

Kynurenine Pathway Regulation at Its Critical Junctions with Fluctuation of Tryptophan


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
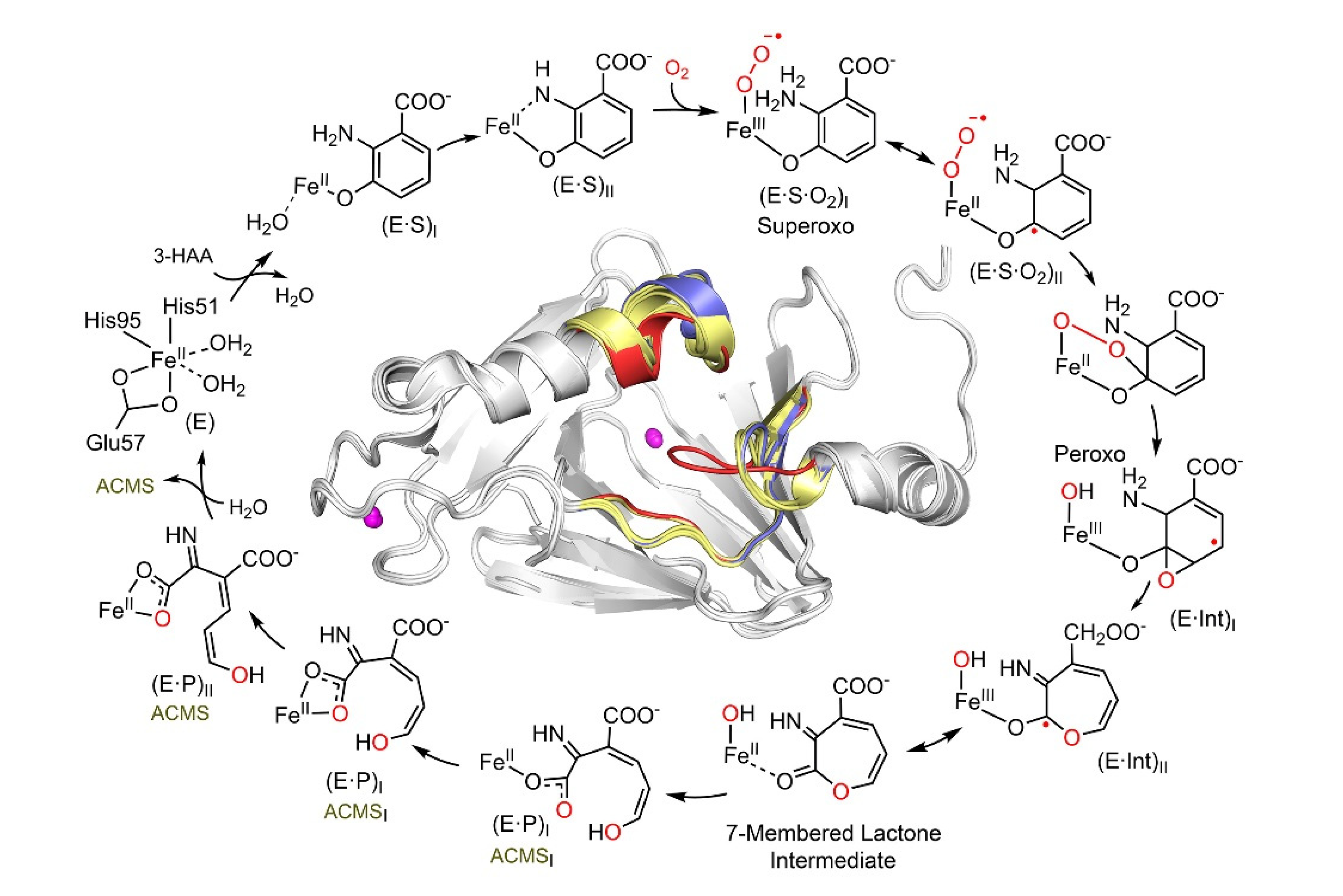
2. HAO: The Phenyl Ring-Breaking Oxygenase in the KP Trio
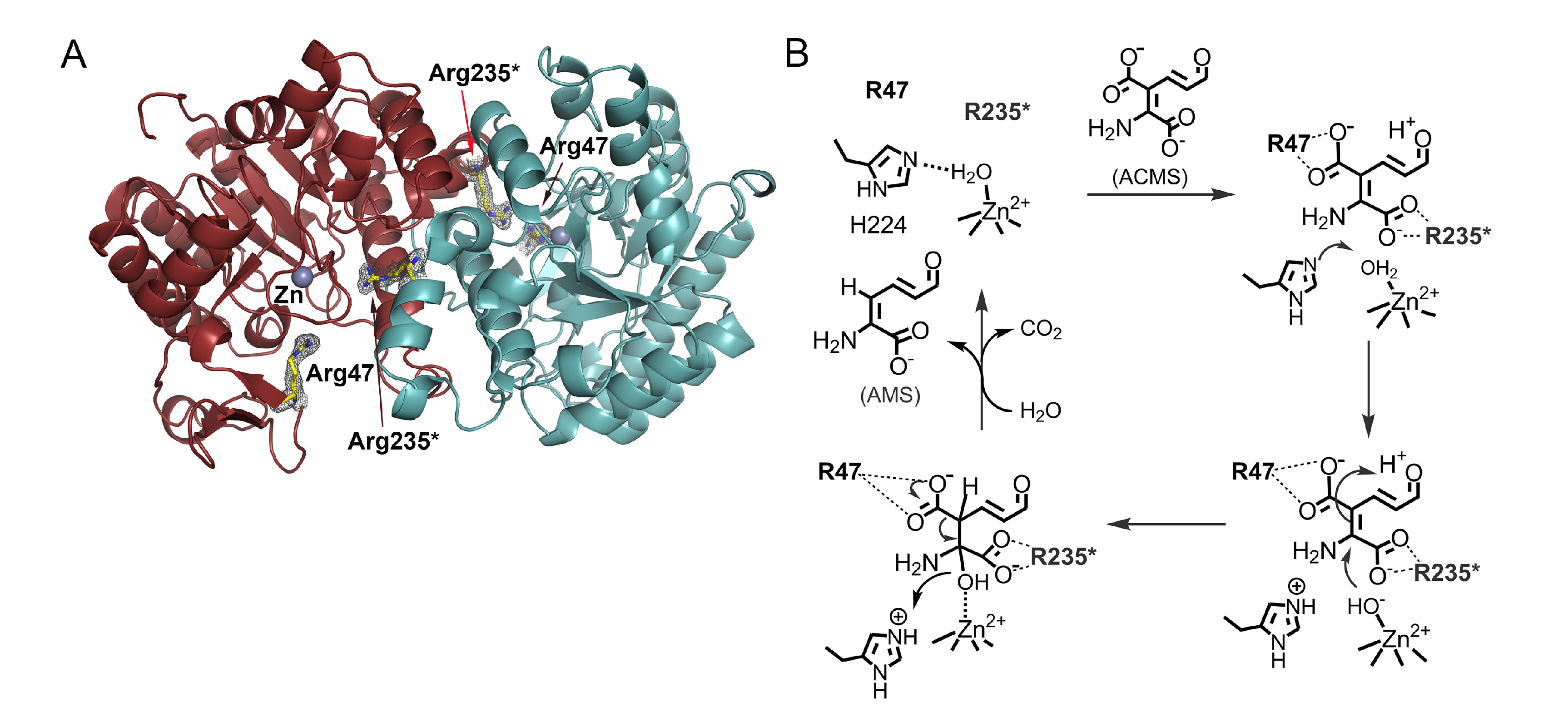
3. ACMSD: A Novel Metal-Dependent Non-Oxidative Decarboxylase
4. AMSDH: An NAD+-Dependent Dehydrogenase in the KP Trio
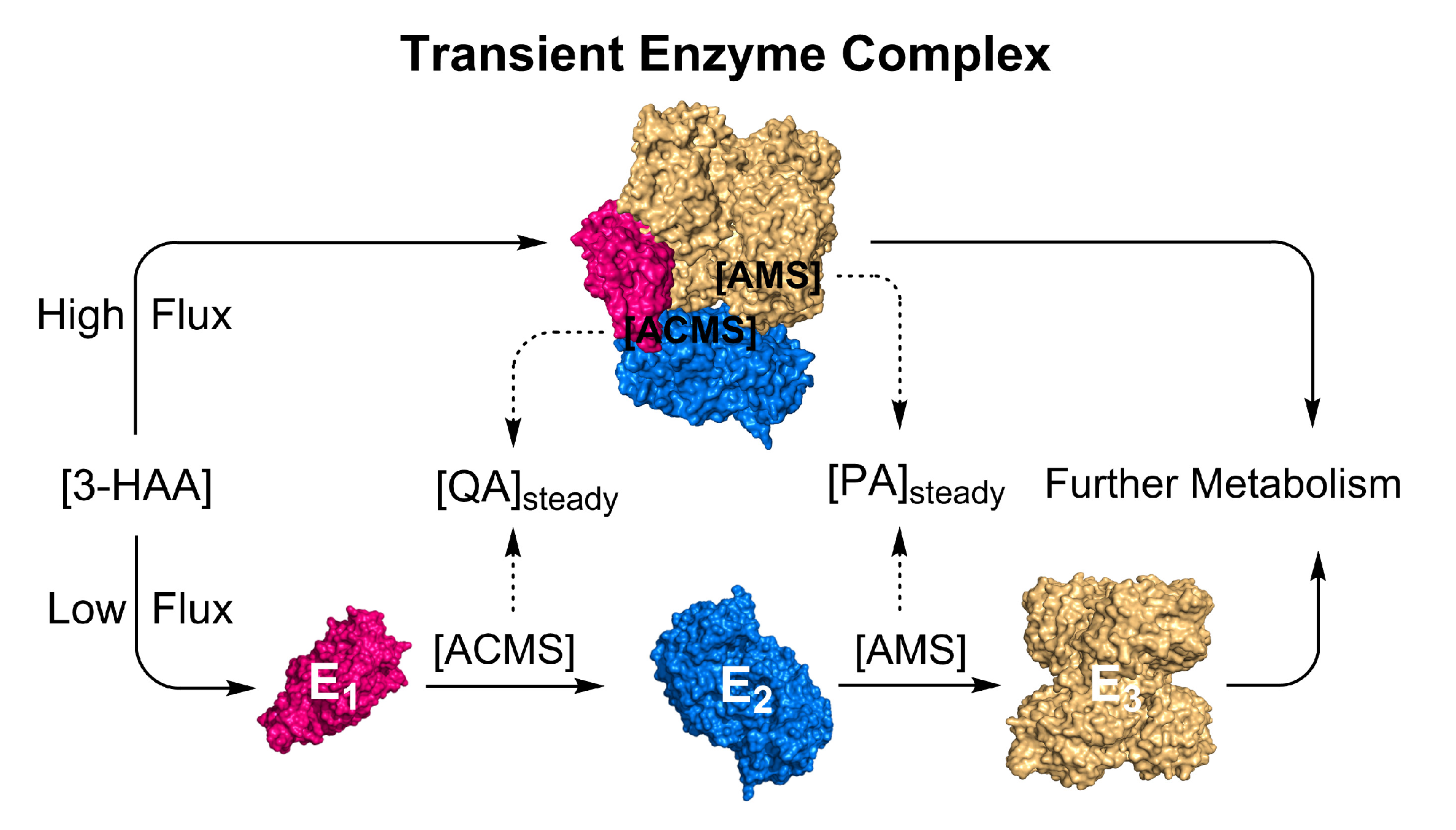
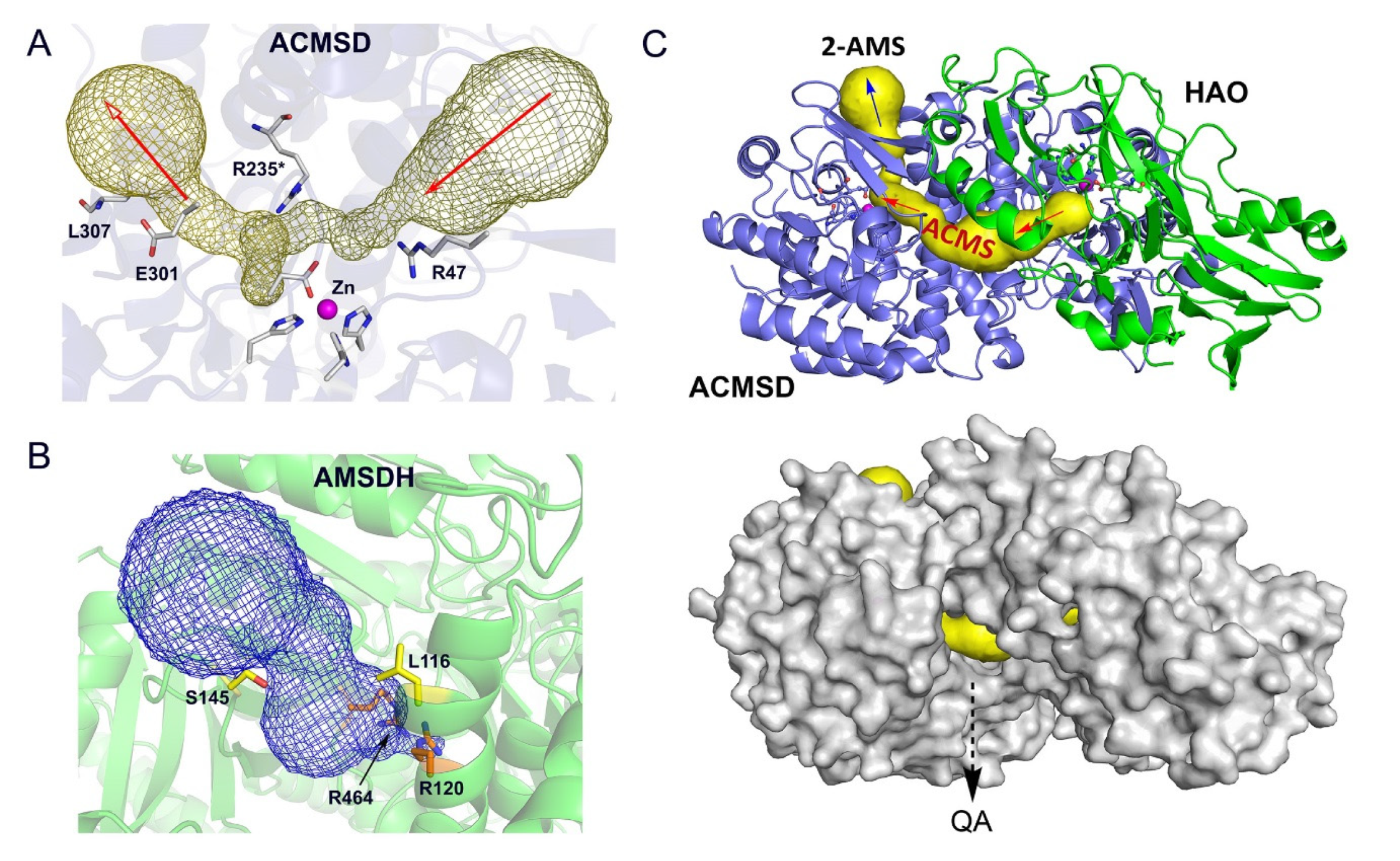
5. Transient Enzyme Complex as a Possible Pathway Regulatory Mechanism
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Lapin, I.P.; Oxenkrug, G.F. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet 1969, 293, 132–136. [Google Scholar] [CrossRef]
- Haroon, E.; Welle, J.R.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.; Patel, T.; Felger, J.C.; Miller, A.H. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology 2020, 45, 998–1007. [Google Scholar] [CrossRef]
- Chen, X.; Beltran, D.J.; Tsygankova, V.D.; Woolwine, B.J.; Patel, T.; Baer, W.; Felger, J.C.; Miller, A.H.; Haroon, E. Kynurenines increase MRS metabolites in basal ganglia and decrease resting-state connectivity in frontostriatal reward circuitry in depression. Transl. Psychiatry 2021, 11, 456. [Google Scholar] [CrossRef]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Miyazaki, T.; Chung, S.; Sakai, H.; Ohata, H.; Obata, Y.; Shiokawa, D.; Mizoguchi, Y.; Kubo, T.; Ichikawa, H.; Taniguchi, H.; et al. Stemness and immune evasion conferred by the TDO2-AHR pathway are associated with liver metastasis of colon cancer. Cancer Sci. 2022, 113, 170–181. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolusic, E.; Frederick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coplan, J.D.; George, R.; Syed, S.A.; Rozenboym, A.V.; Tang, J.E.; Fulton, S.L.; Perera, T.D. Early life stress and the fate of kynurenine pathway metabolites. Front. Hum. Neurosci. 2021, 15, 636144. [Google Scholar] [CrossRef]
