
Identification of Lignan Compounds as New 6-Phosphogluconate Dehydrogenase Inhibitors for Lung Cancer
 , , and
, , and
Abstract
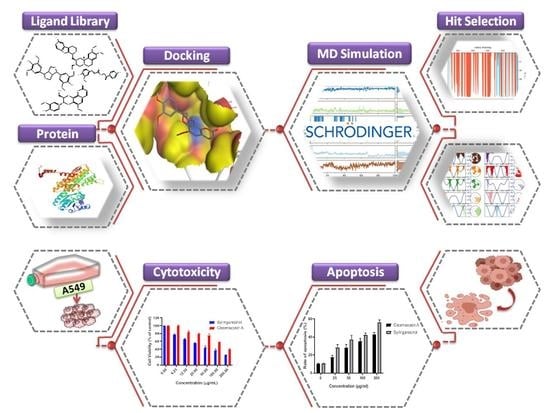
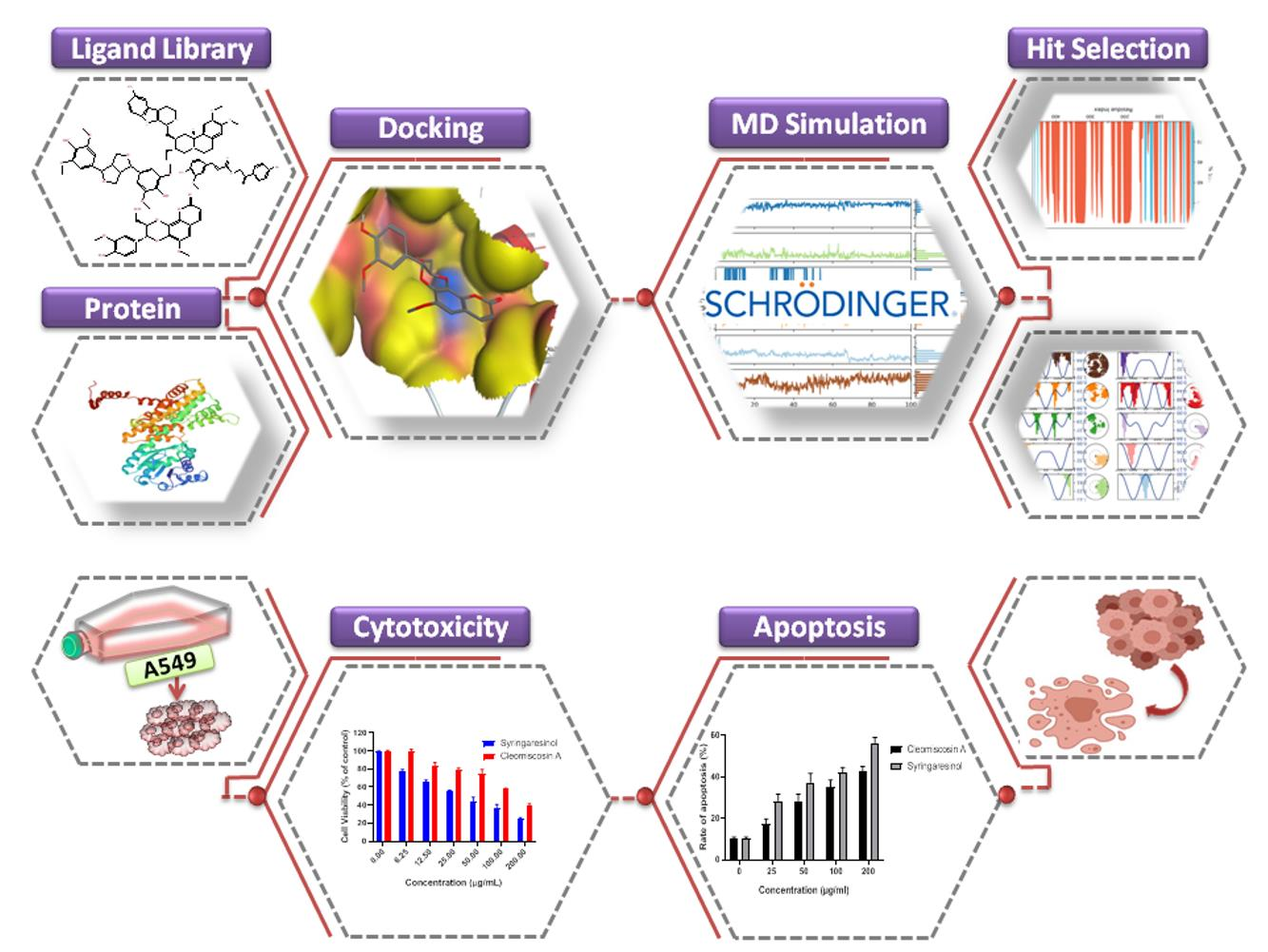
:
1. Introduction
2. Materials and Methods
2.1. Targeted Protein Structure Retrieval and Refinement
2.2. Phytochemical Database Preparation
2.3. Active Site Prediction and Molecular Docking
2.4. In Silico Drug Scan and ADMET Profiling
2.5. Molecular Dynamics Simulation
2.6. In Vitro Enzymatic Assays
2.6.1. 6-PGD Purification Using Human Erythrocytes
2.6.2. Protein Determination and Purity Check
2.6.3. In Vitro Effect of Lignan Compounds Using Erythrocytes Purified 6-PGD
2.6.4. Enzyme Activity Assay Using A549 Cell Lysate Protein
2.6.5. 6-Phosphogluconate Dehydrogenase (6-PGD)
2.7. Cell Line Cytotoxicity Prediction
2.8. Cell Culture
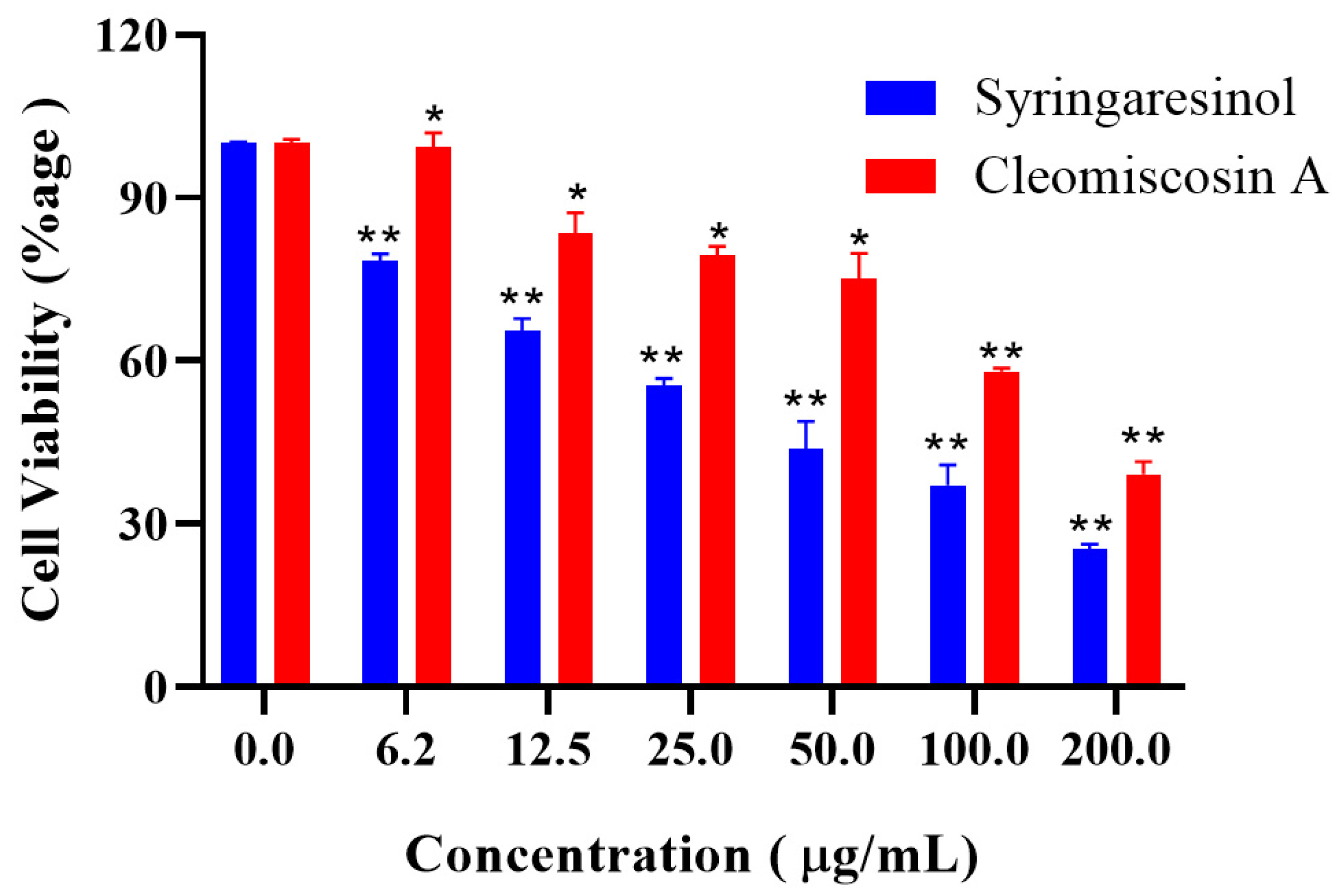
2.9. MTT Cytotoxicity Assay
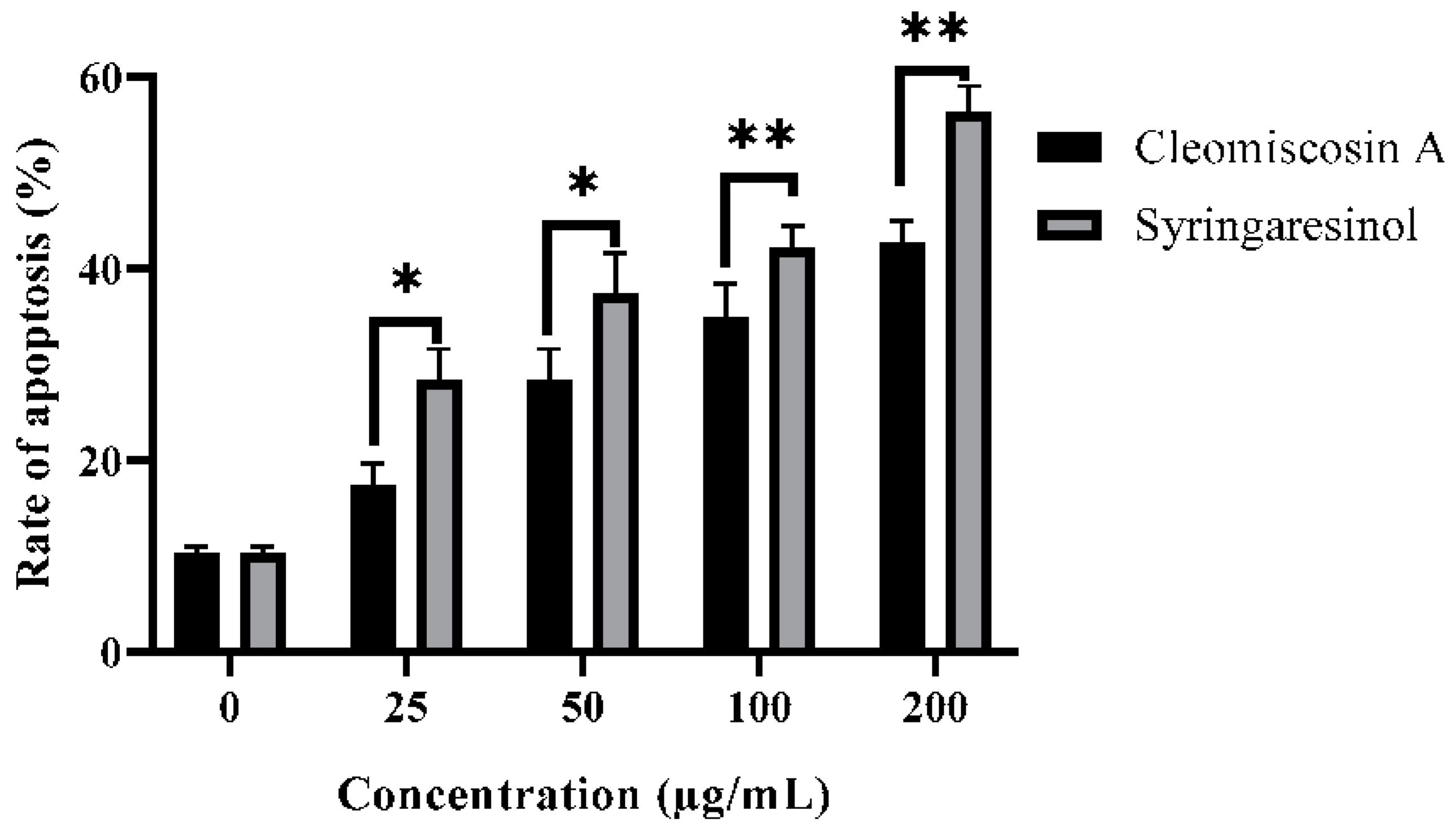
2.10. Flow Cytometric Analysis for Apoptosis
2.11. Statistical Analysis
3. Results
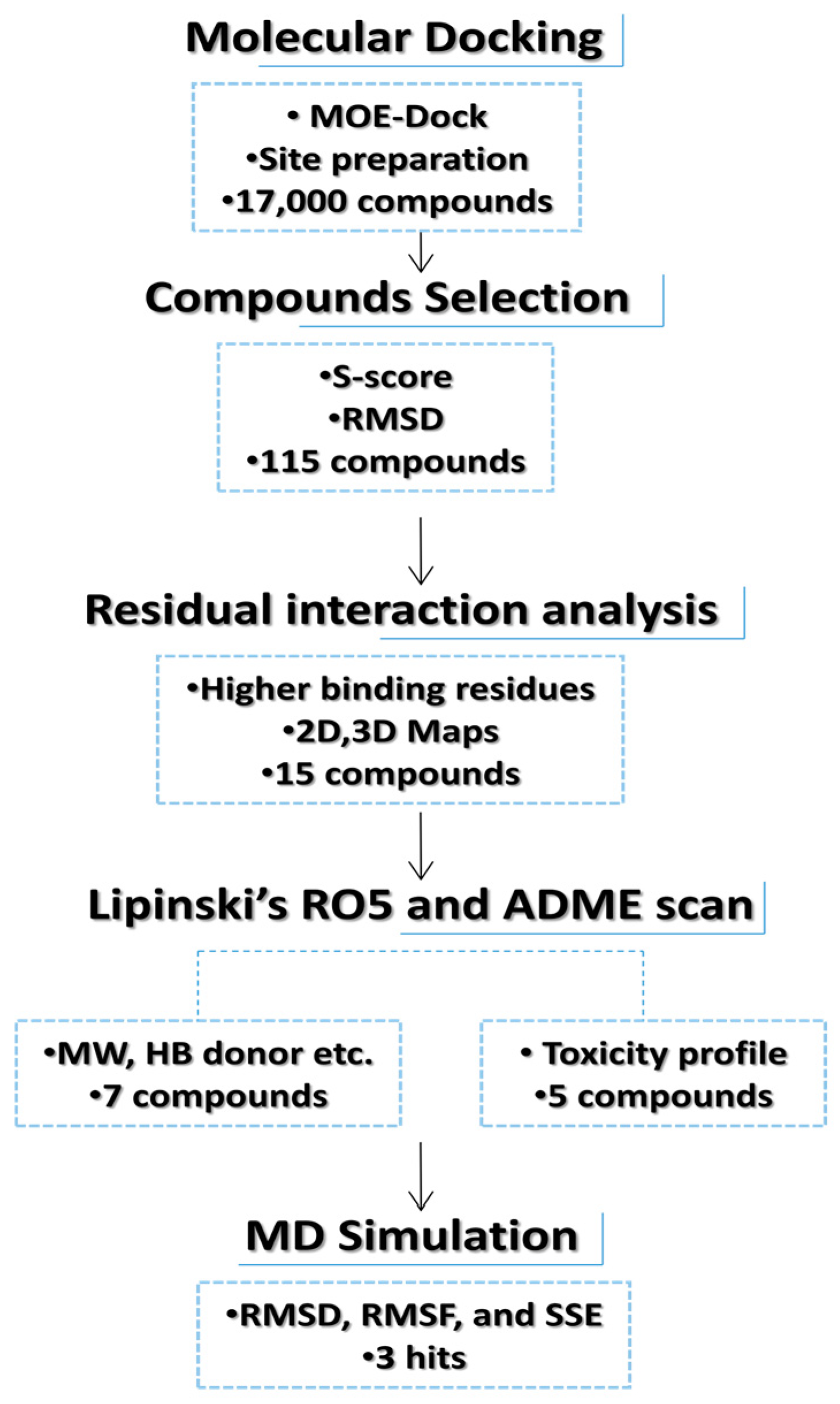
3.1. Database Screening and Docking Study
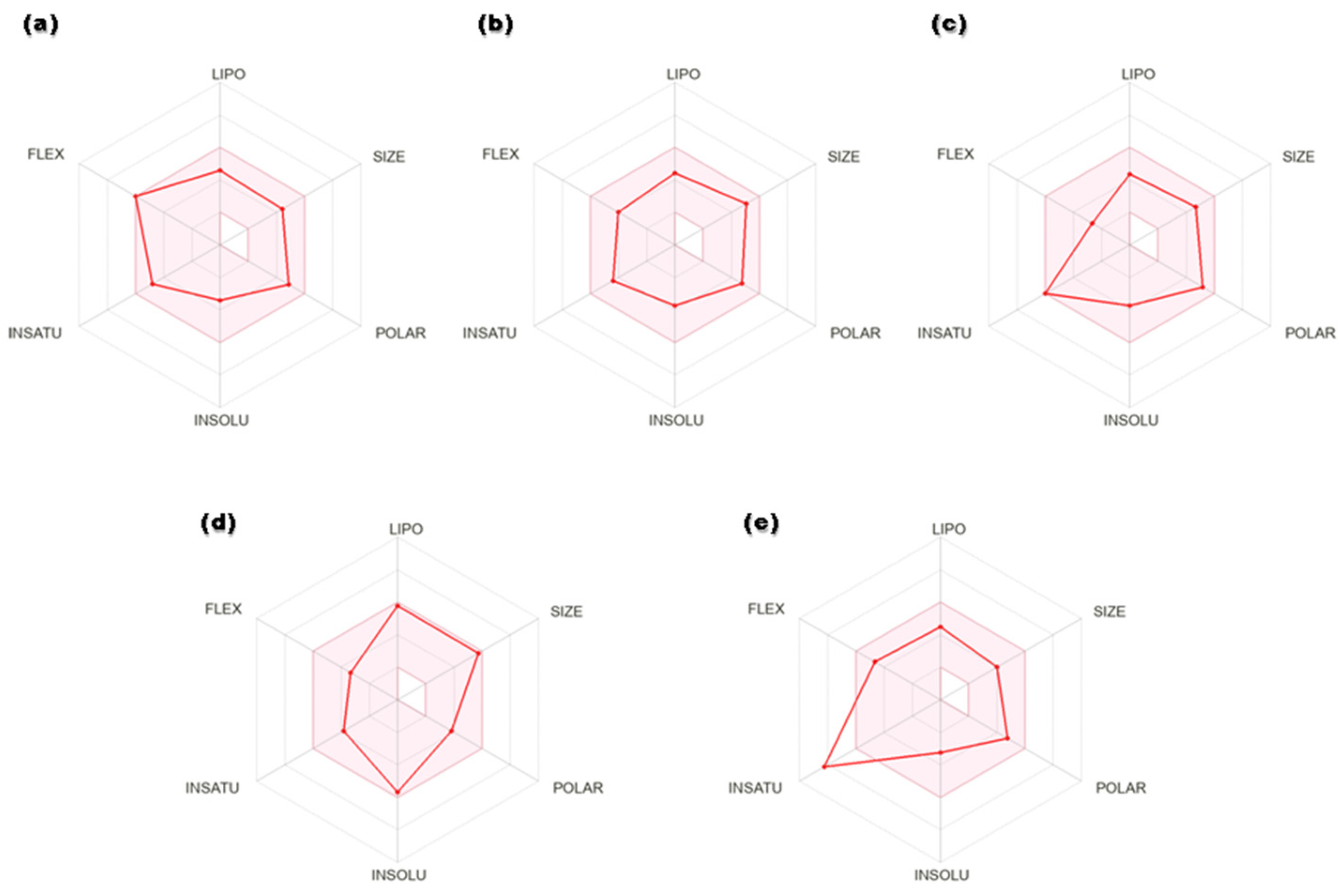
3.2. ADME Predictions and Toxicity Scan
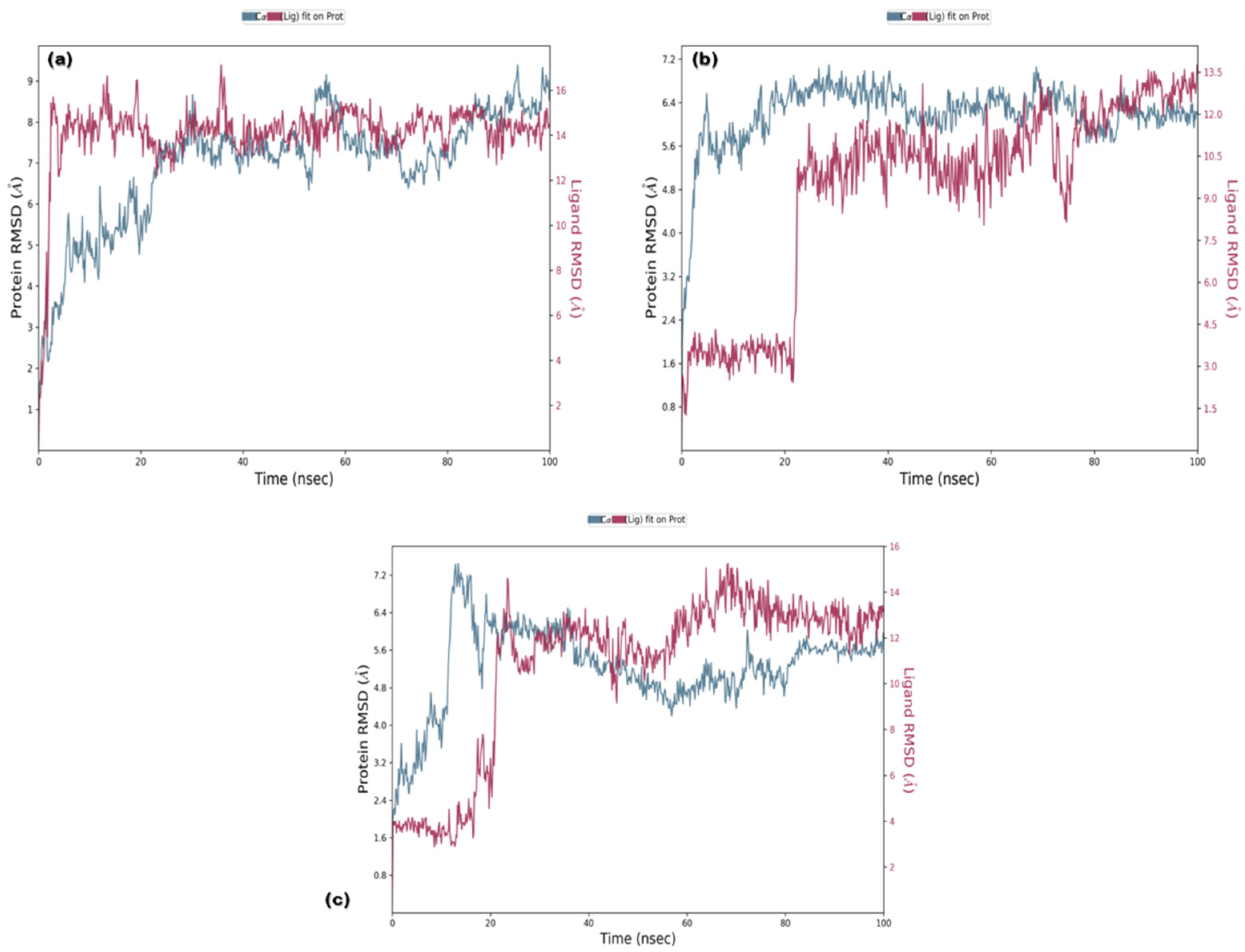
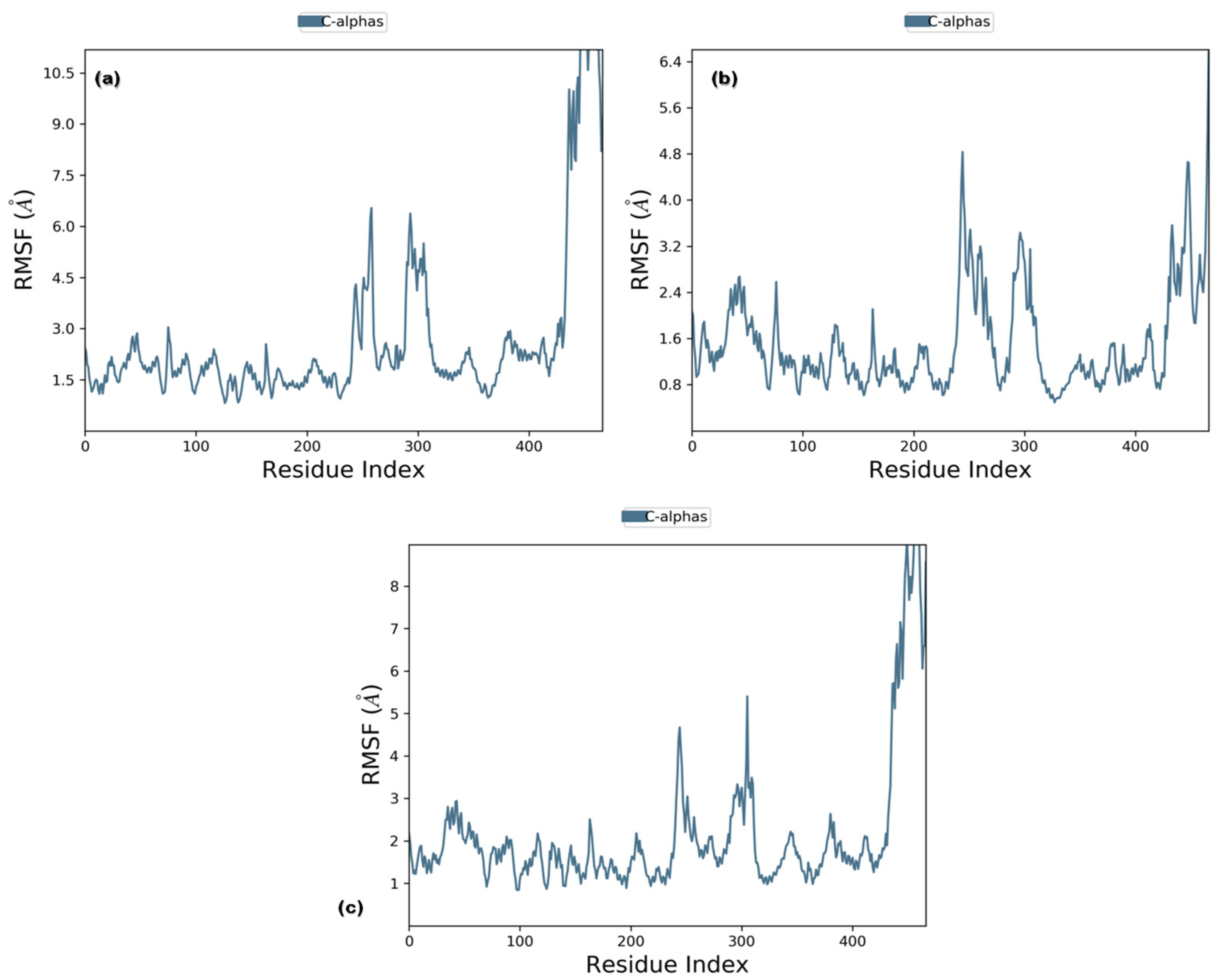
3.3. Molecular Dynamics (MD) Simulations
3.3.1. RMSD and RMSF
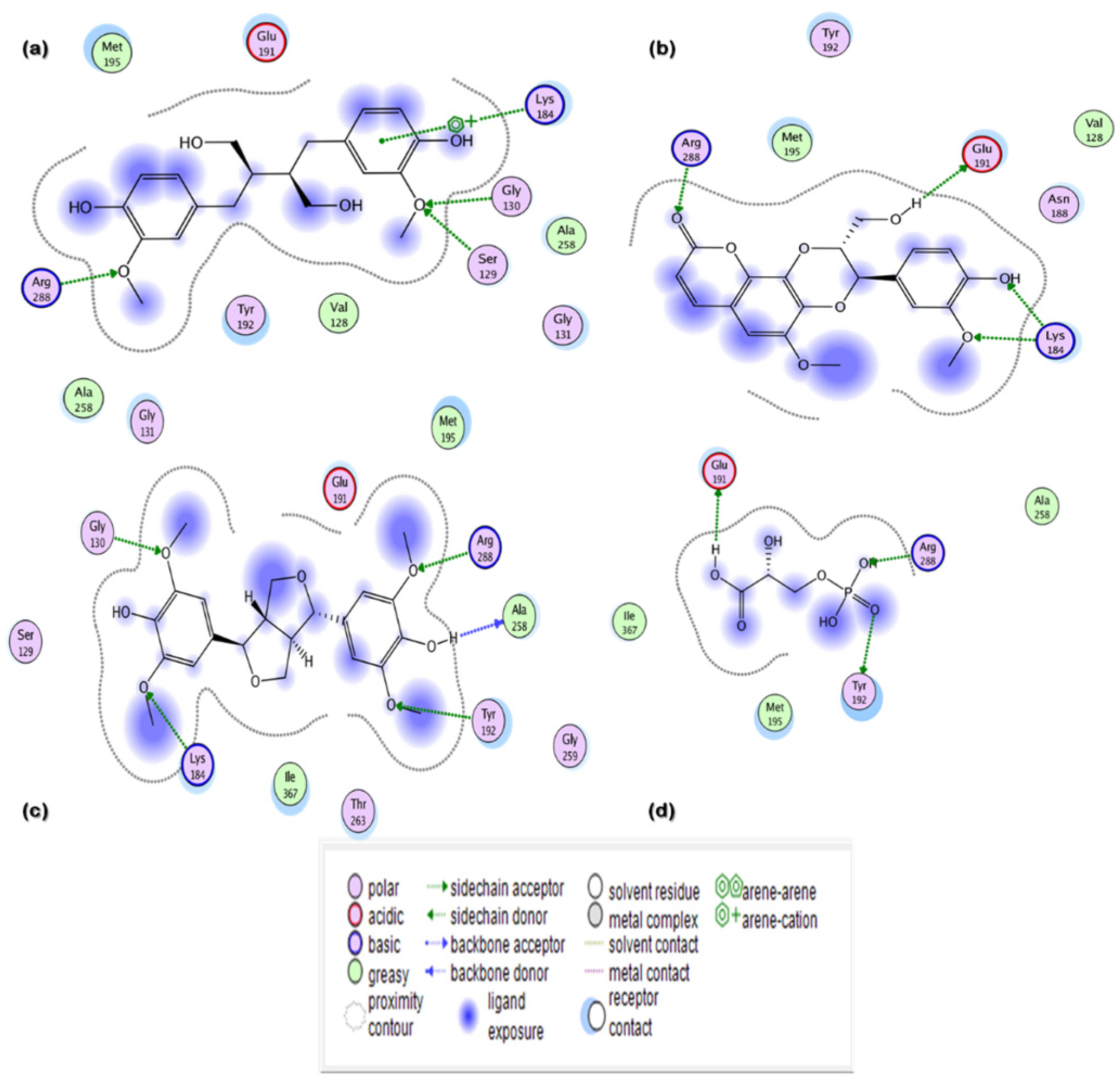
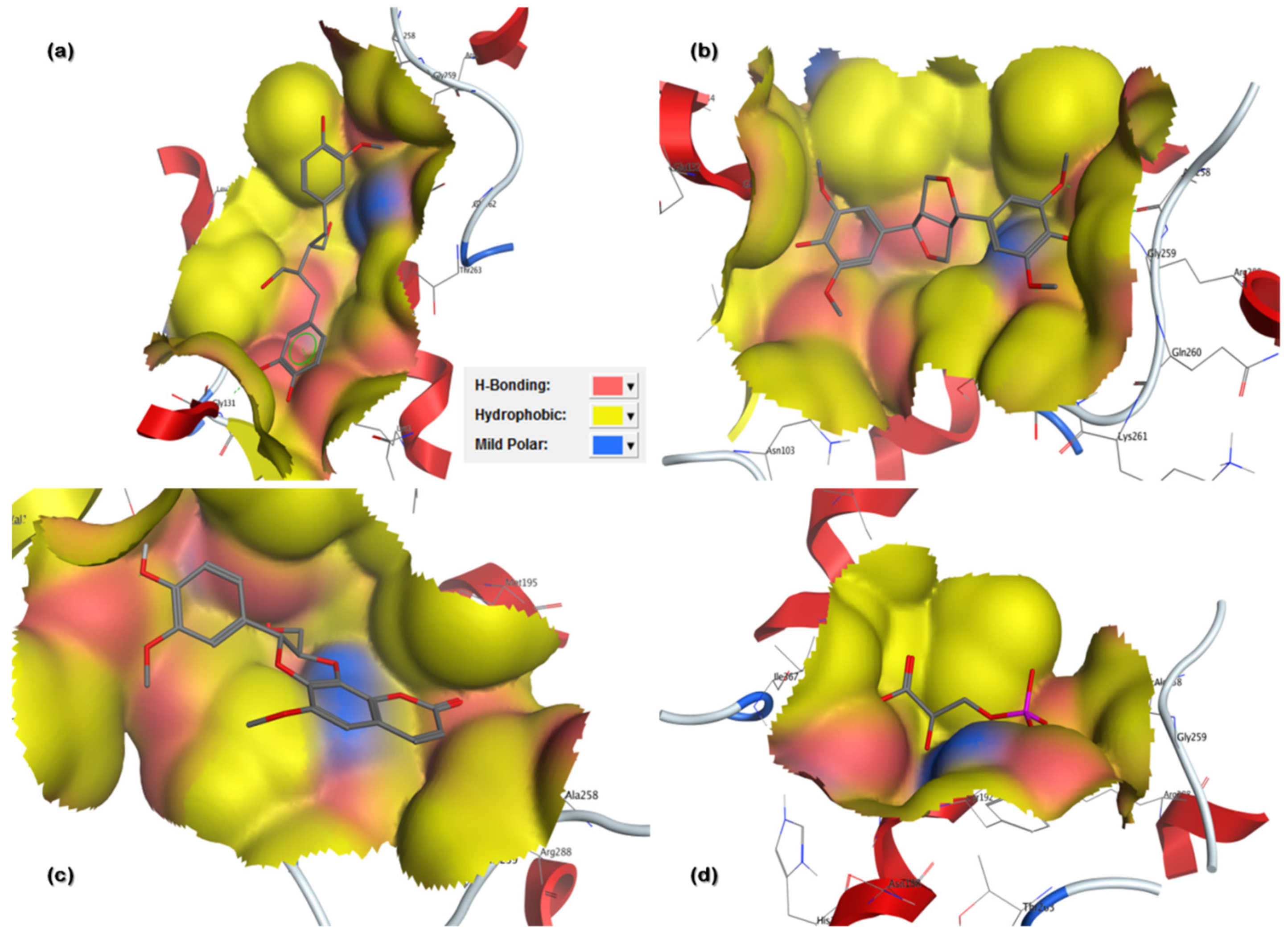
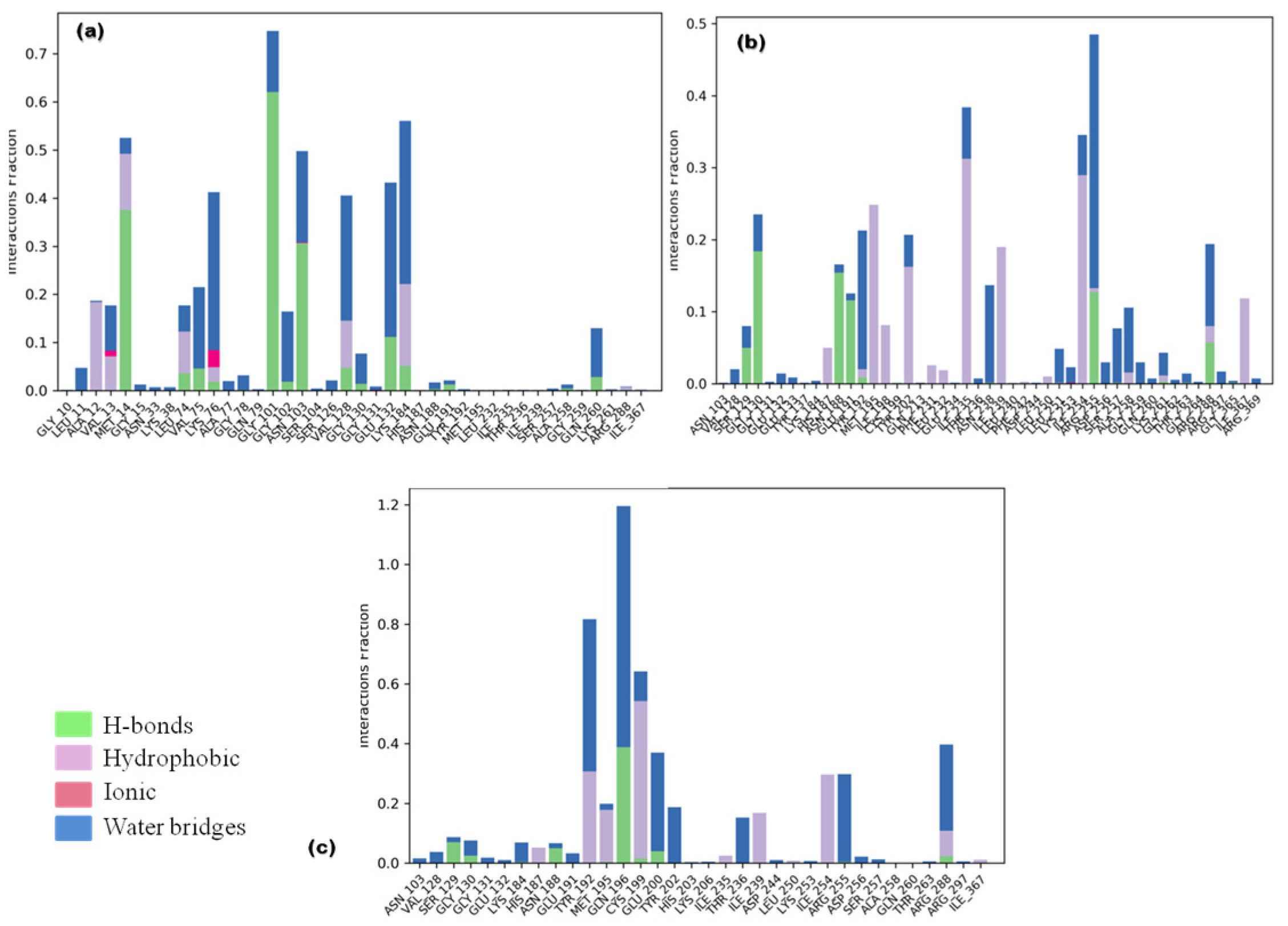
3.3.2. Ligand Contacts and Interactions
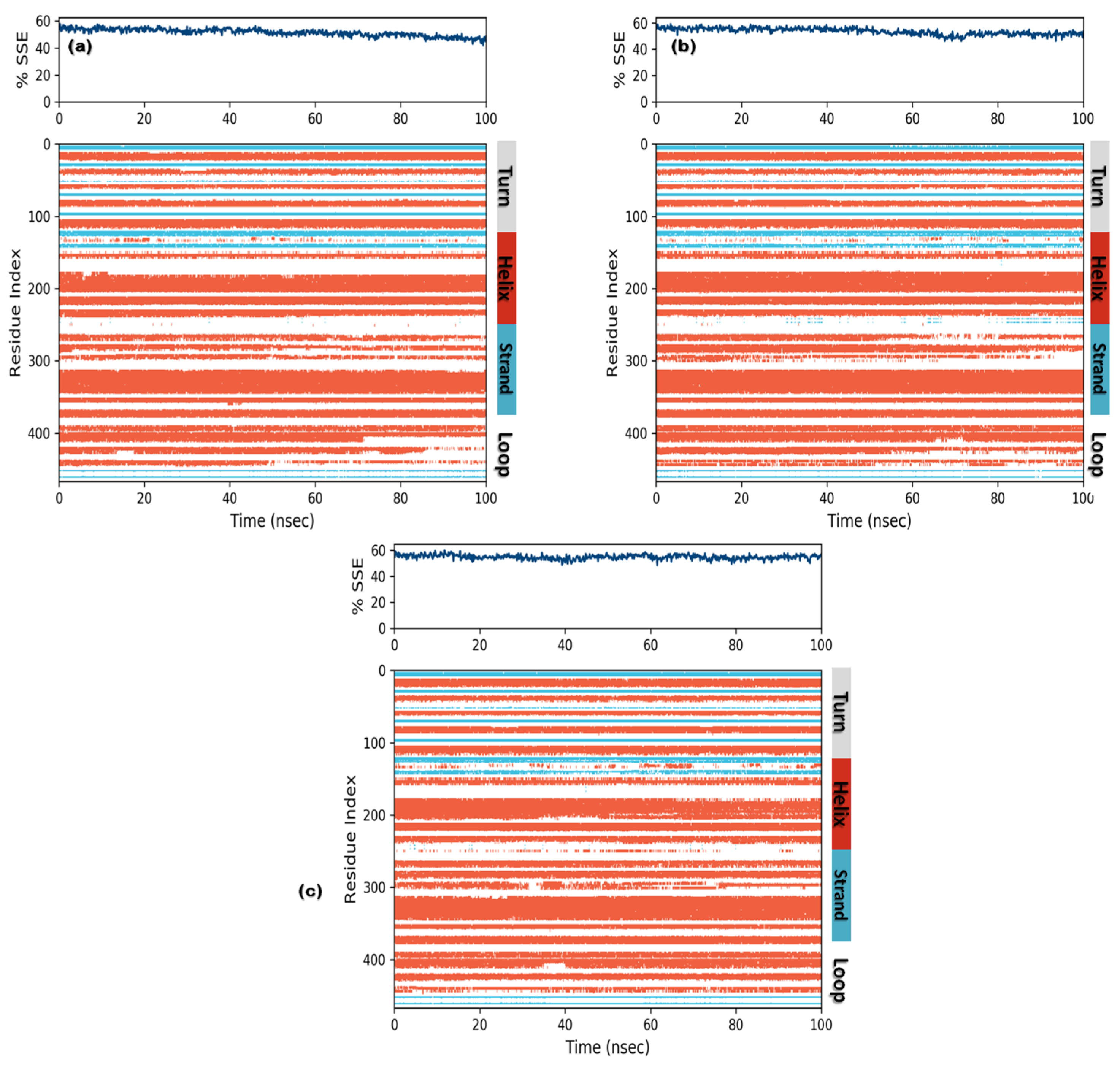
