
Atherosclerosis Calcification: Focus on Lipoproteins


Abstract
:1. Introduction
2. Vascular Calcification Process
3. Lipoproteins and Vascular Calcification
3.1. Lipoproteins and Their Subfractions: Results from Clinical Studies
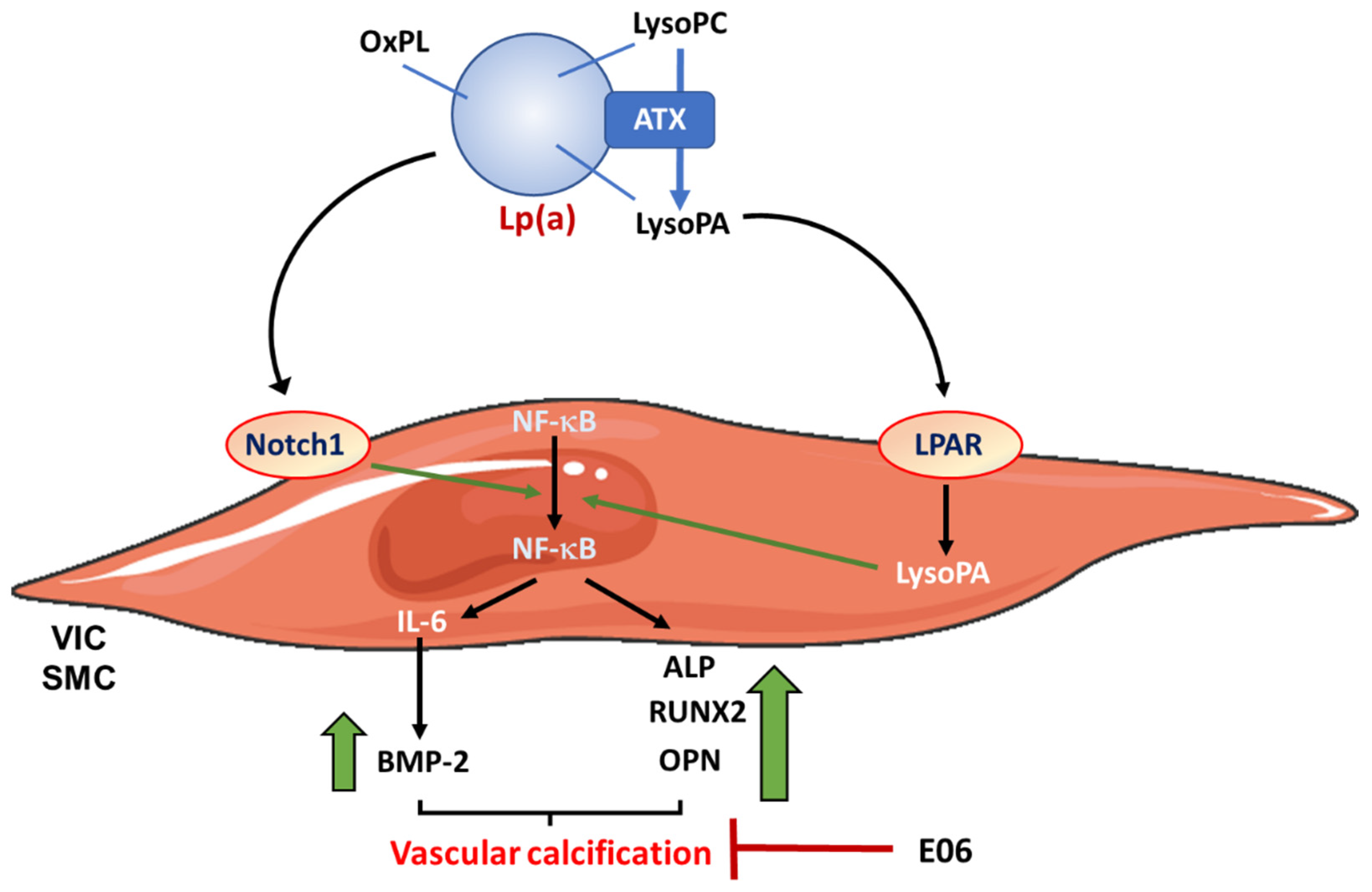
3.1.1. Lipoprotein (a) Lp(a)
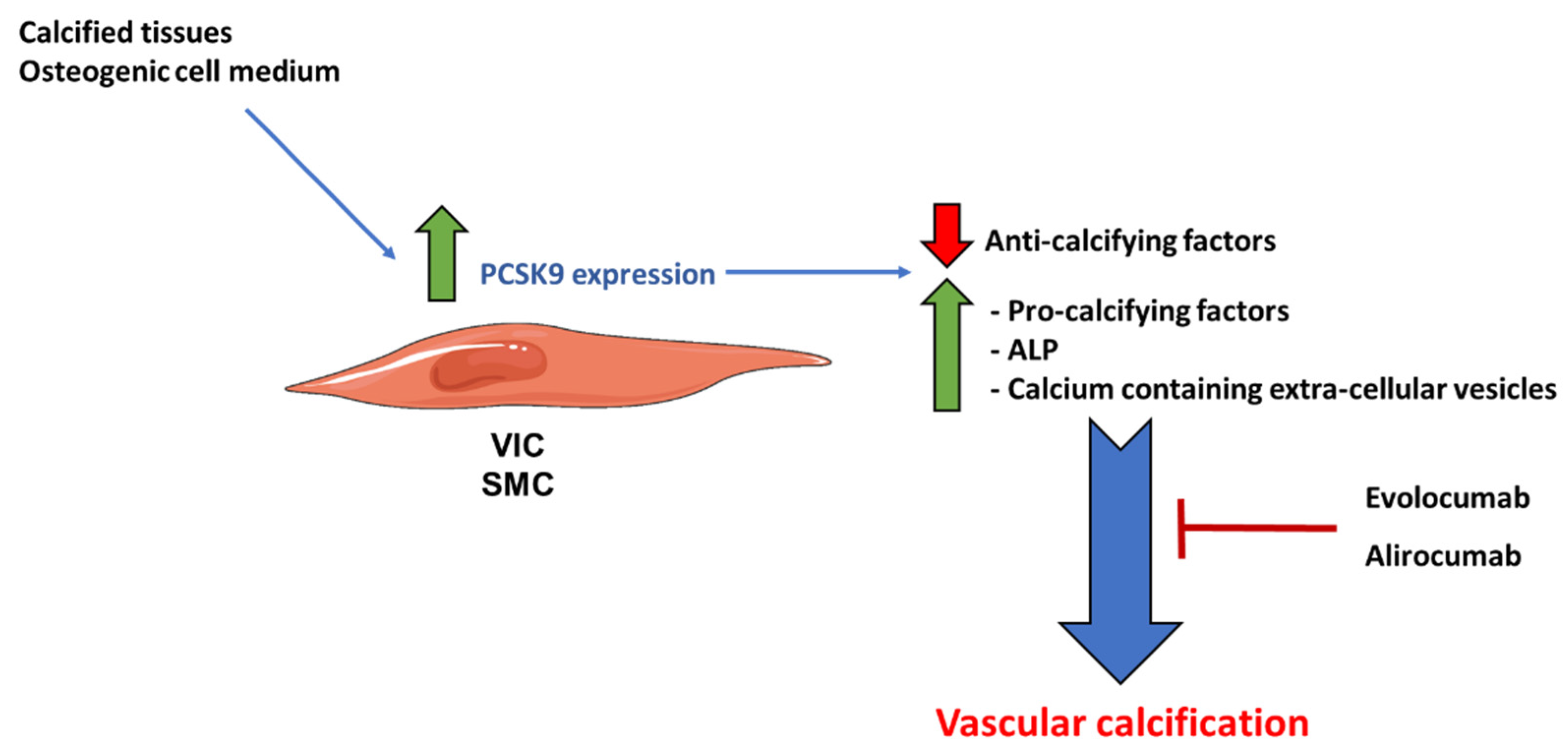
3.1.2. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9)
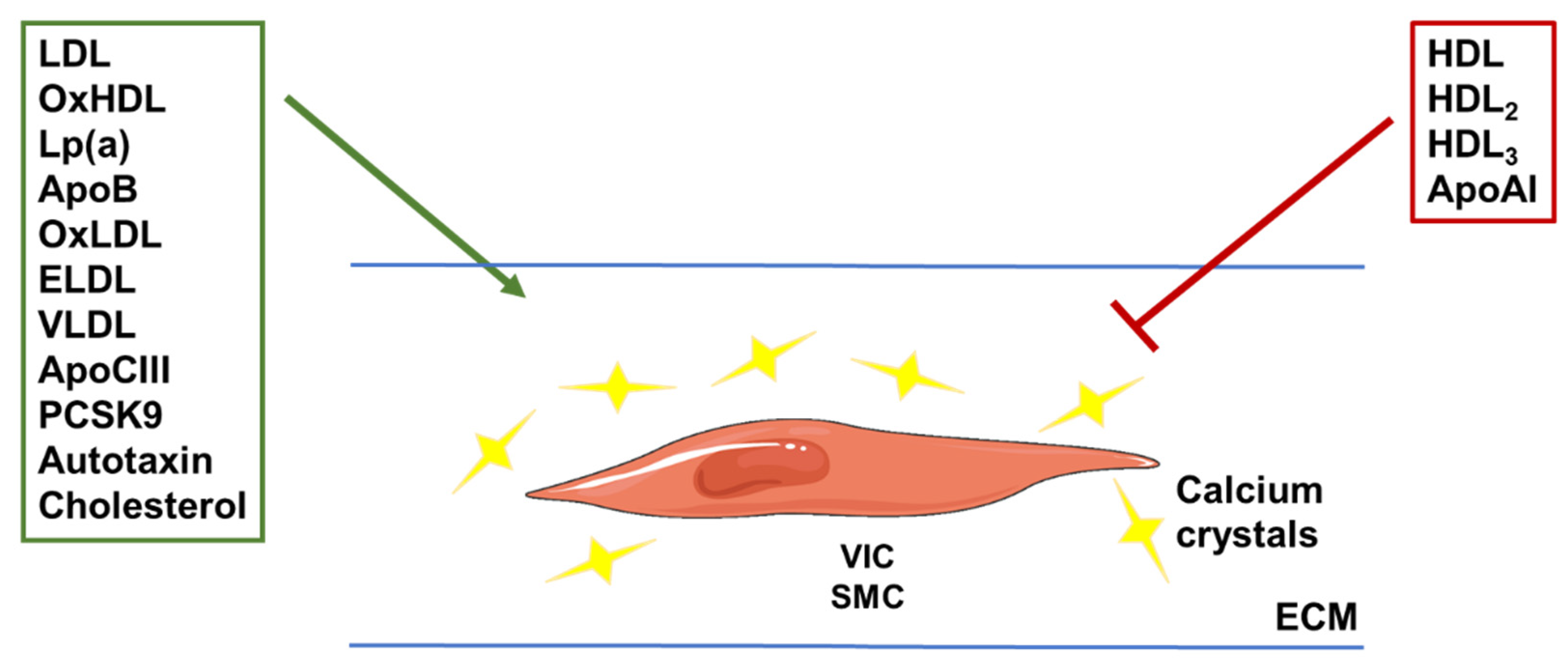
3.2. Lipoproteins and Extra-Cellular Matrix Mineralization: Results from Experimental Studies
3.2.1. Lipoproteins
3.2.2. Apolipoproteins

{kind=link}
{kind=link}
{kind=link}
| Apo and Lipoproteins | Cell or Animal Models | Mechanisms of Action | References |
|---|---|---|---|
| HDL | Bovine VSMCs | Inhibit VSMCs trans-differentiation by reducing ALP activity. Inhibit IL-1β, IL-6 secretion and minimally OxLDL-induced osteogenic activity. | [79] |
| Human THP-1 and U937 monocytic cell lines | Decrease the number of OCN+ monocytes induced by OxLDL by a mechanism involving the SR-B1 receptor. | [30] | |
| OxHDL | Bovine VSMCs | Enhance vascular cell mineralization by increasing ALP activity. | [79] |
| Human VSMCs and VICs | Induce the expression of osteogenic factors (RUNX2, BMP-2, WNT5a, Osterix, etc.). | [51,80] | |
| ELDL | Human coronary artery SMCs | Inhibit the expression of calcification inhibitors such as matrix gla protein and ENPP-1. Up-regulate the expression of genes promoting calcification (RUNX2, ALP, BMP, Osterix…). | [82] |
| OxLDL | Human VICs | Induce the expression of the inorganic phosphate transporter Pit-1 and of BMP-2. | [83] |
| Human SMCs | OxLDL-derived LysoPA promotes mineralization and cellular osteogenic transition. | [84] | |
| Increased the RANKL expression in human SMC, without affecting the RANKL decoy receptor osteoprotegerin (OPG). The lipid extracts of OxLDL reproduce the effects of the whole particle. | [84] | ||
| Cholesterol | Aortic SMCs from LDL-R deficient (LDLR-/-) mice, cultured under pro-calcifying conditions | Lower ALP activity and matrix calcium deposition compared to SMCs isolated from control mice. | [86] |
| Cells from control mice | Treatment with lipoprotein deficient serum (LPDS), reduces matrix calcium deposition, compared to the normal serum. | [86] | |
| Cells from LDLR-/- mice | Mevastatin reduces the matrix calcium deposition. | [86] | |
| Mouse SMCs | 25-hydroxy cholesterol upregulates ALP expression and increases calcification. | [87] | |
| Reduction of circulating cholesterol concentration in ApoE-deficient mice | Reduces aortic root calcification. | [88] | |
| apoCIII | Human VICs | Increases calcium deposition by a mechanism involving mitochondrial dysfunction and inflammatory pathways. | [46] |
| apoAI | Human VICs | Reduces calcification. | [46] |
| Mice and rabbits | Mimetic peptides significantly reduced calcification. | [89,90,91] | |
| apoAI, HDL2, or HDL3 | Human valve myofibroblasts | Increase OPG secretion. | [44] |
3.2.3. Lp(a) and PCSK9


4. Lipid-Lowering Drugs in VC and AVC: The Good and the Bad
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.I.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Aikawa, M.; Schonbeck, U. Cholesterol and atherosclerosis. Biochim. Biophys. Acta 2000, 1529, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2255–2264. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Prim. 2019, 5, 56. [Google Scholar] [CrossRef]
- Gebbers, J.O. Atherosclerosis, cholesterol, nutrition, and statins—A critical review. Ger. Med. Sci. 2007, 5, Doc04. [Google Scholar] [PubMed]