- Davis, I.; Liu, A. What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother. 2015, 15, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Davis, I.; Yang, Y.; Wherritt, D.; Liu, A. Reassignment of the human aldehyde dehydrogenase ALDH8A1 (ALDH12) to the kynurenine pathway in tryptophan catabolism. J. Biol. Chem. 2018, 293, 9594–9603. [Google Scholar] [CrossRef] [Green Version]
- Tauber, E.; Miller-Fleming, L.; Mason, R.P.; Kwan, W.; Clapp, J.; Butler, N.J.; Outeiro, T.F.; Muchowski, P.J.; Giorgini, F. Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J. Biol. Chem. 2011, 286, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Allegri, G.; Ragazzi, E.; Bertazzo, A.; Costa, C.V.L. Enzyme activities along the kynurenine pathway in mice. Adv. Exp. Med. Biol. 2003, 527, 497–510. [Google Scholar]
- Allegri, G.; Ragazzi, E.; Bertazzo, A.; Costa, C.V.L.; Rocchi, R. Tryptophan metabolism along the kynurenine pathway in rats. Adv. Exp. Med. Biol. 2003, 527, 481–496. [Google Scholar] [PubMed]
- Allegri, G.; Ragazzi, E.; Costa, C.V.L.; Caparrotta, L.; Biasiolo, M.; Comai, S.; Bertazzo, A. Tryptophan metabolism along the kynurenine pathway in diet-induced and genetic hypercholesterolemic rabbits. Clin. Chim. Acta 2004, 350, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Allegri, G.; Ragazzi, E.; Costa, C.V.L.; Caparrotta, L.; Biasiolo, M.; Vanin, S. The kynurenine pathway enzymes in healthy and hyperlipidemic rabbits. Adv. Exp. Med. Biol. 2003, 527, 381–386. [Google Scholar]
- Comai, S.; Costa, C.V.L.; Ragazzi, E.; Bertazzo, A.; Allegri, G. The effect of age on the enzyme activities of tryptophan metabolism along the kynurenine pathway in rats. Clin. Chim. Acta 2005, 360, 67–80. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes. Adv. Exp. Med. Biol. 1999, 467, 125–131. [Google Scholar] [PubMed]
- Stone, T.W.; Mackay, G.M.; Forrest, C.M.; Clark, C.J.; Darlington, L.G. Tryptophan metabolites and brain disorders. Clin. Chem. Lab. Med. 2003, 41, 852–859. [Google Scholar] [CrossRef]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171. [Google Scholar] [CrossRef]
