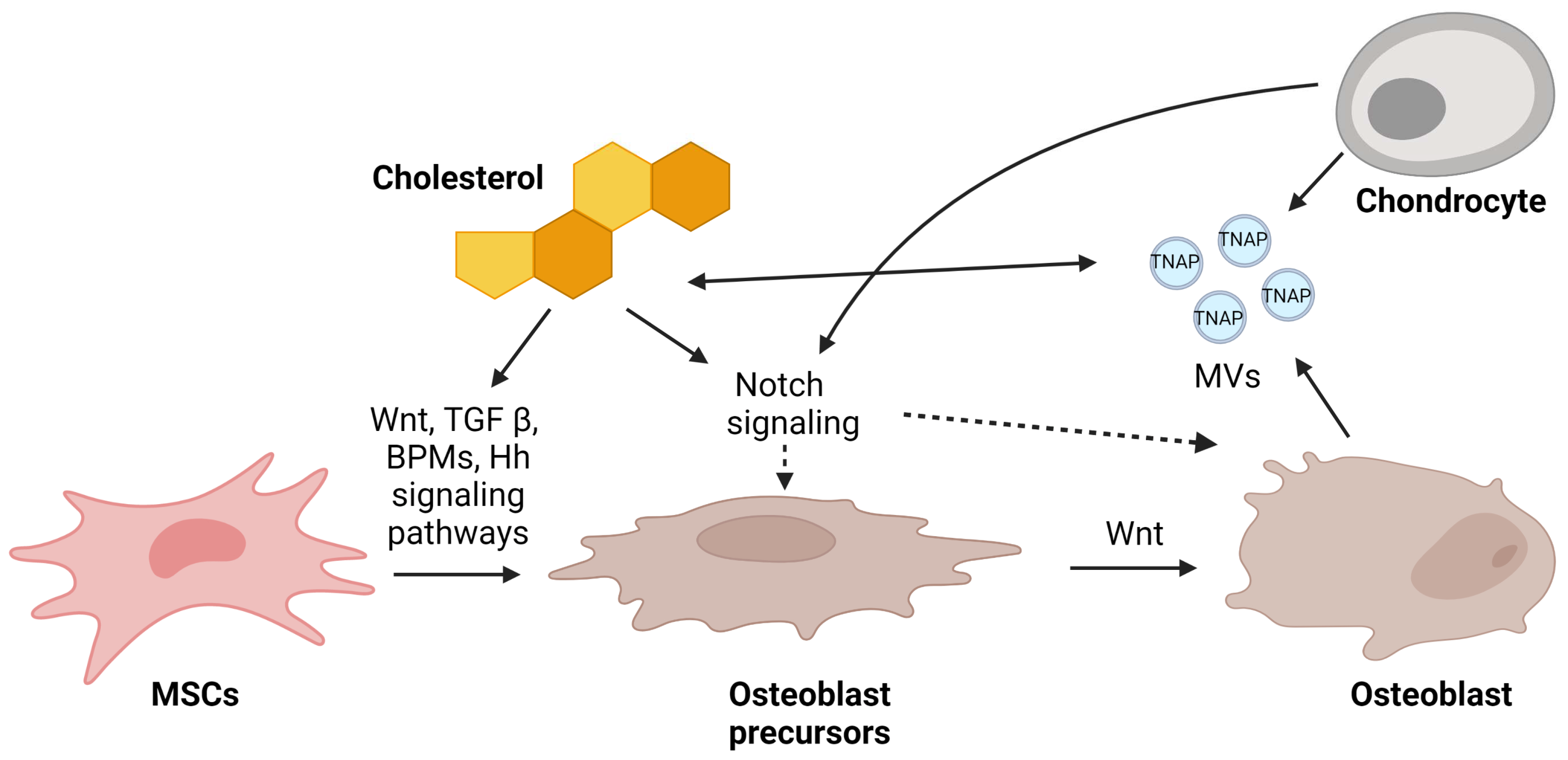
4. Mechanism of Osteoblast Regulation by Cholesterol via Signaling Pathways
4.1. Wnt-Lrp5-β-Catenin
4.2. TGF-β/BMP2
4.3. Notch
4.4. Hedgehog (Hh)
5. Mechanism of Mineralization Regulation by Matrix Vesicles
6. Conclusions
7. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Su, N.; Yang, J.; Xie, Y.; Du, X.; Chen, H.; Zhou, H.; Chen, L. Bone function, dysfunction and its role in diseases including critical illness. Int. J. Biol. Sci. 2019, 15, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Castillo, A.B.; Turner, C.H. Biomechanical and molecular regulation of bone remodeling. Annu. Rev. Biomed. Eng. 2006, 8, 455–498. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L. Bone composition: Relationship to bone fragility and antiosteoporotic drug effects. Bonekey Rep. 2013, 2, 447. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Glimcher, M.J.; Cooper, R.R.; Recker, R. Bone biology. I: Structure, blood supply, cells, matrix, and mineralization. Instr. Course Lect. 1996, 45, 371–386. [Google Scholar]
- Mohan, A.; Girdhar, M.; Kumar, R.; Chaturvedi, H.S.; Vadhel, A.; Solanki, P.R.; Kumar, A.; Kumar, D.; Mamidi, N. Polyhydroxybutyrate-Based Nanocomposites for Bone Tissue Engineering. Pharmaceuticals 2021, 14, 1163. [Google Scholar] [CrossRef]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the Human Skeletal Stem Cell. Cell 2018, 175, 43–56.e21. [Google Scholar] [CrossRef]
- Marie, P.J. Transcription factors controlling osteoblastogenesis. Arch. Biochem. Biophys. 2008, 473, 98–105. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells During Fracture Healing. J. Cell. Physiol. 2017, 232, 913–921. [Google Scholar] [CrossRef]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef]
- Brauer, A.; Pohlemann, T.; Metzger, W. Osteogenic differentiation of immature osteoblasts: Interplay of cell culture media and supplements. Biotech. Histochem. 2016, 91, 161–169. [Google Scholar] [CrossRef]
- Dai, R.; Liu, M.; Xiang, X.; Xi, Z.; Xu, H. Osteoblasts and osteoclasts: An important switch of tumour cell dormancy during bone metastasis. J. Exp. Clin. Cancer Res. 2022, 41, 316. [Google Scholar] [CrossRef]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng. Part. B. Rev. 2017, 23, 268–280. [Google Scholar] [CrossRef]
- Gordon, J.A.R.; Stein, J.L.; Westendorf, J.J.; van Wijnen, A.J. Chromatin modifiers and histone modifications in bone formation, regeneration, and therapeutic intervention for bone-related disease. Bone 2015, 81, 739–745. [Google Scholar] [CrossRef]
- Yi, S.J.; Lee, H.; Lee, J.; Lee, K.; Kim, J.; Kim, Y.; Park, J.I.; Kim, K. Bone Remodeling: Histone Modifications as Fate Determinants of Bone Cell Differentiation. Int. J. Mol. Sci. 2019, 20, 3147. [Google Scholar] [CrossRef]
- Sen, B.; Xie, Z.; Uzer, G.; Thompson, W.R.; Styner, M.; Wu, X.; Rubin, J. Intranuclear Actin Regulates Osteogenesis. Stem Cells 2015, 33, 3065–3076. [Google Scholar] [CrossRef]
- Suzuki, H.; Tatei, K.; Ohshima, N.; Sato, S.; Izumi, T. Regulation of MC3T3-E1 differentiation by actin cytoskeleton through lipid mediators reflecting the cell differentiation stage. Biochem. Biophys. Res. Commun. 2019, 514, 393–400. [Google Scholar] [CrossRef]