- Jakubiak, G.K.; Pawlas, N.; Cieslar, G.; Stanek, A. Pathogenesis and Clinical Significance of In-Stent Restenosis in Patients with Diabetes. Int. J. Environ. Res. Public Health 2021, 18, 11970. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Sutton, N.R.; Malhotra, R.; St Hilaire, C.; Aikawa, E.; Blumenthal, R.S.; Gackenbach, G.; Goyal, P.; Johnson, A.; Nigwekar, S.U.; Shanahan, C.M.; et al. Molecular Mechanisms of Vascular Health: Insights From Vascular Aging and Calcification. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Fortini, F.; Severi, P.; Rizzo, P.; Gardi, I.; Cimaglia, P.; Rapezzi, C.; Tavazzi, L.; Ferrari, R. Cardiac Calcifications: Phenotypes, Mechanisms, Clinical and Prognostic Implications. Biology 2022, 11, 414. [Google Scholar] [CrossRef]
- Dos Santos, V.P.; Pozzan, G.; Castelli, V.; Caffaro, R.A. Arteriosclerosis, atherosclerosis, arteriolosclerosis, and Monckeberg medial calcific sclerosis: What is the difference? J. Vasc. Bras. 2021, 20, e20200211. [Google Scholar] [CrossRef]
- Lee, H.Y.; Lim, S.; Park, S. Role of Inflammation in Arterial Calcification. Korean Circ. J. 2021, 51, 114–125. [Google Scholar] [CrossRef]
- St Hilaire, C. Medial Arterial Calcification: A Significant and Independent Contributor of Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 253–260. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef]
- Aboyans, V.; Lacroix, P.; Tran, M.H.; Salamagne, C.; Galinat, S.; Archambeaud, F.; Criqui, M.H.; Laskar, M. The prognosis of diabetic patients with high ankle-brachial index depends on the coexistence of occlusive peripheral artery disease. J. Vasc. Surg. 2011, 53, 984–991. [Google Scholar] [CrossRef]
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 2012, 14, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Jie, W.; Huang, H. Vascular calcification: Molecular mechanisms and therapeutic interventions. MedComm 2023, 4, e200. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Buers, I.; Nitschke, Y. Hereditary Disorders of Cardiovascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 35–47. [Google Scholar] [CrossRef]
- Montanaro, M.; Scimeca, M.; Anemona, L.; Servadei, F.; Giacobbi, E.; Bonfiglio, R.; Bonanno, E.; Urbano, N.; Ippoliti, A.; Santeusanio, G.; et al. The Paradox Effect of Calcification in Carotid Atherosclerosis: Microcalcification is Correlated with Plaque Instability. Int. J. Mol. Sci. 2021, 22, 395. [Google Scholar] [CrossRef]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasian, N. Vascular Calcification Mechanisms: Updates and Renewed Insight into Signaling Pathways Involved in High Phosphate-Mediated Vascular Smooth Muscle Cell Calcification. Biomedicines 2021, 9, 804. [Google Scholar] [CrossRef]
- Waring, O.J.; Skenteris, N.T.; Biessen, E.A.L.; Donners, M. Two-faced Janus: The dual role of macrophages in atherosclerotic calcification. Cardiovasc. Res. 2022, 118, 2768–2777. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Z.; Zhang, L.; Yan, J.; Shao, C.; Jing, L.; Li, L.; Wang, Z. Role of Macrophages in the Progression and Regression of Vascular Calcification. Front. Pharmacol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Maddaloni, E.; Xia, Y.; Park, K.; D’Eon, S.; Tinsley, L.J.; St-Louis, R.; Khamaisi, M.; Li, Q.; King, G.L.; Keenan, H.A. High density lipoprotein modulates osteocalcin expression in circulating monocytes: A potential protective mechanism for cardiovascular disease in type 1 diabetes. Cardiovasc. Diabetol. 2017, 16, 116. [Google Scholar] [CrossRef] [Green Version]