- Schwarcz, R.; Whetsell, W.O., Jr.; Mangano, R.M. Quinolinic acid: An endogenous metabolite that produces axon-sparing lesions in rat brain. Science 1983, 219, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.P.; Rubinow, D.; Lane, C.; Markey, S.P. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Ann. Neurol. 1989, 26, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Heyes, M.P. Kynurenine pathway enzymes in brain. Properties of enzymes and regulation of quinolinic acid synthesis. Adv. Exp. Med. Biol. 1996, 398, 485–492. [Google Scholar]
- Patterson, J.I.; Brown, R.R.; Linkswiler, H.; Harper, A.E. Excretion of tryptophan-niacin metabolites by young men: Effects of tryptophan, leucine, and vitamin B6 intakes. Am. J. Clin. Nutr. 1980, 33, 2157–2167. [Google Scholar] [CrossRef]
- Kurnasov, O.; Goral, V.; Colabroy, K.; Gerdes, S.; Anantha, S.; Osterman, A.; Begley, T.P. NAD biosynthesis: Identification of the tryptophan to quinolinate pathway in bacteria. Chem. Biol. 2003, 10, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Colabroy, K.L.; Begley, T.P. Tryptophan catabolism: Identification and characterization of a new degradative pathway. J. Bacteriol. 2005, 187, 7866–7869. [Google Scholar] [CrossRef] [Green Version]
- Muraki, T.; Taki, M.; Hasegawa, Y.; Iwaki, H.; Lau, P.C. Prokaryotic homologs of the eukaryotic 3-hydroxyanthranilate 3,4-dioxygenase and 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase in the 2-nitrobenzoate degradation pathway of Pseudomonas fluorescens strain KU-7. Appl. Environ. Microbiol. 2003, 69, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Ma, J.; Hosler, J.P.; Davidson, V.L.; Liu, A. Detection of transient intermediates in the metal-dependent non-oxidative decarboxylation catalyzed by α-amino-β-carboxymuconic-ε-semialdehyde decarboxylase. J. Am. Chem. Soc. 2007, 129, 9278–9279. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, K.F.; Yang, Y.; Davis, I.; Liu, A. Observing 3-hydroxyanthranilate-3,4-dioxygenase in action through a crystalline lens. Proc. Natl. Acad. Sci. USA 2020, 117, 19720–19730. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, S.; Ishiguro, K.; Yanagihara, K.; Tanabe, A.; Egashira, Y.; Sanada, H.; Shibata, K. Identification and expression of a cDNA encoding human α-Amino-β-carboxymuconic-ε-semialdehyde decarboxylase (ACMSD). A key enzyme for the tryptophan-niacine pathway and “quinolinate hypothesis”. J. Biol. Chem. 2002, 277, 35162–35167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Walker, A.L.; Iwaki, H.; Hasegawa, Y.; Liu, A. Kinetic and spectroscopic characterization of ACMSD from Pseudomonas fluorescens reveals a pentacoordinate mononuclear metallocofactor. J. Am. Chem. Soc. 2005, 127, 12282–12290. [Google Scholar] [CrossRef]
- Notarangelo, F.M.; Wu, H.Q.; Macherone, A.; Graham, D.R.; Schwarcz, R. Gas chromatography/tandem mass spectrometry detection of extracellular kynurenine and related metabolites in normal and lesioned rat brain. Anal. Biochem. 2012, 421, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Stone, T.W.; Stoy, N.; Darlington, L.G. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci. 2013, 34, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Okuno, E.; White, R.J.; Bird, E.D.; Whetsell, W.O., Jr. 3-Hydroxyanthranilate oxygenase activity is increased in the brains of Huntington disease victims. Proc. Natl. Acad. Sci. USA 1988, 85, 4079–4081. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, H.-C.; Sudek, S.; Caro, S.; Rosa, S.; Perovic, S.; Steffen, R.; Mueller, I.M.; Mueller, W.E.G. Synthesis of the neurotoxin quinolinic acid in apoptotic tissue from Suberites domuncula: Cell biological, molecular biological, and chemical analyses. J. Mar. Biotechnol. 2002, 4, 546–558. [Google Scholar] [CrossRef]
- Hayes, M.P.; Saito, K.; Lackner, A.; Wiley, C.A.; Achim, C.L.; Markey, S.P. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques. FASEB J. 1998, 12, 881–896. [Google Scholar]
- Nakano, K.; Takahashi, S.; Mizobuchi, M.; Kuroda, T.; Masuda, K.; Kitoh, J. High levels of quinolinic acid in brain of epilepsy-prone E1 mice. Brain Res. 1993, 619, 195–198. [Google Scholar] [CrossRef]