3.3.3. Protein Secondary Structure Elements (SSE)
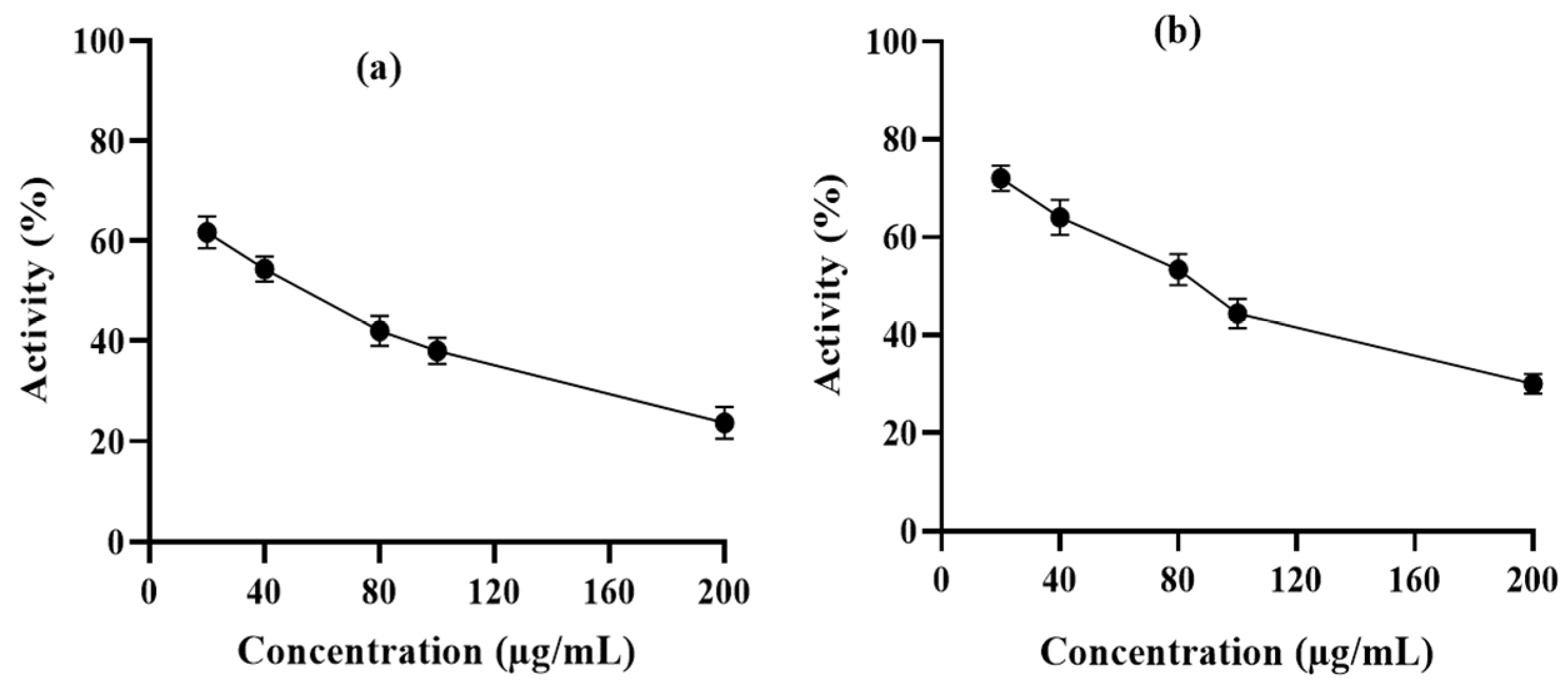
3.4. Enzymatic Assay Using Human Erythrocyte Purified 6-PGD
3.5. Syringaresinol and Cleomiscosin A Are 6-PGD Inhibitors
3.6. Cytotoxicity Prediction
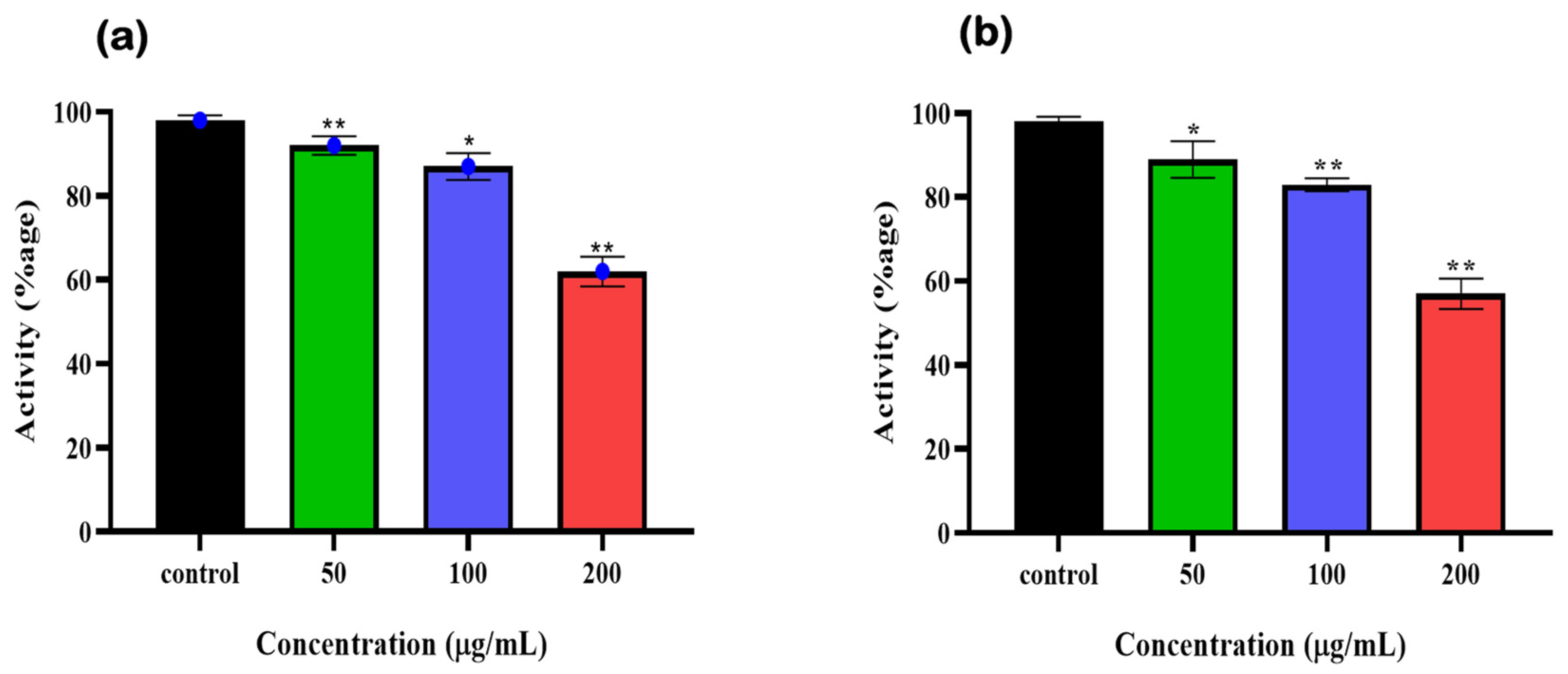
3.7. Cytotoxicity Assessment of Final Hits
3.8. Flow Cytometry for Apoptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, X.; Yu, H. Cancer issue: Global burden of cancer. Yale J. Biol. Med. 2006, 79, 85. [Google Scholar] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The pentose phosphate pathway and its involvement in cisplatin resistance. Int. J. Mol. Sci. 2020, 21, 937. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Zhou, Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.C.S. Microbial nutrition and basic metabolism. In Handbook of Water and Wastewater Microbiology; Academic Press: Cambridge, MA, USA, 2003; pp. 3–33. [Google Scholar]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free. Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Elf, S.; Lin, R.; Xia, S.; Pan, Y.; Shan, C.; Wu, S.; Lonial, S.; Gaddh, M.; Arellano, M.L.; Khoury, H.J. Targeting 6-phosphogluconate dehydrogenase in the oxidative PPP sensitizes leukemia cells to antimalarial agent dihydroartemisinin. Oncogene 2017, 36, 254–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Wu, D.; Bao, L.; Yin, T.; Lei, D.; Yu, J.; Tong, X. 6PGD inhibition sensitizes hepatocellular carcinoma to chemotherapy via AMPK activation and metabolic reprogramming. Biomed. Pharmacother. 2019, 111, 1353–1358. [Google Scholar] [CrossRef]