- Ansari, S.; de Wildt, B.W.M.; Vis, M.A.M.; de Korte, C.E.; Ito, K.; Hofmann, S.; Yuana, Y. Matrix Vesicles: Role in Bone Mineralization and Potential Use as Therapeutics. Pharmaceuticals 2021, 14, 289. [Google Scholar] [CrossRef]
- Cui, L.; Houston, D.A.; Farquharson, C.; MacRae, V.E. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone 2016, 87, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Riddle, R.C.; Clemens, T.L. Bone Cell Bioenergetics and Skeletal Energy Homeostasis. Physiol. Rev. 2017, 97, 667–698. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Xiao, J.; Wang, J.; Ma, Y.; Zhang, Y.; Zhang, Q.; Zhang, Z.; Yin, H. The Interaction Between Intracellular Energy Metabolism and Signaling Pathways During Osteogenesis. Front. Mol. Biosci. 2021, 8, 807487. [Google Scholar] [CrossRef] [PubMed]
- Sautchuk, R., Jr.; Eliseev, R.A. Cell energy metabolism and bone formation. Bone Rep. 2022, 16, 101594. [Google Scholar] [CrossRef] [PubMed]
- Guntur, A.R.; Le, P.T.; Farber, C.R.; Rosen, C.J. Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 2014, 155, 1589–1595. [Google Scholar] [CrossRef]
- Ansari, S.; Ito, K.; Hofmann, S. Alkaline Phosphatase Activity of Serum Affects Osteogenic Differentiation Cultures. ACS Omega 2022, 7, 12724–12733. [Google Scholar] [CrossRef]
- Zhang, Q.; Riddle, R.C.; Clemens, T.L. Bone and the regulation of global energy balance. J. Intern. Med. 2015, 277, 681–689. [Google Scholar] [CrossRef]
- Dirckx, N.; Moorer, M.C.; Clemens, T.L.; Riddle, R.C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 2019, 15, 651–665. [Google Scholar] [CrossRef]
- Shum, L.C.; White, N.S.; Mills, B.N.; Bentley, K.L.; Eliseev, R.A. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells Dev. 2016, 25, 114–122. [Google Scholar] [CrossRef]
- Motyl, K.J.; Guntur, A.R.; Carvalho, A.L.; Rosen, C.J. Energy Metabolism of Bone. Toxicol. Pathol. 2017, 45, 887–893. [Google Scholar] [CrossRef]
- Jayapalan, S.; Nandy, A.; Rendina-Ruedy, E. Using Real-Time Cell Metabolic Flux Analyzer to Monitor Osteoblast Bioenergetics. J. Vis. Exp. 2022, e63142. [Google Scholar] [CrossRef]
- Misra, B.B.; Jayapalan, S.; Richards, A.K.; Helderman, R.C.M.; Rendina-Ruedy, E. Untargeted metabolomics in primary murine bone marrow stromal cells reveals distinct profile throughout osteoblast differentiation. Metabolomics 2021, 17, 86. [Google Scholar] [CrossRef]
- Tencerova, M.; Rendina-Ruedy, E.; Neess, D.; Faergeman, N.; Figeac, F.; Ali, D.; Danielsen, M.; Haakonsson, A.; Rosen, C.J.; Kassem, M. Metabolic programming determines the lineage-differentiation fate of murine bone marrow stromal progenitor cells. Bone Res. 2019, 7, 35. [Google Scholar] [CrossRef]
- Guntur, A.R.; Gerencser, A.A.; Le, P.T.; DeMambro, V.E.; Bornstein, S.A.; Mookerjee, S.A.; Maridas, D.E.; Clemmons, D.E.; Brand, M.D.; Rosen, C.J. Osteoblast-like MC3T3-E1 Cells Prefer Glycolysis for ATP Production but Adipocyte-like 3T3-L1 Cells Prefer Oxidative Phosphorylation. J. Bone Miner. Res. 2018, 33, 1052–1065. [Google Scholar] [CrossRef]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef]
- Frey, J.L.; Li, Z.; Ellis, J.M.; Zhang, Q.; Farber, C.R.; Aja, S.; Wolfgang, M.J.; Clemens, T.L.; Riddle, R.C. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol. Cell. Biol. 2015, 35, 1979–1991. [Google Scholar] [CrossRef]