- Mackey, R.H.; Kuller, L.H.; Sutton-Tyrrell, K.; Evans, R.W.; Holubkov, R.; Matthews, K.A. Lipoprotein subclasses and coronary artery calcium in postmenopausal women from the healthy women study. Am. J. Cardiol. 2002, 90, 71i–76i. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Long, Q.; Li, J.; Li, G.; Ding, Y.; Cui, Q.; Liu, Z. Small dense low-density lipoprotein cholesterol is strongly associated with NIHSS score and intracranial arterial calcification in acute ischemic stroke subjects. Sci. Rep. 2020, 10, 7645. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, W.C.; Otvos, J.D.; Keyes, M.J.; Pencina, M.J.; Sullivan, L.; Vasan, R.S.; Wilson, P.W.; D’Agostino, R.B. LDL Particle Number and Risk of Future Cardiovascular Disease in the Framingham Offspring Study—Implications for LDL Management. J. Clin. Lipidol. 2007, 1, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Prado, K.B.; Shugg, S.; Backstrand, J.R. Low-density lipoprotein particle number predicts coronary artery calcification in asymptomatic adults at intermediate risk of cardiovascular disease. J. Clin. Lipidol. 2011, 5, 408–413. [Google Scholar] [CrossRef]
- Zaid, M.; Miura, K.; Fujiyoshi, A.; Abbott, R.D.; Hisamatsu, T.; Kadota, A.; Arima, H.; Kadowaki, S.; Torii, S.; Miyagawa, N.; et al. Associations of serum LDL particle concentration with carotid intima-media thickness and coronary artery calcification. J. Clin. Lipidol. 2016, 10, 1195–1202.e1. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Nomura, S.O.; Steffen, B.T.; Guan, W.; Remaley, A.T.; Karger, A.B.; Ouyang, P.; Michos, E.D.; Tsai, M.Y. Apolipoprotein B discordance with low-density lipoprotein cholesterol and non-high-density lipoprotein cholesterol in relation to coronary artery calcification in the Multi-Ethnic Study of Atherosclerosis (MESA). J. Clin. Lipidol. 2020, 14, 109–121.e5. [Google Scholar] [CrossRef]
- Chang, T.Y.; Chen, J.D. Low-density lipoprotein cholesterol/apolipoprotein B ratio is superior to apolipoprotein B alone in the diagnosis of coronary artery calcification. Coron. Artery Dis. 2021, 32, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Sandesara, P.B.; Mehta, A.; O’Neal, W.T.; Mohamed Kelli, H.; Sathiyakumar, V.; Martin, S.S.; Blaha, M.J.; Blumenthal, R.S.; Sperling, L.S. Association of Elevated High-Density Lipoprotein Cholesterol and Particle Concentration With Coronary Artery Calcium: The Multi-Ethnic Study of Atherosclerosis. Circ. Cardiovasc. Imaging 2020, 13, e010473. [Google Scholar] [CrossRef]
- Mackey, R.H.; Greenland, P.; Goff, D.C., Jr.; Lloyd-Jones, D.; Sibley, C.T.; Mora, S. High-density lipoprotein cholesterol and particle concentrations, carotid atherosclerosis, and coronary events: MESA (multi-ethnic study of atherosclerosis). J. Am. Coll. Cardiol. 2012, 60, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Voros, S.; Joshi, P.; Qian, Z.; Rinehart, S.; Vazquez-Figueroa, J.G.; Anderson, H.; Elashoff, M.; Murrieta, L.; Karmpaliotis, D.; Kalynych, A.; et al. Apoprotein B, small-dense LDL and impaired HDL remodeling is associated with larger plaque burden and more noncalcified plaque as assessed by coronary CT angiography and intravascular ultrasound with radiofrequency backscatter: Results from the ATLANTA I study. J. Am. Heart Assoc. 2013, 2, e000344. [Google Scholar]