- Sarfraz, I.; Rasul, A.; Hussain, G.; Shah, M.A.; Zahoor, A.F.; Asrar, M.; Selamoglu, Z.; Ji, X.Y.; Adem, Ş.; Sarker, S.D.J.B. 6-Phosphogluconate dehydrogenase fuels multiple aspects of cancer cells: From cancer initiation to metastasis and chemoresistance. BioFactors 2020, 46, 550–562. [Google Scholar] [CrossRef]
- Yang, X.; Peng, X.; Huang, J. Inhibiting 6-phosphogluconate dehydrogenase selectively targets breast cancer through AMPK activation. Clin. Transl. Oncol. 2018, 20, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Elf, S.; Shan, C.; Kang, H.-B.; Ji, Q.; Zhou, L.; Hitosugi, T.; Zhang, L.; Zhang, S.; Seo, J.H. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat. Cell Biol. 2015, 17, 1484–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Xiang, Z.; Zhang, Y.; Sun, D. Inhibiting 6-phosphogluconate dehydrogenase enhances chemotherapy efficacy in cervical cancer via AMPK-independent inhibition of RhoA and Rac1. Clin. Transl. Oncol. 2019, 21, 404–411. [Google Scholar] [CrossRef]
- Alvarez, J.C. High-throughput docking as a source of novel drug leads. Curr. Opin. Chem. Biol. 2004, 8, 365–370. [Google Scholar] [CrossRef]
- Berry, M.; Fielding, B.; Gamieldien, J. Practical considerations in virtual screening and molecular docking. In Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 487–502. [Google Scholar]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Perez, S.; Tvaroška, I. Carbohydrate–protein interactions: Molecular modeling insights. In Advances in Carbohydrate Chemistry and Biochemistry; Elsevier: Amsterdam, The Netherlands, 2014; Volume 71, pp. 9–136. [Google Scholar]