- Shen, L.; Hu, G.; Karner, C.M. Bioenergetic Metabolism In Osteoblast Differentiation. Curr. Osteoporos. Rep. 2022, 20, 53–64. [Google Scholar] [CrossRef]
- Zampelas, A.; Magriplis, E. New Insights into Cholesterol Functions: A Friend or an Enemy? Nutrients 2019, 11, 1645. [Google Scholar] [CrossRef]
- During, A.; Penel, G.; Hardouin, P. Understanding the local actions of lipids in bone physiology. Prog. Lipid Res. 2015, 59, 126–146. [Google Scholar] [CrossRef]
- Duan, Y.; Gong, K.; Xu, S.; Zhang, F.; Meng, X.; Han, J. Regulation of cholesterol homeostasis in health and diseases: From mechanisms to targeted therapeutics. Signal. Transduct. Target. Ther. 2022, 7, 265. [Google Scholar] [CrossRef]
- Osborne, J.C.; Brewer, H.B. The Plasma Lipoproteins. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 1977; pp. 253–337. [Google Scholar]
- Parhami, F.; Mody, N.; Gharavi, N.; Ballard, A.J.; Tintut, Y.; Demer, L.L. Role of the cholesterol biosynthetic pathway in osteoblastic differentiation of marrow stromal cells. J. Bone Miner. Res. 2002, 17, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Aghaloo, T.L.; Amantea, C.M.; Cowan, C.M.; Richardson, J.A.; Wu, B.M.; Parhami, F.; Tetradis, S. Oxysterols enhance osteoblast differentiation in vitro and bone healing in vivo. J. Orthop. Res. 2007, 25, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, H.; Li, H. Cholesterol loading affects osteoblastic differentiation in mouse mesenchymal stem cells. Steroids 2013, 78, 426–433. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Sheng, Z.Y.; Tang, C.L.; Chen, L.; Pan, L.; Chen, J.Y. High cholesterol diet increases osteoporosis risk via inhibiting bone formation in rats. Acta Pharmacol. Sin. 2011, 32, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Jackson, S.M.; Tintut, Y.; Le, V.; Balucan, J.P.; Territo, M.; Demer, L.L. Atherogenic diet and minimally oxidized low density lipoprotein inhibit osteogenic and promote adipogenic differentiation of marrow stromal cells. J. Bone Miner. Res. 1999, 14, 2067–2078. [Google Scholar] [CrossRef]
- Mangaraj, M.; Nanda, R.; Panda, S. Apolipoprotein A-I: A Molecule of Diverse Function. Indian. J. Clin. Biochem. 2016, 31, 253–259. [Google Scholar] [CrossRef]
- Blair, H.C.; Kalyvioti, E.; Papachristou, N.I.; Tourkova, I.L.; Syggelos, S.A.; Deligianni, D.; Orkoula, M.G.; Kontoyannis, C.G.; Karavia, E.A.; Kypreos, K.E.; et al. Apolipoprotein A-1 regulates osteoblast and lipoblast precursor cells in mice. Lab. Investig. 2016, 96, 763–772. [Google Scholar] [CrossRef]
- Shouhed, D.; Kha, H.T.; Richardson, J.A.; Amantea, C.M.; Hahn, T.J.; Parhami, F. Osteogenic oxysterols inhibit the adverse effects of oxidative stress on osteogenic differentiation of marrow stromal cells. J. Cell. Biochem. 2005, 95, 1276–1283. [Google Scholar] [CrossRef]
- Maziere, C.; Salle, V.; Gomila, C.; Maziere, J.C. Oxidized low density lipoprotein enhanced RANKL expression in human osteoblast-like cells. Involvement of ERK, NFkappaB and NFAT. Biochim. Biophys. Acta 2013, 1832, 1756–1764. [Google Scholar] [CrossRef]
- Maziere, C.; Savitsky, V.; Galmiche, A.; Gomila, C.; Massy, Z.; Maziere, J.C. Oxidized low density lipoprotein inhibits phosphate signaling and phosphate-induced mineralization in osteoblasts. Involvement of oxidative stress. Biochim. Biophys. Acta 2010, 1802, 1013–1019. [Google Scholar] [CrossRef]