- Armstrong, M.K.; Fraser, B.J.; Hartiala, O.; Buscot, M.J.; Juonala, M.; Wu, F.; Koskinen, J.; Hutri-Kahonen, N.; Kahonen, M.; Laitinen, T.P.; et al. Association of Non-High-Density Lipoprotein Cholesterol Measured in Adolescence, Young Adulthood, and Mid-Adulthood With Coronary Artery Calcification Measured in Mid-Adulthood. JAMA Cardiol. 2021, 6, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.K.; Blaha, M.J.; Loprinzi, P.D. Atherogenic Index of Plasma and Triglyceride/High-Density Lipoprotein Cholesterol Ratio Predict Mortality Risk Better Than Individual Cholesterol Risk Factors, Among an Older Adult Population. Mayo Clin. Proc. 2017, 92, 680–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, K.B.; Han, D.; Lee, J.H.; Choi, S.Y.; Chun, E.J.; Park, S.H.; Han, H.W.; Sung, J.; Jung, H.O.; Chang, H.J. Atherogenic index of plasma and coronary artery calcification progression beyond traditional risk factors according to baseline coronary artery calcium score. Sci. Rep. 2020, 10, 21324. [Google Scholar] [CrossRef]
- Lommi, J.I.; Kovanen, P.T.; Jauhiainen, M.; Lee-Rueckert, M.; Kupari, M.; Helske, S. High-density lipoproteins (HDL) are present in stenotic aortic valves and may interfere with the mechanisms of valvular calcification. Atherosclerosis 2011, 219, 538–544. [Google Scholar] [CrossRef]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; Franke, U.F.W.; et al. Lipoprotein(a) Associated Molecules are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef]
- Schlotter, F.; de Freitas, R.C.C.; Rogers, M.A.; Blaser, M.C.; Wu, P.J.; Higashi, H.; Halu, A.; Iqbal, F.; Andraski, A.B.; Rodia, C.N.; et al. ApoC-III is a novel inducer of calcification in human aortic valves. J. Biol. Chem. 2021, 296, 100193. [Google Scholar] [CrossRef]
- Gerber, Y.; Goldbourt, U.; Feinberg, M.S.; Segev, S.; Harats, D. Are triglyceride-rich lipoproteins associated with aortic valve sclerosis? A preliminary report. Atherosclerosis 2003, 170, 301–305. [Google Scholar] [CrossRef]
- Pollin, T.I.; Damcott, C.M.; Shen, H.; Ott, S.H.; Shelton, J.; Horenstein, R.B.; Post, W.; McLenithan, J.C.; Bielak, L.F.; Peyser, P.A.; et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008, 322, 1702–1705. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Torzewski, M.; Mayr, M.; Chan, K.L.; Mathieu, P.; Bosse, Y.; Dumesnil, J.G.; Tam, J.; Teo, K.K.; Burnap, S.A.; et al. ApoCIII-Lp(a) complexes in conjunction with Lp(a)-OxPL predict rapid progression of aortic stenosis. Heart 2020, 106, 738–745. [Google Scholar] [CrossRef]
- Qiao, Y.N.; Zou, Y.L.; Guo, S.D. Low-density lipoprotein particles in atherosclerosis. Front. Physiol. 2022, 13, 931931. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.T.; Chen, Y.Y.; Mao, J.Y.; Wang, Y.P.; Chen, Y.F.; Hu, X.; Yang, K.; Liu, Y. Oxidized HDL, as a Novel Biomarker for Calcific Aortic Valve Disease, Promotes the Calcification of Aortic Valve Interstitial Cells. J. Cardiovasc. Transl. Res. 2019, 12, 560–568. [Google Scholar] [CrossRef]
- Miki, T.; Miyoshi, T.; Kotani, K.; Kohno, K.; Asonuma, H.; Sakuragi, S.; Koyama, Y.; Nakamura, K.; Ito, H. Decrease in oxidized high-density lipoprotein is associated with slowed progression of coronary artery calcification: Subanalysis of a prospective multicenter study. Atherosclerosis 2019, 283, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakino, A.; Usami, Y.; Horiuchi, S.; Fujita, Y.; Kotani, K.; Chen, C.H.; Okamura, T.; Sawamura, T. A Novel Cell-Free, Non-Fluorescent Method to Measure LOX-1-Binding Activity Corresponding to The Functional Activity of HDL. J. Atheroscler. Thromb. 