- Ashfaq, U.A.; Mumtaz, A.; ul Qamar, T.; Fatima, T. MAPS Database: Medicinal plant activities, phytochemical and structural database. Bioinformation 2013, 9, 993. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2008; Volume 4, pp. 217–241. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Mumtaz, A.; Ashfaq, U.A.; Ul Qamar, M.T.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- ul Qamar, M.T.; Bari, A.; Adeel, M.M.; Maryam, A.; Ashfaq, U.A.; Du, X.; Muneer, I.; Ahmad, H.I.; Wang, J. Peptide vaccine against chikungunya virus: Immuno-informatics combined with molecular docking approach. J. Transl. Med. 2018, 16, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.-B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef] [Green Version]
- ul Qamar, M.T.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1433. [Google Scholar] [CrossRef] [Green Version]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Brice Landry, K.; Tariq, S.; Malik, A.; Sufyan, M.; Ashfaq, U.A.; Ijaz, B.; Shahid, A.A. Berberis lyceum and Fumaria indica: In vitro cytotoxicity, antioxidant activity, and in silico screening of their selected phytochemicals as novel hepatitis C virus nonstructural protein 5A inhibitors. J. Biomol. Struct. Dyn. 2021, 40, 7829–7851. [Google Scholar] [CrossRef] [PubMed]
- Adem, S.; Comakli, V.; Kuzu, M.; Demirdag, R. Investigation of the effects of some phenolic compounds on the Activities of glucose-6-phosphate dehydrogenase and 6-Phosphogluconate dehydrogenase from human erythrocytes. J. Biochem. Mol. Toxicol. 2014, 28, 510–514. [Google Scholar] [CrossRef]