- Harun, N.H.; Froemming, G.R.A.; Mohd Ismail, A.; Nawawi, H.; Mokhtar, S.S.; Abd Muid, S. Osteoblast Demineralization Induced by Oxidized High-Density Lipoprotein via the Inflammatory Pathway Is Suppressed by Adiponectin. Int. J. Mol. Sci. 2022, 23, 14616. [Google Scholar] [CrossRef]
- Pelton, K.; Krieder, J.; Joiner, D.; Freeman, M.R.; Goldstein, S.A.; Solomon, K.R. Hypercholesterolemia promotes an osteoporotic phenotype. Am. J. Pathol. 2012, 181, 928–936. [Google Scholar] [CrossRef]
- Demigne, C.; Bloch-Faure, M.; Picard, N.; Sabboh, H.; Besson, C.; Remesy, C.; Geoffroy, V.; Gaston, A.T.; Nicoletti, A.; Hagege, A.; et al. Mice chronically fed a westernized experimental diet as a model of obesity, metabolic syndrome and osteoporosis. Eur. J. Nutr. 2006, 45, 298–306. [Google Scholar] [CrossRef]
- Parhami, F.; Tintut, Y.; Beamer, W.G.; Gharavi, N.; Goodman, W.; Demer, L.L. Atherogenic high-fat diet reduces bone mineralization in mice. J. Bone Miner. Res. 2001, 16, 182–188. [Google Scholar] [CrossRef]
- Chen, X.; Wang, C.; Zhang, K.; Xie, Y.; Ji, X.; Huang, H.; Yu, X. Reduced femoral bone mass in both diet-induced and genetic hyperlipidemia mice. Bone 2016, 93, 104–112. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, Y.; Lin, J.; Qiu, X.; Chen, L.; Pan, X.; Lu, Y.; Zhang, J.; Wang, Y.; Li, D.; et al. Low-density lipoprotein receptor deficiency impaired mice osteoblastogenesis in vitro. Biosci. Trends 2018, 11, 658–666. [Google Scholar] [CrossRef]
- Martineau, C.; Martin-Falstrault, L.; Brissette, L.; Moreau, R. The atherogenic Scarb1 null mouse model shows a high bone mass phenotype. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E48–E57. [Google Scholar] [CrossRef]
- Isales, C.M.; Zaidi, M.; Blair, H.C. ACTH is a novel regulator of bone mass. Ann. N. Acad. Sci. 2010, 1192, 110–116. [Google Scholar] [CrossRef]
- Tourkova, I.L.; Dobrowolski, S.F.; Secunda, C.; Zaidi, M.; Papadimitriou-Olivgeri, I.; Papachristou, D.J.; Blair, H.C. The high-density lipoprotein receptor Scarb1 is required for normal bone differentiation in vivo and in vitro. Lab. Investig. 2019, 99, 1850–1860. [Google Scholar] [CrossRef]
- Alekos, N.S.; Moorer, M.C.; Riddle, R.C. Dual Effects of Lipid Metabolism on Osteoblast Function. Front. Endocrinol. 2020, 11, 578194. [Google Scholar] [CrossRef]
- Helderman, R.C.; Whitney, D.G.; Duta-Mare, M.; Akhmetshina, A.; Vujic, N.; Jayapalan, S.; Nyman, J.S.; Misra, B.B.; Rosen, C.J.; Czech, M.P.; et al. Loss of function of lysosomal acid lipase (LAL) profoundly impacts osteoblastogenesis and increases fracture risk in humans. Bone 2021, 148, 115946. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H. Lysosomal Acid Lipase in Lipid Metabolism and Beyond. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Mangu, S.R.; Patel, K.; Sukhdeo, S.V.; Savitha, M.R.; Sharan, K. Maternal high-cholesterol diet negatively programs offspring bone development and downregulates hedgehog signaling in osteoblasts. J. Biol. Chem. 2022, 298, 102324. [Google Scholar] [CrossRef] [PubMed]
- DuSell, C.D.; Nelson, E.R.; Wang, X.; Abdo, J.; Modder, U.I.; Umetani, M.; Gesty-Palmer, D.; Javitt, N.B.; Khosla, S.; McDonnell, D.P. The endogenous selective estrogen receptor modulator 27-hydroxycholesterol is a negative regulator of bone homeostasis. Endocrinology 2010, 151, 3675–3685. [Google Scholar] [CrossRef]