- Reinhard, J.F., Jr.; Flanagan, E.M. The neurotoxin quinolinic acid is increased in spinal cords of mice with herpes simplex virus encephalitis. Adv. Exp. Med. Biol. 1996, 398, 241–246. [Google Scholar]
- Blight, A.R.; Cohen, T.I.; Saito, K.; Heyes, M.P. Quinolinic acid accumulation and functional deficits following experimental spinal cord injury. Brain 1995, 118, 735–752. [Google Scholar] [CrossRef]
- Eastman, C.L.; Urbanska, E.; Love, A.; Kristensson, K.; Schwarcz, R. Increased brain quinolinic acid production in mice infected with a hamster neurotropic measles virus. Exp. Neurol. 1994, 125, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lahdou, I.; Sadeghi, M.; Oweira, H.; Fusch, G.; Daniel, V.; Mehrabi, A.; Jung, G.; Elhadedy, H.; Schmidt, J.; Sandra-Petrescu, F.; et al. Increased serum levels of quinolinic acid indicate enhanced severity of hepatic dysfunction in patients with liver cirrhosis. Hum. Immunol. 2013, 74, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.C.; Varani, A.M.; Menck, C.F.M. NAD biosynthesis evolution in bacteria: Lateral gene transfer of kynurenine pathway in Xanthomonadales and Flavobacteriales. Mol. Biol. Evol. 2009, 26, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Geng, J.; Gumpper, R.H.; Barman, A.; Davis, I.; Ozarowski, A.; Hamelberg, D.; Liu, A. An iron reservoir to the catalytic metal: The rubredoxin iron in an extradiol dioxygenase. J. Biol. Chem. 2015, 290, 15621–15634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, F.; Liu, A. Adapting to oxygen: 3-Hydroxyanthrinilate 3,4-dioxygenase employs loop dynamics to accommodate two substrates with disparate polarities. J. Biol. Chem. 2018, 293, 10415–10424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colabroy, K.L.; Zhai, H.; Li, T.; Ge, Y.; Zhang, Y.; Liu, A.; Ealick, S.E.; McLafferty, F.W.; Begley, T.P. The mechanism of inactivation of 3-hydroxyanthranilate-3,4-dioxygenase by 4-chloro-3-hydroxyanthranilate. Biochemistry 2005, 44, 7623–7631. [Google Scholar] [CrossRef]