2019, 26, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, A.; Kakino, A.; Okamura, T.; Usami, Y.; Fujita, Y.; Kadota, A.; Fujiyoshi, A.; Hisamatsu, T.; Kondo, K.; Segawa, H.; et al. The relationship between serum levels of LOX-1 ligand containing ApoAI as a novel marker of dysfunctional HDL and coronary artery calcification in middle-aged Japanese men. Atherosclerosis 2020, 313, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ten Kate, G.R.; Bos, S.; Dedic, A.; Neefjes, L.A.; Kurata, A.; Langendonk, J.G.; Liem, A.; Moelker, A.; Krestin, G.P.; de Feyter, P.J.; et al. Increased Aortic Valve Calcification in Familial Hypercholesterolemia: Prevalence, Extent, and Associated Risk Factors. J. Am. Coll. Cardiol. 2015, 66, 2687–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Kindi, M.; Belanger, A.M.; Sayegh, K.; Senouci, S.; Aljenedil, S.; Sivakumaran, L.; Ruel, I.; Al Rasadi, K.; Al Waili, K.; Awan, Z.; et al. Aortic Calcification Progression in Heterozygote Familial Hypercholesterolemia. Can. J. Cardiol. 2017, 33, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, M.L.; Marcovina, S.M. Structure-function relationships in apolipoprotein(a): Insights into lipoprotein(a) assembly and pathogenicity. Curr. Opin. Lipidol. 2004, 15, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and its Significance in Cardiovascular Disease: A Review. JAMA Cardiol 2022, 7, 760–769. [Google Scholar] [CrossRef]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Tsimikas, S. Potential Causality and Emerging Medical Therapies for Lipoprotein(a) and Its Associated Oxidized Phospholipids in Calcific Aortic Valve Stenosis. Circ. Res. 2019, 124, 405–415. [Google Scholar] [CrossRef]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Liu, M.M.; Liu, H.H.; Xu, R.X.; Zhu, C.G.; Guo, Y.L.; Wu, N.Q.; Dong, Q.; Cui, C.J.; Li, J.J. Lipoprotein (a)-mediated vascular calcification: Population-based and in vitro studies. Metabolism 2022, 127, 154960. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.A.; Doris, M.K.; White, A.C.; Timmers, N.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Despres, A.A.; Perrot, N.; Poulin, A.; Tastet, L.; Shen, M.; Chen, H.Y.; Bourgeois, R.; Trottier, M.; Tessier, M.; Guimond, J.; et al. Lipoprotein(a), Oxidized Phospholipids, and Aortic Valve Microcalcification Assessed by 18F-Sodium Fluoride Positron Emission Tomography and Computed Tomography. CJC Open 2019, 1, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechlivanis, S.; Mahabadi, A.A.; Hoffmann, P.; Nothen, M.M.; Broecker-Preuss, M.; Erbel, R.; Moebus, S.; Stang, A.; Jockel, K.H. Association between lipoprotein(a) (Lp(a)) levels and Lp(a) genetic variants with coronary artery calcification. BMC Med. Genet. 2020, 21, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, B.J.; Boekholdt, S.M.; Dube, M.P.; Rheaume, E.; Wareham, N.J.; Khaw, K.T.; Sandhu, M.S.; Tardif, J.C. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: A prospective Mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Bergmark, B.A.; O’Donoghue, M.L.; Murphy, S.A.; Kuder, J.F.; Ezhov, M.V.; Ceska, R.; Gouni-Berthold, I.; Jensen, H.K.; Tokgozoglu, S.L.; Mach, F.; et al. An Exploratory Analysis of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibition and Aortic Stenosis in the FOURIER Trial. JAMA Cardiol. 2020, 5, 709–713. [Google Scholar] [CrossRef]
- Wodaje, T.; Littmann, K.; Habel, H.; Bottai, M.; Back, M.; Parini, P.; Brinck, J. Plasma Lipoprotein(a) measured in routine clinical care and the association with incident calcified aortic valve stenosis during a 14-year observational period. Atherosclerosis 2022, 349, 175–182. [Google Scholar] [CrossRef]