- Nelson, E.R.; DuSell, C.D.; Wang, X.; Howe, M.K.; Evans, G.; Michalek, R.D.; Umetani, M.; Rathmell, J.C.; Khosla, S.; Gesty-Palmer, D.; et al. The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver X receptors. Endocrinology 2011, 152, 4691–4705. [Google Scholar] [CrossRef]
- Tarakida, A.; Iino, K.; Abe, K.; Taniguchi, R.; Higuchi, T.; Mizunuma, H.; Nakaji, S. Hypercholesterolemia accelerates bone loss in postmenopausal women. Climacteric 2011, 14, 105–111. [Google Scholar] [CrossRef]
- Go, J.H.; Song, Y.M.; Park, J.H.; Park, J.Y.; Choi, Y.H. Association between Serum Cholesterol Level and Bone Mineral Density at Lumbar Spine and Femur Neck in Postmenopausal Korean Women. Korean J. Fam. Med. 2012, 33, 166–173. [Google Scholar] [CrossRef]
- Tanko, L.B.; Bagger, Y.Z.; Nielsen, S.B.; Christiansen, C. Does serum cholesterol contribute to vertebral bone loss in postmenopausal women? Bone 2003, 32, 8–14. [Google Scholar] [CrossRef]
- Yamauchi, M.; Yamaguchi, T.; Nawata, K.; Tanaka, K.; Takaoka, S.; Sugimoto, T. Increased low-density lipoprotein cholesterol level is associated with non-vertebral fractures in postmenopausal women. Endocrine 2015, 48, 279–286. [Google Scholar] [CrossRef]
- Uyama, O.; Yoshimoto, Y.; Yamamoto, Y.; Kawai, A. Bone changes and carotid atherosclerosis in postmenopausal women. Stroke 1997, 28, 1730–1732. [Google Scholar] [CrossRef]
- Bagger, Y.Z.; Rasmussen, H.B.; Alexandersen, P.; Werge, T.; Christiansen, C.; Tanko, L.B.; PERF Study Group. Links between cardiovascular disease and osteoporosis in postmenopausal women: Serum lipids or atherosclerosis per se? Osteoporos. Int. 2007, 18, 505–512. [Google Scholar] [CrossRef]
- Majima, T.; Komatsu, Y.; Fukao, A.; Ninomiya, K.; Matsumura, T.; Nakao, K. Short-term effects of atorvastatin on bone turnover in male patients with hypercholesterolemia. Endocr. J. 2007, 54, 145–151. [Google Scholar] [CrossRef]
- Majima, T.; Shimatsu, A.; Komatsu, Y.; Satoh, N.; Fukao, A.; Ninomiya, K.; Matsumura, T.; Nakao, K. Short-term effects of pitavastatin on biochemical markers of bone turnover in patients with hypercholesterolemia. Intern. Med. 2007, 46, 1967–1973. [Google Scholar] [CrossRef]
- Yerges-Armstrong, L.M.; Shen, H.; Ryan, K.A.; Streeten, E.A.; Shuldiner, A.R.; Mitchell, B.D. Decreased bone mineral density in subjects carrying familial defective apolipoprotein B-100. J. Clin. Endocrinol. Metab. 2013, 98, E1999–E2005. [Google Scholar] [CrossRef]
- Awan, Z.; Alwaili, K.; Alshahrani, A.; Langsetmo, L.; Goltzman, D.; Genest, J. Calcium homeostasis and skeletal integrity in individuals with familial hypercholesterolemia and aortic calcification. Clin. Chem. 2010, 56, 1599–1607. [Google Scholar] [CrossRef]
- Hernandez, J.L.; Olmos, J.M.; Ramos, C.; Martinez, J.; de Juan, J.; Valero, C.; Nan, D.; Gonzalez-Macias, J. Serum lipids and bone metabolism in Spanish men: The Camargo cohort study. Endocr. J. 2010, 57, 51–60. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Zhang, Y.; Xu, G.; Yi, X.; Liang, J.; Zhao, C.; Liang, J.; Ma, C.; Ye, Y.; et al. Association Between Bone Mineral Density, Bone Turnover Markers, and Serum Cholesterol Levels in Type 2 Diabetes. Front. Endocrinol. 2018, 9, 646. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Bloch, K. The Biological Synthesis of Cholesterol. Science 1965, 150, 19–28. [Google Scholar] [CrossRef]
