
Quality Control of Targeted Plasma Lipids in a Large-Scale Cohort Study Using Liquid Chromatography–Tandem Mass Spectrometry
 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. Extraction of Target Lipids
2.3. Targeted Lipid Analysis
2.4. Method Validation
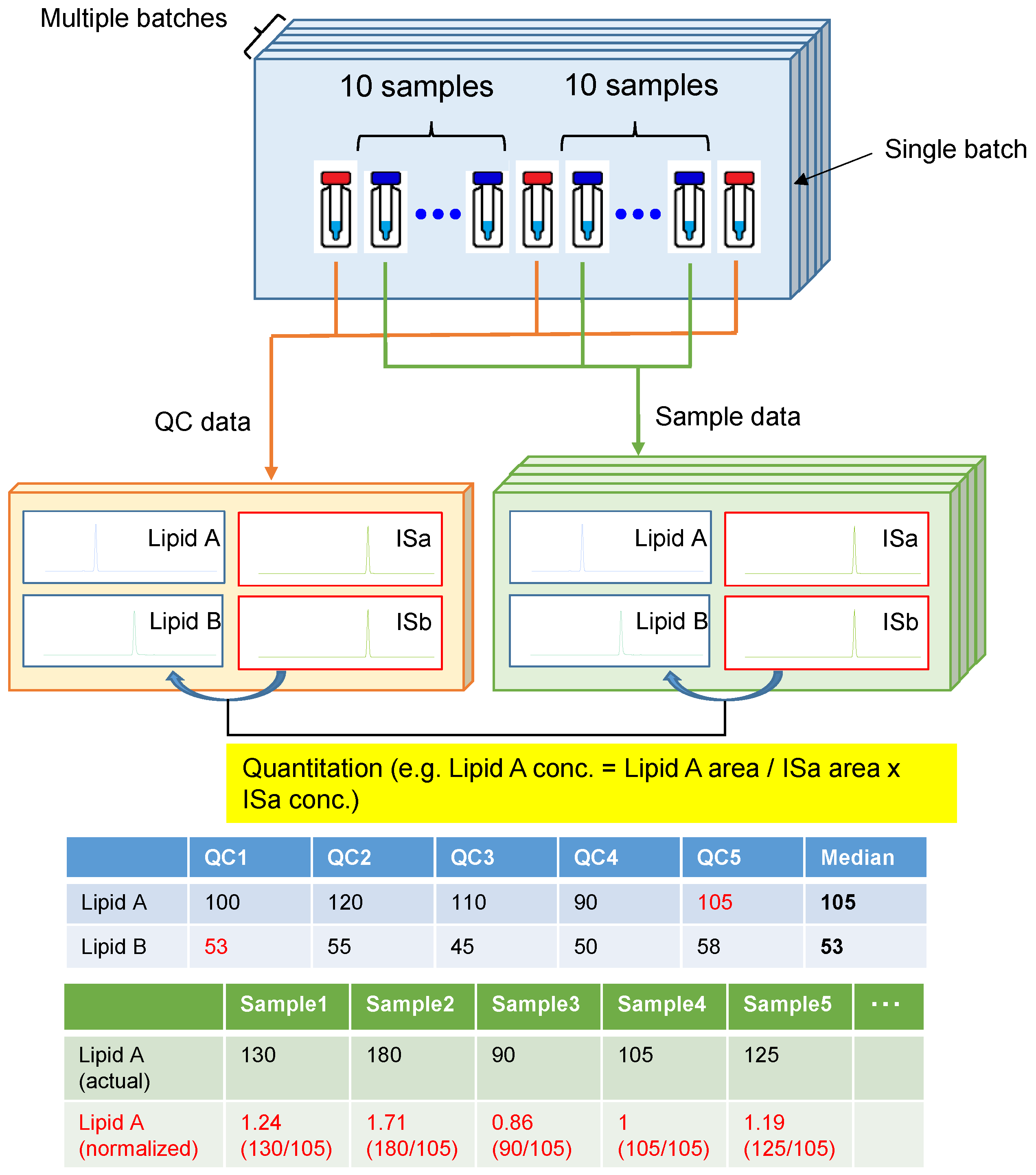
2.5. Quantitative and Normalization Method of Metabolomic Profile
2.6. Statistical Analysis
3. Results and Discussion
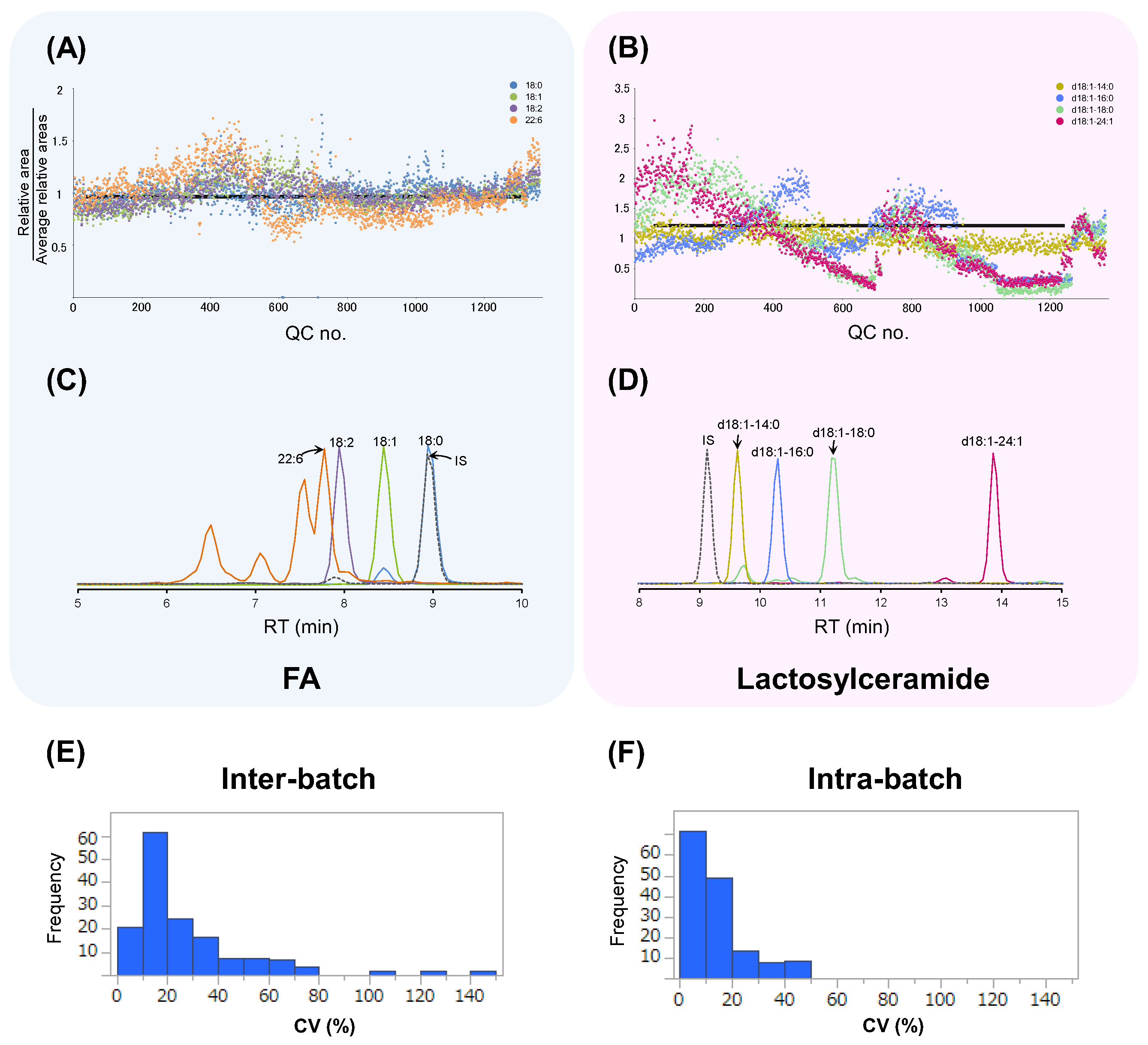
3.1. Method Validation
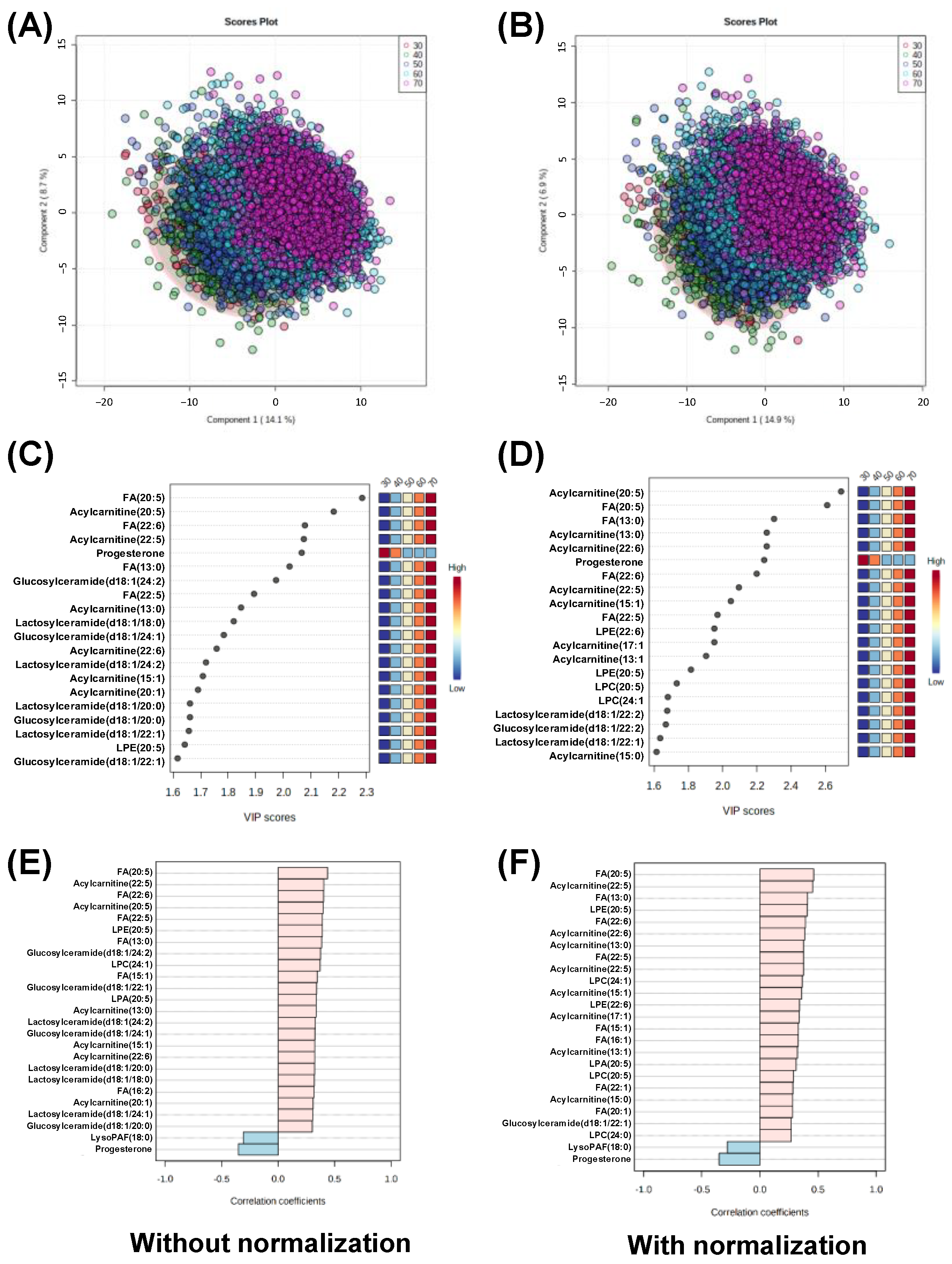
3.2. Comparison of Analytical Results with and without Normalization
3.3. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Floegel, A.; Stefan, N.; Yu, Z.; Mühlenbruch, K.; Drogan, D.; Joost, H.G.; Fritsche, A.; Häring, H.U.; Hrabě de Angelis, M.; Peters, A.; et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 2013, 62, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Akbaraly, T.; Würtz, P.; Singh-Manoux, A.; Shipley, M.J.; Haapakoski, R.; Lehto, M.; Desrumaux, C.; Kähönen, M.; Lehtimäki, T.; Mikkilä, V.; et al. Association of circulating metabolites with healthy diet and risk of cardiovascular disease: Analysis of two cohort studies. Sci. Rep. 2018, 8, 8620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Würtz, P.; Havulinna, A.S.; Soininen, P.; Tynkkynen, T.; Prieto-Merino, D.; Tillin, T.; Ghorbani, A.; Artati, A.; Wang, Q.; Tiainen, M.; et