- Vongpromek, R.; Bos, S.; Ten Kate, G.J.; Yahya, R.; Verhoeven, A.J.; de Feyter, P.J.; Kronenberg, F.; Roeters van Lennep, J.E.; Sijbrands, E.J.; Mulder, M.T. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J. Intern. Med. 2015, 278, 166–173. [Google Scholar] [CrossRef]
- Kaiser, Y.; Singh, S.S.; Zheng, K.H.; Verbeek, R.; Kavousi, M.; Pinto, S.J.; Vernooij, M.W.; Sijbrands, E.J.G.; Boekholdt, S.M.; de Rijke, Y.B.; et al. Lipoprotein(a) is robustly associated with aortic valve calcium. Heart 2021, 107, 1422–1428. [Google Scholar] [CrossRef]
- Kaiser, Y.; van der Toorn, J.E.; Singh, S.S.; Zheng, K.H.; Kavousi, M.; Sijbrands, E.J.G.; Stroes, E.S.G.; Vernooij, M.W.; de Rijke, Y.B.; Boekholdt, S.M.; et al. Lipoprotein(a) is associated with the onset but not the progression of aortic valve calcification. Eur. Heart J. 2022, 43, 3960–3967. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bosse, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nsaibia, M.J.; Mahmut, A.; Boulanger, M.C.; Arsenault, B.J.; Bouchareb, R.; Simard, S.; Witztum, J.L.; Clavel, M.A.; Pibarot, P.; Bosse, Y.; et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J. Intern. Med. 2016, 280, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Poggio, P.; Songia, P.; Cavallotti, L.; Barbieri, S.S.; Zanotti, I.; Arsenault, B.J.; Valerio, V.; Ferri, N.; Capoulade, R.; Camera, M. PCSK9 Involvement in Aortic Valve Calcification. J. Am. Coll. Cardiol. 2018, 72, 3225–3227. [Google Scholar] [CrossRef]
- Perrot, N.; Valerio, V.; Moschetta, D.; Boekholdt, S.M.; Dina, C.; Chen, H.Y.; Abner, E.; Martinsson, A.; Manikpurage, H.D.; Rigade, S.; et al. Genetic and In Vitro Inhibition of PCSK9 and Calcific Aortic Valve Stenosis. JACC Basic Transl. Sci. 2020, 5, 649–661. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Benn, M.; Tybjaerg-Hansen, A.; Kamstrup, P.R. PCSK9 R46L Loss-of-Function Mutation Reduces Lipoprotein(a), LDL Cholesterol, and Risk of Aortic Valve Stenosis. J. Clin. Endocrinol. Metab. 2016, 101, 3281–3287. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Li, Y.P.; Ma, X.T.; Wang, Z.J.; Shi, D.M.; Zhou, Y.J. Effect of Alirocumab on Coronary Calcification in Patients With Coronary Artery Disease. Front. Cardiovasc. Med. 2022, 9, 907662. [Google Scholar] [CrossRef]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification: A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2022, 118, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Basseri, B.; Hwang, J.; Tintut, Y.; Demer, L.L. High-density lipoprotein regulates calcification of vascular cells. Circ. Res. 2002, 91, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Harun, N.H.; Froemming, G.R.A.; Nawawi, H.M.; Muid, S.A. Inflammation and Vascular Calcification Causing Effects of Oxidized HDL are Attenuated by Adiponectin in Human Vascular Smooth Muscle Cells. Int. J. Mol. Cell. Med. 2019, 8, 39–55. [Google Scholar]
- Twardowski, L.; Cheng, F.; Michaelsen, J.; Winter, S.; Hofmann, U.; Schaeffeler, E.; Muller, S.; Sonnenberg, M.; Steuer, K.; Ott, G.; et al. Enzymatically Modified Low-Density Lipoprotein Is Present in All Stages of Aortic Valve Sclerosis: Implications for Pathogenesis of the Disease. J. Am. Heart Assoc. 2015, 4, e002156. [Google Scholar] [CrossRef] [Green Version]