- Ogihara, N.; Haley-Vicente, D. Protein target discovery and characterization-DS modeling and discovery studio streamline target discovery. Genet. Eng. News 2002, 22, 77. [Google Scholar]
- Polat, I.H.; Tarrado-Castellarnau, M.; Bharat, R.; Perarnau, J.; Benito, A.; Cortés, R.; Sabatier, P.; Cascante, M. Oxidative pentose phosphate pathway enzyme 6-phosphogluconate dehydrogenase plays a key role in breast cancer metabolism. Biology 2021, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.M.; Rampogu, S.; Zeb, A.; Son, M.; Park, C.; Lee, G.; Yoon, S.; Baek, A.; Parameswaran, S.; Park, S.J. In Silico Study Probes Potential Inhibitors of Human Dihydrofolate Reductase for Cancer Therapeutics. J. Clin. Med. 2019, 8, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Di, J.; Rasul, A.; Zhao, C.; Millimouno, F.M.; Tsuji, I.; Iqal, F.; Malhi, M.; Li, X.; Li, J. Dracorhodin perchlorate induces apoptosis in bladder cancer cells through Bcl-2, Bcl-XL, survivin down-regulation and caspase-3 activation. Bangladesh J. Pharm. 2013, 8, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Hampel, R.; Ibald-Mulli, A.; Zareba, W.; Schmidt, G.; Schneider, R.; Rückerl, R.; Couderc, J.P.; Mykins, B.; Oberdörster, G.J.P.; et al. Changes in deceleration capacity of heart rate and heart rate variability induced by ambient air pollution in individuals with coronary artery disease. Part. Fibre Toxicol. 2010, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Krokhotin, A.; Dokholyan, N.V. Computational methods toward accurate RNA structure prediction using coarse-grained and all-atom models. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 553, pp. 65–89. [Google Scholar]