al. Metabolite profiling and cardiovascular event risk: A prospective study of 3 population-based cohorts. Circulation 2015, 131, 774–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekula, P.; Goek, O.N.; Quaye, L.; Barrios, C.; Levey, A.S.; Römisch-Margl, W.; Menni, C.; Yet, I.; Gieger, C.; Inker, L.A.; et al. A metabolome-wide association study of kidney function and disease in the general population. J. Am. Soc. Nephrol. 2016, 27, 1175–1188. [Google Scholar] [CrossRef] [Green Version]
- Toledo, J.B.; Arnold, M.; Kastenmüller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimer’s Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.C.; Matthews, C.E.; Sampson, J.N.; Stolzenberg-Solomon, R.Z.; Zheng, W.; Cai, Q.; Tan, Y.T.; Chow, W.H.; Ji, B.T.; Liu, D.K.; et al. Human metabolic correlates of body mass index. Metabolomics 2014, 10, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Menni, C.; Graham, D.; Kastenmüller, G.; Alharbi, N.H.; Alsanosi, S.M.; McBride, M.; Mangino, M.; Titcombe, P.; Shin, S.Y.; Psatha, M.; et al. Metabolomic identification of a novel pathway of blood pressure regulation involving hexadecanedioate. Hypertension 2015, 66, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Zanetti, K.A.; Temprosa, M.; Albanes, D.; Appel, N.; Barrera, C.B.; Ben-Shlomo, Y.; Boerwinkle, E.; Casas, J.P.; Clish, C.; et al. The consortium of metabolomics studies (COMETS): Metabolomics in 47 prospective cohort studies. Am. J. Epidemiol. 