- Chellan, B.; Rojas, E.; Zhang, C.; Hofmann Bowman, M.A. Enzyme-modified non-oxidized LDL (ELDL) induces human coronary artery smooth muscle cell transformation to a migratory and osteoblast-like phenotype. Sci. Rep. 2018, 8, 11954. [Google Scholar] [CrossRef]
- Nadlonek, N.A.; Lee, J.H.; Weyant, M.J.; Meng, X.; Fullerton, D.A. ox-LDL induces PiT-1 expression in human aortic valve interstitial cells. J. Surg. Res. 2013, 184, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Maziere, C.; Salle, V.; Gomila, C.; Maziere, J.C. Oxidized low density lipoprotein increases RANKL level in human vascular cells. Involvement of oxidative stress. Biochem. Biophys. Res. Commun. 2013, 440, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Nsaibia, M.J.; Boulanger, M.C.; Bouchareb, R.; Mkannez, G.; Le Quang, K.; Hadji, F.; Argaud, D.; Dahou, A.; Bosse, Y.; Koschinsky, M.L.; et al. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-kappaB pathway. Cardiovasc. Res. 2017, 113, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Hsu, J.J.; Lu, J.; Ting, T.C.; Miyazaki, M.; Demer, L.L.; Tintut, Y. Role of cellular cholesterol metabolism in vascular cell calcification. J. Biol. Chem. 2011, 286, 33701–33706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.E.; Bostrom, K.; Ravindranath, R.; Lam, T.; Norton, B.; Demer, L.L. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J. Clin. Investig. 1994, 93, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Schutkowski, A.; Hirche, F.; Geissler, S.; Radtke, J.; Stangl, G.I. Additive effects of lupin protein and phytic acid on aortic calcification in ApoE deficient mice. J. Clin. Transl. Endocrinol. 2015, 2, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speidl, W.S.; Cimmino, G.; Ibanez, B.; Elmariah, S.; Hutter, R.; Garcia, M.J.; Fuster, V.; Goldman, M.E.; Badimon, J.J. Recombinant apolipoprotein A-I Milano rapidly reverses aortic valve stenosis and decreases leaflet inflammation in an experimental rabbit model. Eur. Heart J. 2010, 31, 2049–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapeaux, J.; Busseuil, D.; Shi, Y.; Nobari, S.; Shustik, D.; Mecteau, M.; El-Hamamsy, I.; Lebel, M.; Mongrain, R.; Rheaume, E.; et al. Improvement of aortic valve stenosis by ApoA-I mimetic therapy is associated with decreased aortic root and valve remodelling in mice. Br. J. Pharmacol. 2013, 169, 1587–1599. [Google Scholar] [CrossRef]
- Busseuil, D.; Shi, Y.; Mecteau, M.; Brand, G.; Kernaleguen, A.E.; Thorin, E.; Latour, J.G.; Rheaume, E.; Tardif, J.C. Regression of aortic valve stenosis by ApoA-I mimetic peptide infusions in rabbits. Br. J. Pharmacol. 2008, 154, 765–773. [Google Scholar] [CrossRef]
- Rogers, M.A.; Atkins, S.K.; Zheng, K.H.; Singh, S.A.; Chelvanambi, S.; Pham, T.H.; Kuraoka, S.; Stroes, E.S.G.; Aikawa, M.; Aikawa, E. Lipoprotein(a) Induces Vesicular Cardiovascular Calcification Revealed With Single-Extracellular Vesicle Analysis. Front. Cardiovasc. Med. 2022, 9, 778919. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Maatta, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Lupo, M.G.; Bressan, A.; Donato, M.; Canzano, P.; Camera, M.; Poggio, P.; Greco, M.F.; Garofalo, M.; De Martin, S.; Panighel, G.; et al. PCSK9 promotes arterial medial calcification. Atherosclerosis 2022, 346, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Chang, H.J.; Sung, J.M.; Park, H.B.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, Y.J.; et al. Effects of Statins on Coronary Atherosclerotic Plaques: The PARADIGM Study. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, J.H. Involvement of inflammatory responses in the early development of calcific aortic valve disease: Lessons from statin therapy. Anim. Cells Syst. 2018, 22, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Qureshi, A.R.; Parini, P.; Hurt-Camejo, E.; Ripsweden, J.; Brismar, T.B.; Barany, P.; Jaminon, A.M.; Schurgers, L.J.; Heimburger, O.; et al. Does statins promote vascular calcification in chronic kidney disease? Eur. J. Clin. Investig. 2017, 47, 137–148. [Google Scholar] [CrossRef]