- Rasul, A.; Riaz, A.; Wei, W.; Sarfraz, I.; Hassan, M.; Li, J.; Asif, F.; Adem, Ş.; Bukhari, S.A.; Asrar, M.; et al. Mangifera indica extracts as novel PKM2 inhibitors for treatment of triple negative breast cancer. BioMed Res. Int. 2021, 2021, 5514669. [Google Scholar] [CrossRef]
- Rasul, A.; Yu, B.; Zhong, L.; Khan, M.; Yang, H.; Ma, T. Cytotoxic effect of evodiamine in SGC-7901 human gastric adenocarcinoma cells via simultaneous induction of apoptosis and autophagy. Oncol. Rep. 2012, 27, 1481–1487. [Google Scholar]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Corrigendum: Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharmacol. 2020, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Yi, E.H.; Li, Y.C.; Park, I.C.; Park, J.Y.; Ye, S.K. Anticancer activity of tubulosine through suppression of interleukin-6-induced janus kinase 2/signal transducer and activation of transcription 3 signaling. J. Breast Cancer 2019, 22, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Kadir, N.A.A.A.; Azlan, A.; Abas, F.; Ismail, I.S. Beneficial effect of supercritical carbon dioxide extracted (SC-CO2) Dabai (Canarium odontophyllum) pulp oil in hypercholesterolemia-induced SPF sprague-dawley rats. Nat. Prod. Commun. 2018, 13, 1934578X1801301205. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-X.; Shi, Q.; Xiong, Q.-B.; Prasain, J.K.; Tezuka, Y.; Hareyama, T.; Wang, Z.