2019, 188, 991–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, S.; Hirayama, A.; Chan, Q.; Kurihara, A.; Fukai, K.; Iida, M.; Kato, S.; Sugiyama, D.; Kuwabara, K.; Takeuchi, A.; et al. Reliability of plasma polar metabolite concentrations in a large-scale cohort study using capillary electrophoresis-mass spectrometry. PLoS ONE 2018, 13, e0191230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, K.; Harada, S.; Takeuchi, A.; Kurihara, A.; Iida, M.; Fukai, K.; Kuwabara, K.; Kato, S.; Matsumoto, M.; Hirata, A.; et al. Association between dyslipidemia and plasma levels of branched-chain amino acids in the Japanese population without diabetes mellitus. J. Clin. Lipidol. 2019, 13, 932–939.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, S.; Takebayashi, T.; Kurihara, A.; Akiyama, M.; Suzuki, A.; Hatakeyama, Y.; Sugiyama, D.; Kuwabara, K.; Takeuchi, A.; Okamura, T.; et al. Metabolomic profiling reveals novel biomarkers of alcohol intake and alcohol-induced liver injury in community-dwelling men. Environ. Health Prev. Med. 2016, 21, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, M.; Harada, S.; Kurihara, A.; Fukai, K.; Kuwabara, K.; Sugiyama, D.; Takeuchi, A.; Okamura, T.; Akiyama, M.; Nishiwaki, Y.; et