- Mundy, G.; Garrett, R.; Harris, S.; Chan, J.; Chen, D.; Rossini, G.; Boyce, B.; Zhao, M.; Gutierrez, G. Stimulation of bone formation in vitro and in rodents by statins. Science 1999, 286, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C. High Cholesterol Deteriorates Bone Health: New Insights into Molecular Mechanisms. Front. Endocrinol. 2015, 6, 165. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, N.; Mandal, C.C.; Choudhury, G.G. Statin-induced Ras activation integrates the phosphatidylinositol 3-kinase signal to Akt and MAPK for bone morphogenetic protein-2 expression in osteoblast differentiation. J. Biol. Chem. 2007, 282, 4983–4993. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bertl, K.; Sun, H.; Liu, Z.H.; Andrukhov, O.; Rausch-Fan, X. Effect of simvastatin on the osteogenetic behavior of alveolar osteoblasts and periodontal ligament cells. Hum. Cell. 2012, 25, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Matsunuma, A.; Kurahashi, I.; Yanagawa, T.; Yoshida, H.; Horiuchi, N. Induction of osteoblast differentiation indices by statins in MC3T3-E1 cells. J. Cell. Biochem. 2004, 92, 458–471. [Google Scholar] [CrossRef]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yanae, M.; Kato, C.; Takagoshi, R.; Komai, M.; Nishida, S. Bisphosphonate- and statin-induced enhancement of OPG expression and inhibition of CD9, M-CSF, and RANKL expressions via inhibition of the Ras/MEK/ERK pathway and activation of p38MAPK in mouse bone marrow stromal cell line ST2. Mol. Cell. Endocrinol. 2012, 361, 219–231. [Google Scholar] [CrossRef]
- Ruan, F.; Zheng, Q.; Wang, J. Mechanisms of bone anabolism regulated by statins. Biosci. Rep. 2012, 32, 511–519. [Google Scholar] [CrossRef]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef]
- Lappalainen, S.; Saarinen, A.; Utriainen, P.; Voutilainen, R.; Jaaskelainen, J.; Makitie, O. LRP5 in premature adrenarche and in metabolic characteristics of prepubertal children. Clin. Endocrinol. 2009, 70, 725–731. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kobayashi, E.; Uetani, M.; Miura, K.; Morikawa, Y.; Ishizaki, M.; Kido, T.; Nakagawa, H.; Nogawa, K. G-protein beta 3 subunit polymorphism C1429T and low-density lipoprotein receptor-related protein 5 polymorphism A1330V are risk factors for hypercholesterolemia in Japanese males--a prospective study over 5 years. Metabolism 2006, 55, 751–757. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kobayashi, E.; Uetani, M.; Miura, K.; Morikawa, Y.; Ishizaki, M.; Kido, T.; Nakagawa, H.; Nogawa, K. Low-density lipoprotein receptor-related protein 5 variant Q89R is associated with hypertension in Japanese females. Blood Press. 2006, 15, 80–87. [Google Scholar] [CrossRef]
- Guo, Y.F.; Xiong, D.H.; Shen, H.; Zhao, L.J.; Xiao, P.; Guo, Y.; Wang, W.; Yang, T.L.; Recker, R.R.; Deng, H.W. Polymorphisms of the low-density lipoprotein receptor-related protein 5 (LRP5) gene are associated with obesity phenotypes in a large family-based association study. J. Med. Genet. 2006, 43, 798–803. [Google Scholar] [CrossRef]