-T.; Tanaka, K.; Namba, T.; Kadota, S. Tribulusamide A and B, new hepatoprotective lignanamides from the fruits of Tribulus terrestris: Indications of cytoprotective activity in murine hepatocyte culture. Planta Med. 1998, 64, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, J.W.; Jang, H.; Le, T.P.L.; Kim, J.G.; Lee, M.S.; Hong, J.T.; Lee, M.K.; Hwang, B.Y. Phenolic amides from Tribulus terrestris and their inhibitory effects on nitric oxide production in RAW 264.7 cells. Arch. Pharmacal Res. 2018, 41, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Thuong, P.T.; Su, N.D.; Min, B.S.; Son, K.H.; Chang, H.W.; Kim, H.P.; Kang, S.S.; Sok, D.E.; Bae, K. Antioxidant activity of cleomiscosins A and C isolated fromAcer okamotoanum. Arch. Pharmacal Res. 2007, 30, 275–281. [Google Scholar] [CrossRef] [PubMed]
- David, J.; Barreiros, A.; David, J. Antioxidant phenylpropanoid esters of triterpenes from Dioclea lasiophylla. Pharm. Biol. 2004, 42, 36–38. [Google Scholar] [CrossRef]
- Céspedes, C.L.; Avila, J.G.; Garcıa, A.M.; Becerra, J.; Flores, C.; Aqueveque, P.; Bittner, M.; Hoeneisen, M.; Martinez, M.; Silva, M. Antifungal and antibacterial activities of Araucaria araucana (Mol.) K. Koch heartwood lignans. Z. Nat. C 2006, 61, 35–43. [Google Scholar] [CrossRef]
- Panyo, J.; Matsunami, K.; Panichayupakaranant, P. Bioassay-guided isolation and evaluation of antimicrobial compounds from Ixora megalophylla against some oral pathogens. Pharm. Biol. 2016, 54, 1522–1527. [Google Scholar] [CrossRef]