al. Profiling of plasma metabolites in postmenopausal women with metabolic syndrome. Menopause 2016, 23, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, K.; Harada, S.; Iida, M.; Kurihara, A.; Takeuchi, A.; Kuwabara, K.; Sugiyama, D.; Okamura, T.; Akiyama, M.; Nishiwaki, Y.; et al. Metabolic profiling of total physical activity and sedentary behavior in community-dwelling men. PLoS ONE 2016, 11, e0164877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibutami, E.; Ishii, R.; Harada, S.; Kurihara, A.; Kuwabara, K.; Kato, S.; Iida, M.; Akiyama, M.; Sugiyama, D.; Hirayama, A.; et al. Charged metabolite biomarkers of food intake assessed via plasma metabolomics in a population-based observational study in Japan. PLoS ONE 2021, 16, e0246456. [Google Scholar] [CrossRef]
- Luo, P.; Yin, P.; Zhang, W.; Zhou, L.; Lu, X.; Lin, X.; Xu, G. Optimization of large-scale pseudotargeted metabolomics method based on liquid chromatography-mass spectrometry. J. Chromatogr. A 2016, 1437, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Brunius, C.; Shi, L.; Landberg, R. Large-scale untargeted LC-MS metabolomics data correction using between-batch feature alignment and cluster-based within-batch signal intensity drift correction. Metabolomics 2016, 12, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Harada, S.; Kawasaki, Y.; Watanabe, M.; Ito, H.; Tanaka, H.; Takeuchi, A.; Tsubota, K.; Takebayashi, T.; Nishiwaki, Y.; et al. Gender-specific association of early age-related macular degeneration with systemic and genetic factors in a Japanese population. Sci. Rep. 2018, 8, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, K.T.; Uehara, T.; Aoshima, K.; Oda, Y. Polar anionic metabolome analysis by nano-LC/MS with a metal chelating agent. Anal. Chem. 2009, 81, 7766–7772. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
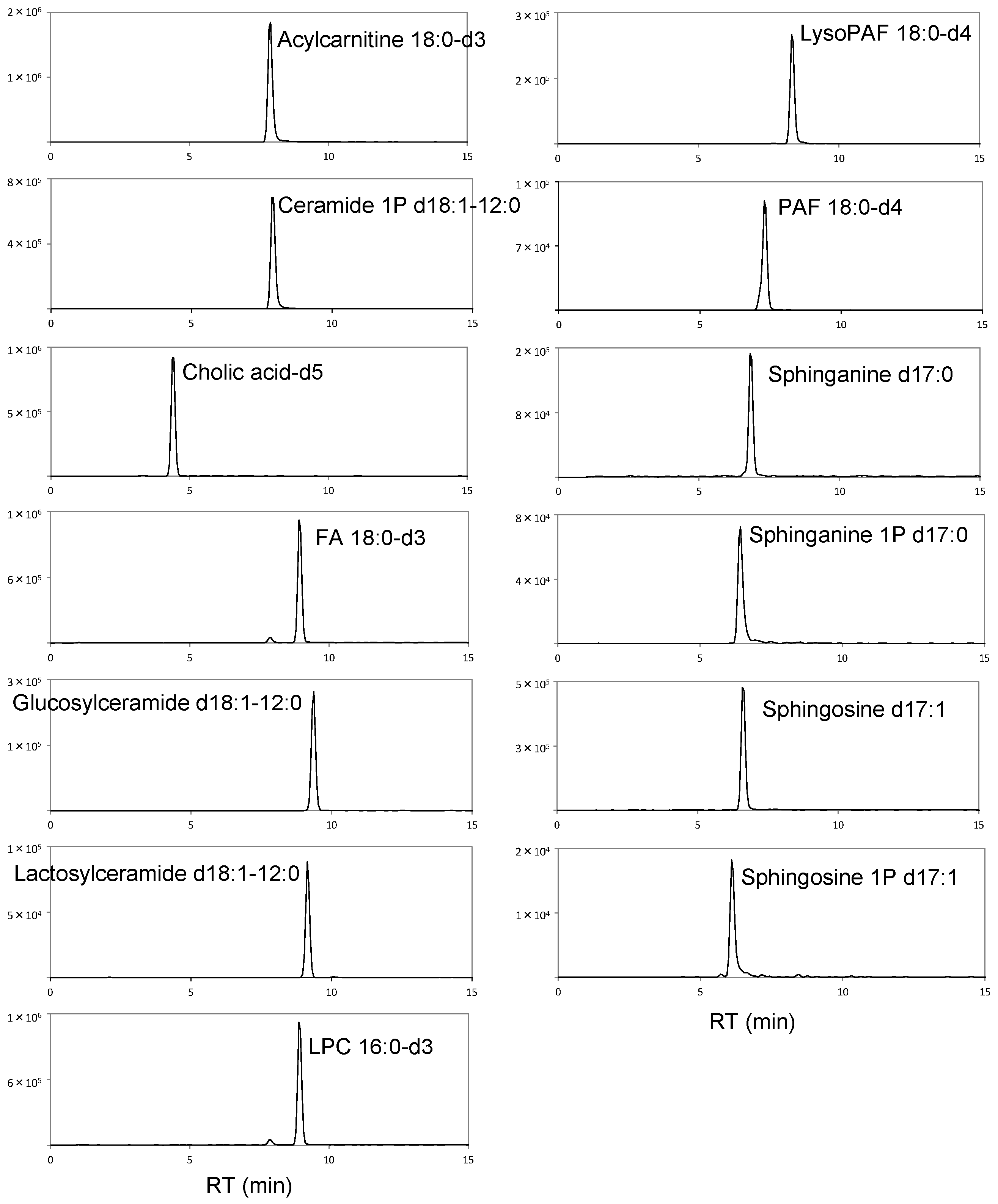
| Compound | Calibration Curve | R2 Value | Limit of Detection (nmol/L) | Linear Dynamic Range (μmol/L) |
|---|---|---|---|---|
| Acylcarnitine 18:0-d3 | y = (3.81 × 107)x − 1.81 × 103 | 0.994 | 0.41 | 0.0005–2 |
| Ceramide 1P d18:1-12:0 | y = (6.51 × 106)x − 1.68 × 102 | 0.988 | 1.13 | 0.001–5 |
| Cholic acid-d5 | y = (3.54 × 106)x + 1.86 × 103 | 0.994 | 3.48 | 0.005–10 |
| FA 18:0-d3 | y = (1.94 × 106)x + 3.36 × 104 | 0.979 | 23.05 | 0.02–20 |
| Glucosylceramide d18:1-12:0 | y = (1.09 × 107)x − 3.16 × 103 | 0.991 | 2.28 | 0.0025–5 |
| Lactosylceramide d18:1-12:0 | y = (3.87 × 106)x − 4.48 × 103 | 0.991 | 5.09 | 0.01–1 |
| LPC 16:0-d3 | y = (7.51 × 106)x − 1.40 × 103 | 0.991 | 2.03 | 0.0025–5 |
| PAF 18:0-d4 | y = (2.18 × 107)x − 7.87 × 102 | 0.990 | 0.45 | 0.0005–5 |