- Walsh, J.L.; Todd, W.P.; Carpenter, B.K.; Schwarcz, R. 4-Halo-3-hydroxyanthranilates are potent inhibitors of 3-hydroxyanthranilate oxygenase in the rat brain in vitro and in vivo. Kynurenine Serotonin Pathway. 1991, 294, 579–582. [Google Scholar]
- Martynowski, D.; Eyobo, Y.; Li, T.; Yang, K.; Liu, A.; Zhang, H. Crystal structure of α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase: Insight into the active site and catalytic mechanism of a novel decarboxylation reaction. Biochemistry 2006, 45, 10412–10421. [Google Scholar] [CrossRef]
- Li, T.; Iwaki, H.; Fu, R.; Hasegawa, Y.; Zhang, H.; Liu, A. α-Amino-β-carboxymuconic-ε-semialdehyde decarboxylase (ACMSD) is a new member of the amidohydrolase superfamily. Biochemistry 2006, 45, 6628–6634. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, H. Transition metal-catalyzed non-oxidative decarboxylation reactions. Biochemistry 2006, 45, 10407–10411. [Google Scholar] [CrossRef]
- Li, T.; Huo, L.; Pulley, C.; Liu, A. Decarboxylation mechanisms in the biological system. Bioorg. Chem. 2012, 43, 2–14. [Google Scholar] [CrossRef]
- Pellicciari, R.; Liscio, P.; Giacchè, N.; De Franco, F.; Carotti, A.; Robertson, J.; Cialabrini, L.; Katsyuba, E.; Raffaelli, N.; Auwerx, J. α-Amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD) inhibitors as novel modulators of de novo nicotinamide adenine dinucleotide (NAD+) biosynthesis. J. Med. Chem. 2018, 61, 745–759. [Google Scholar] [CrossRef]
- Yang, Y.; Borel, T.; de Azambuja, F.; Johnson, D.; Sorrentino, J.P.; Udokwu, C.; Davis, I.; Liu, A.; Altman, R.A. Diflunisal derivatives as modulators of ACMS decarboxylase targeting the tryptophan–kynurenine pathway. J. Med. Chem. 2021, 64, 797–811. [Google Scholar] [CrossRef]
- Mehler, A.H.; Yano, K.; May, E.L. Nicotinic acid biosynthesis: Control by an enzyme that competes with a spontaneous reaction. Science 1964, 145, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Du, F. Quinolinic acid and kynurenic acid in the mammalian brain. Kynurenine Serotonin Pathway. 1991, 294, 185–199. [Google Scholar]