- Fujino, T.; Asaba, H.; Kang, M.J.; Ikeda, Y.; Sone, H.; Takada, S.; Kim, D.H.; Ioka, R.X.; Ono, M.; Tomoyori, H.; et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 229–234. [Google Scholar] [CrossRef]
- Little, R.D.; Carulli, J.P.; Del Mastro, R.G.; Dupuis, J.; Osborne, M.; Folz, C.; Manning, S.P.; Swain, P.M.; Zhao, S.C.; Eustace, B.; et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002, 70, 11–19. [Google Scholar] [CrossRef]
- Wang, B.; Wang, H.; Li, Y.; Song, L. Lipid metabolism within the bone micro-environment is closely associated with bone metabolism in physiological and pathophysiological stages. Lipids Health Dis. 2022, 21, 5. [Google Scholar] [CrossRef]
- Shen, G.; Ren, H.; Shang, Q.; Zhao, W.; Zhang, Z.; Yu, X.; Tang, K.; Tang, J.; Yang, Z.; Liang, D.; et al. Foxf1 knockdown promotes BMSC osteogenesis in part by activating the Wnt/beta-catenin signalling pathway and prevents ovariectomy-induced bone loss. EBioMedicine 2020, 52, 102626. [Google Scholar] [CrossRef]
- Foldi, J.; Chung, A.Y.; Xu, H.; Zhu, J.; Outtz, H.H.; Kitajewski, J.; Li, Y.; Hu, X.; Ivashkiv, L.B. Autoamplification of Notch signaling in macrophages by TLR-induced and RBP-J-dependent induction of Jagged1. J. Immunol. 2010, 185, 5023–5031. [Google Scholar] [CrossRef]
- Bjornson, C.R.; Cheung, T.H.; Liu, L.; Tripathi, P.V.; Steeper, K.M.; Rando, T.A. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 2012, 30, 232–242. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, D.; Guo, D.; Li, J.; Xu, S.; Wei, J.; Xie, J.; Zhou, X. Osteoblasts impair cholesterol synthesis in chondrocytes via Notch1 signalling. Cell. Prolif. 2021, 54, e13156. [Google Scholar] [CrossRef]
- Colombo, M.; Platonova, N.; Giannandrea, D.; Palano, M.T.; Basile, A.; Chiaramonte, R. Re-establishing Apoptosis Competence in Bone Associated Cancers via Communicative Reprogramming Induced Through Notch Signaling Inhibition. Front. Pharmacol. 2019, 10, 145. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Xiu, C.; Zhou, Q.; Ni, L.; Du, J.; Gong, T.; Li, M.; Saijilafu; Yang, H.; Chen, J. A dual role of cholesterol in osteogenic differentiation of bone marrow stromal cells. J. Cell. Physiol. 2019, 234, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Golub, E.E. Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta 2009, 1790, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.L. The role of phosphatases in the initiation of skeletal mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef]
- Favarin, B.Z.; Andrade, M.A.R.; Bolean, M.; Simao, A.M.S.; Ramos, A.P.; Hoylaerts, M.F.; Millan, J.L.; Ciancaglini, P. Effect of the presence of cholesterol in the interfacial microenvironment on the modulation of the alkaline phosphatase activity during in vitro mineralization. Colloids Surf. B Biointerfaces 2017, 155, 466–476. [Google Scholar] [CrossRef]
- Vimalraj, S. Alkaline phosphatase: Structure, expression and its function in bone mineralization. Gene 2020, 754, 144855. [Google Scholar] [CrossRef]
- Luu, W.; Sharpe, L.J.; Gelissen, I.C.; Brown, A.J. The role of signalling in cellular cholesterol homeostasis. IUBMB Life 2013, 65, 675–684. [Google Scholar] [CrossRef]