| LysoPAF 18:0-d4 | y = (1.21 × 107)x − 3.45 × 103 | 0.991 | 1.87 | 0.0025–5 |
| Sphinganine d17:0 | y = (6.41 × 106)x + 7.79 × 103 | 0.985 | 4.90 | 0.005–5 |
| Sphinganine 1P d17:0 | y = (3.93 × 106)x − 1.30 × 103 | 0.983 | 2.21 | 0.0025–5 |
| Sphingosine d17:1 | y = (2.12 × 107)x + 6.83 × 103 | 0.991 | 1.12 | 0.0025–2.5 |
| Sphingosine 1P d17:1 | y = (9.83 × 105)x − 5.92 × 103 | 0.983 | 29.17 | 0.025–5 |
| Compound | Low (n = 5, %) | Middle (n = 5, %) | High (n = 5, %) | Extraction Recovery (n = 3, %) | |||
|---|---|---|---|---|---|---|---|
| Accuracy | Precision | Accuracy | Precision | Accuracy | Precision | ||
| Acylcarnitine 18:0-d3 | 101.2 | 22.1 | 95.6 | 9.3 | 99.5 | 1.4 | 100.2 ± 10.0 |
| Ceramide 1P d18:1-12:0 | 96.6 | 30.3 | 92.8 | 3.9 | 102.8 | 3.3 | 103.2 ± 8.1 |
| Cholic acid-d5 | 98.3 | 18.6 | 105.0 | 4.4 | 102.2 | 5.6 | 111.1 ± 10.3 |
| FA 18:0-d3 | 101.0 | 30.8 | 96.3 | 4.8 | 93.8 | 2.3 | 104.0 ± 7.4 |
| Glucosylceramide d18:1-12:0 | 104.3 | 24.4 | 90.3 | 5.4 | 109.6 | 4.5 | 104.5 ± 12.1 |
| Lactosylceramide d18:1-12:0 | 106.1 | 13.6 | 92.8 | 5.2 | 110.5 | 5.5 | 110.4 ± 8.0 |
| LPC 16:0-d3 | 99.8 | 21.7 | 106.3 | 3.9 | 86.7 | 1.7 | 63.0 ± 3.8 |
| PAF 18:0-d4 | 97.6 | 23.9 | 104.5 | 5.6 | 80.0 | 2.5 | 105.2 ± 8.8 |
| LysoPAF 18:0-d4 | 97.4 | 19.9 | 100.1 | 10.7 | 85.5 | 3.9 | 106.9 ± 3.0 |
| Sphinganine d17:0 | 99.3 | 26.2 | 103.3 | 7.0 | 87.1 | 4.8 | 102.9 ± 10.2 |