- Jhamandas, K.H.; Boegman, R.J.; Beninger, R.J.; Miranda, A.F.; Lipic, K.A. Excitotoxicity of quinolinic acid: Modulation by endogenous antagonists. Neurotox. Res. 2000, 2, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Huo, L.; Fielding, A.J.; Chen, Y.; Li, T.; Iwaki, H.; Hosler, J.P.; Chen, L.; Hasegawa, Y.; Que, L.; Liu, A. Evidence for a dual role of an active site histidine in α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase. Biochemistry 2012, 51, 5811–5821. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Davis, I.; Chen, L.; Liu, A. The power of two: Arginine 51 and arginine 239* from a neighboring subunit are essential for catalysis in α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase. J. Biol. Chem. 2013, 288, 30862–30871. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Davis, I.; Matsui, T.; Rubalcava, I.; Liu, A. Quaternary structure of α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD) controls its activity. J. Biol. Chem. 2019, 294, 11609–11621. [Google Scholar] [CrossRef]
- Huo, L.; Liu, F.; Iwaki, H.; Li, T.; Hasegawa, Y.; Liu, A. Human α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD): A structural and mechanistic unveiling. Proteins 2015, 83, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianci, M.; Giacchè, N.; Cialabrini, L.; Carotti, A.; Liscio, P.; Rosatelli, E.; De Franco, F.; Gasparrini, M.; Robertson, J.; Amici, A.; et al. Structural basis of human dimeric α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase inhibition with TES-1025. Front. Mol. Biosci. 2022, 9, 834700. [Google Scholar] [CrossRef]
- Garavaglia, S.; Perozzi, S.; Galeazzi, L.; Raffaelli, N.; Rizzi, M. The crystal structure of human α-amino-β-carboxymuconic-ε-semialdehyde decarboxylase in complex with 1,3-dihydroxyacetonephosphate suggests a regulatory link between NAD synthesis and glycolysis. FEBS J. 2009, 276, 6615–6623. [Google Scholar] [CrossRef]