- Nelson, D.L.; Hoskins, A.A.; Cox, M.M.; Lehninger, A.L. Lehninger Principles of Biochemistry, 8th ed.; Macmillan Learning: Austin, TX, USA, 2021. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Research Object | Treatment/Diet | Cholesterol/Cholesterol Derivatives Concentration | Effect | Reference |
|---|---|---|---|---|
| M2-10B4 (mouse stromal cell line) | 22(S)-hydroxycholesterol 20(S)-hydroxycholesterol (SS) | 5 mM | ↑ALP activity ↑mineralization ↑OCN mRNA | [43] |
| MSCs (bone marrow- derived mesenchymal stem cells) | Chol:MbCD (‘‘water-soluble cholesterol’’ containing 30 mg of cholesterol/g solid) | 5, 10 and 15 μg/mL | ↑differentiation ↑ALP activity ↑mineralized nodules | [44] |
| MC3T3-E1 (mouse cell line of immature osteoblasts) | Cholesterol | 0, 12.5, 25, and 50 μg/mL | ↓proliferation ↓differentiation ↑oxidative injury | [45] |
| M2–10B4 | Oxidized low-density lipoprotein (MM-LDL) | 150 µg/mL | ↓osteogenic differentiation ↑adipogenic differentiation | [46] |
| M2-10B4, Primary mouse bone marrow stromal cells | Xanthine/xanthine oxidase (XXO) minimally oxidized LDL (MM-LDL) Osteogenic oxysterol combination 22(S)- and 20(S)-hydroxycholesterol | 50 mM/40 mU/mL 200 mg/mL 0.1–5 µM | ↓markers of osteogenic differentiation blocked and reversed the inhibition of osteogenic differentiation | [49] |
| UMR106 (rat osteoblast-like cell line) | Oxidized LDL (oxLDL) | 10–50 μg protein/mL | ↓mineralization | [51] |
| MG63 (human osteosarcoma cell line) | Oxidized LDL (oxLDL) | 10–50 μg/mL | ↑cell-associated and extracellular RANKL levels | [50] |
| HOBs (primary human osteoblast cells) | Oxidized HDL (oxHDL) oxHDL with adiponectin | 100 μg/mL protein 100 μg/mL; 5, 10, and 15 μg/mL | ↓mineralization, ↓calcium incorporation. ↑expression of mineralization markers ↓inflammatory markers | [52] |
| Rat | Poly (lactic-co-glycolic acid) (PLGA) scaffolds alone or oxysterol cocktail | 140 ng (low dose) 1400 ng (high dose) | slight bone healing ↑bone formation | [43] |
| Rat | High-cholesterol diet | 77% normal diet food, 3% cholesterol and 20% lard | ↓femur BMD ↓osteocalcin ↑carboxy-terminal collagen crosslinks | [45] |
| C57BL/6 and C.B-17/Icr-SCID/Sed-Prkdcscid male mice | High-fat/high-cholesterol (HFHC) diet | 1.25% cholesterol | ↓cortical and trabecular bone in the femurs and vertebrae ↓bone mineral density (BMD) | [53] |
| OF1 female mice | Westerntype diet | 1.1 mg cholesterol/g diet | ↓BMD | [54] |
| C57BL/6 and C3H/HeJ male mice | High-fat (atherogenic) diet | 1.25% cholesterol | ↓femoral and vertebral mineral content ↓BMD | [55] |
| C57BL6/J and Swiss Albino mice | High-cholesterol (HC) diet | 0.5% cholesterol | ↓osteoblast cell activity ↑osteoclast cell population delayed skeletal ossification | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhmetshina, A.; Kratky, D.; Rendina-Ruedy, E. Influence of Cholesterol on the Regulation of Osteoblast Function. Metabolites 2023, 13, 578. https://doi.org/10.3390/metabo13040578
Akhmetshina A, Kratky D, Rendina-Ruedy E. Influence of Cholesterol on the Regulation of Osteoblast Function. Metabolites. 2023; 13(4):578. https://doi.org/10.3390/metabo13040578
Chicago/Turabian StyleAkhmetshina, Alena, Dagmar Kratky, and Elizabeth Rendina-Ruedy. 2023. "Influence of Cholesterol on the Regulation of Osteoblast Function" Metabolites 13, no. 4: 578. https://doi.org/10.3390/metabo13040578