| Sphinganine 1P d17:0 | 112.9 | 23.6 | 94.0 | 2.9 | 118.0 | 3.1 | 103.8 ± 5.4 |
| Sphingosine d17:1 | 93.9 | 12.0 | 105.2 | 9.1 | 86.3 | 2.1 | 111.5 ± 5.9 |
| Sphingosine 1P d17:1 | 109.5 | 31.1 | 92.7 | 5.1 | 112.1 | 2.3 | 102.6 ± 12.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirayama, A.; Ishikawa, T.; Takahashi, H.; Yamanaka, S.; Ikeda, S.; Hirata, A.; Harada, S.; Sugimoto, M.; Soga, T.; Tomita, M.; et al. Quality Control of Targeted Plasma Lipids in a Large-Scale Cohort Study Using Liquid Chromatography–Tandem Mass Spectrometry. Metabolites 2023, 13, 558. https://doi.org/10.3390/metabo13040558
Hirayama A, Ishikawa T, Takahashi H, Yamanaka S, Ikeda S, Hirata A, Harada S, Sugimoto M, Soga T, Tomita M, et al. Quality Control of Targeted Plasma Lipids in a Large-Scale Cohort Study Using Liquid Chromatography–Tandem Mass Spectrometry. Metabolites. 2023; 13(4):558. https://doi.org/10.3390/metabo13040558
Chicago/Turabian StyleHirayama, Akiyoshi, Takamasa Ishikawa, Haruka Takahashi, Sanae Yamanaka, Satsuki Ikeda, Aya Hirata, Sei Harada, Masahiro Sugimoto, Tomoyoshi Soga, Masaru Tomita, and et al. 2023. "Quality Control of Targeted Plasma Lipids in a Large-Scale Cohort Study Using Liquid Chromatography–Tandem Mass Spectrometry" Metabolites 13, no. 4: 558. https://doi.org/10.3390/metabo13040558