- Shibata, K.; Fukuwatari, T. Large amounts of picolinic acid are lethal but small amounts increase the conversion of tryptophan-nicotinamide in rats. J. Nutr. Sci. Vitaminol. 2014, 60, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Davis, I.; Liu, F.; Andi, B.; Esaki, S.; Iwaki, H.; Hasegawa, Y.; Orville, A.M.; Liu, A. Crystallographic and spectroscopic snapshots reveal a dehydrogenase in action. Nat. Commun. 2015, 6, 5935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Davis, I.; Ha, U.; Wang, Y.; Shin, I.; Liu, A. A pitcher-and-catcher mechanism drives endogenous substrate isomerization by a dehydrogenase in kynurenine metabolism. J. Biol. Chem. 2016, 291, 26252–26261. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Davis, I.; Liu, A.; Shamsi, S.A. Development of a CZE-ESI-MS assay with a sulfonated capillary for profiling picolinic acid and quinolinic acid formation in multienzyme system. Electrophoresis 2013, 34, 1828–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawdhury, A.; Shamsi, S.A.; Miller, A.; Liu, A. Capillary electrochromatography-mass spectrometry of kynurenine pathway metabolites. J. Chromatogr. A 2021, 1651, 462294. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, A. PDB entry 4IGM: 2.39 Angstrom ligand-free X-ray crystal structure of human ACMSD. RCSB PDB 2012. [Google Scholar] [CrossRef]
- Bitto, E.; Bingman, C.A.; Wesenberg, G.E.; Phillips, G.N., Jr. PDB entry 2QNK: Crystal structure of human 3-hydroxyanthranilate 3,4-dioxygenase. RCSB PDB 2017. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweetlove, L.J.; Fernie, A.R. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 2018, 9, 2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newton, A.; McCann, L.; Huo, L.; Liu, A. Kynurenine Pathway Regulation at Its Critical Junctions with Fluctuation of Tryptophan. Metabolites 2023, 13, 500. https://doi.org/10.3390/metabo13040500
Newton A, McCann L, Huo L, Liu A. Kynurenine Pathway Regulation at Its Critical Junctions with Fluctuation of Tryptophan. Metabolites. 2023; 13(4):500. https://doi.org/10.3390/metabo13040500
Chicago/Turabian StyleNewton, Ashley, Luree McCann, Lu Huo, and Aimin Liu. 2023. "Kynurenine Pathway Regulation at Its Critical Junctions with Fluctuation of Tryptophan" Metabolites 13, no. 4: 500. https://doi.org/10.3390/metabo13040500