- Saremi, A.; Bahn, G.; Reaven, P.D.; Investigators, V. Progression of vascular calcification is increased with statin use in the Veterans Affairs Diabetes Trial (VADT). Diabetes Care 2012, 35, 2390–2392. [Google Scholar] [CrossRef] [Green Version]
- Hermans, H.; Herijgers, P.; Holvoet, P.; Verbeken, E.; Meuris, B.; Flameng, W.; Herregods, M.C. Statins for calcific aortic valve stenosis: Into oblivion after SALTIRE and SEAS? An extensive review from bench to bedside. Curr. Probl. Cardiol. 2010, 35, 284–306. [Google Scholar] [CrossRef]
- Zhao, Y.; Nicoll, R.; He, Y.H.; Henein, M.Y. The effect of statins on valve function and calcification in aortic stenosis: A meta-analysis. Atherosclerosis 2016, 246, 318–324. [Google Scholar] [CrossRef]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of statins on serial coronary calcification during atheroma progression and regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, A.; Clemente, A.; Chiappino, D.; Berti, S.; Vassalle, C. Double Face of Statins at the Crossroad of Coronary Atherosclerotic Plaque and Aortic Valve Calcification? JACC Cardiovasc. Imaging 2018, 11, 1930–1931. [Google Scholar] [CrossRef] [PubMed]
- Trion, A.; Schutte-Bart, C.; Bax, W.H.; Jukema, J.W.; van der Laarse, A. Modulation of calcification of vascular smooth muscle cells in culture by calcium antagonists, statins, and their combination. Mol. Cell. Biochem. 2008, 308, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmut, A.; Boulanger, M.C.; Bouchareb, R.; Hadji, F.; Mathieu, P. Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc. Res. 2015, 106, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Zhelyazkova-Savova, M.D.; Yotov, Y.T.; Nikolova, M.N.; Nazifova-Tasinova, N.F.; Vankova, D.G.; Atanasov, A.A.; Galunska, B.T. Statins, vascular calcification, and vitamin K-dependent proteins: Is there a relation? Kaohsiung J. Med. Sci. 2021, 37, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, Y.; Inoue, I.; Inoue, K.; Shinoda, Y.; Iida, S.; Goto, S.; Nakano, T.; Shimada, A.; Noda, M. The annual rate of coronary artery calcification with combination therapy with a PCSK9 inhibitor and a statin is lower than that with statin monotherapy. NPJ Aging Mech. Dis. 2018, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Li, C.; Tu, Y.; Li, Z.; Zhang, M. Additive effects of ezetimibe, evolocumab, and alirocumab on plaque burden and lipid content as assessed by intravascular ultrasound: A PRISMA-compliant meta-analysis. Medicine 2022, 101, e31199. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neels, J.G.; Leftheriotis, G.; Chinetti, G. Atherosclerosis Calcification: Focus on Lipoproteins. Metabolites 2023, 13, 457. https://doi.org/10.3390/metabo13030457
Neels JG, Leftheriotis G, Chinetti G. Atherosclerosis Calcification: Focus on Lipoproteins. Metabolites. 2023; 13(3):457. https://doi.org/10.3390/metabo13030457
Chicago/Turabian StyleNeels, Jaap G., Georges Leftheriotis, and Giulia Chinetti. 2023. "Atherosclerosis Calcification: Focus on Lipoproteins" Metabolites 13, no. 3: 457. https://doi.org/10.3390/metabo13030457