- Prasad, K. Antioxidant activity of secoisolariciresinol diglucoside-derived metabolites, secoisolariciresinol, enterodiol, and enterolactone. Int. J. Angiol. 2000, 9, 220–225. [Google Scholar] [CrossRef]
- Iizuka, T.; Nagumo, S.; Yotsumoto, H.; Moriyama, H.; Nagai, M.J.B.; Bulletin, P. Vasorelaxant effects of Acer nikoense extract and isolated coumarinolignans on rat aortic rings. Biol. Pharm. Bull. 2007, 30, 1164–1166. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Cohen, K.; Mama, Y.; Rosca, P.; Pinhasov, A.; Weinstein, A. 430 Executive functions and emotional processing deficits among synthetic cannabinoids users. Eur. Neuropsychopharmacol. 2019, 29, S305–S306. [Google Scholar] [CrossRef]
- Bayindir, S.; Temel, Y.; Ayna, A.; Ciftci, M. The synthesis of N-benzoylindoles as inhibitors of rat erythrocyte glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase. J. Biochem. Mol. Toxicol. 2018, 32, e22193. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Song, W.S.; Yoon, S.H.; Park, K.-Y.; Kim, M.-H. Syringaresinol suppresses excitatory synaptic transmission and picrotoxin-induced epileptic activity in the hippocampus through presynaptic mechanisms. Neuropharmacology 2018, 131, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.S.; Kumar, S.S.; Gottapu, S.; Hira, K. J O-Glucoside of natural cleomiscosin-A: An attenuator of pro-inflammatory cytokine production. Phytochem. Lett. 2018, 26, 83–87. [Google Scholar] [CrossRef]
- Gao, X.-X.; Gao, Y.-N.; Wang, D.-D.; Hu, G.-S.; Yan, T.; Jia, J.-M.; Wang, A.-H. Six novel lignanoids with complex structures from Sigesbeckia glabrescens Makino with their cytotoxic activities. Fitoterapia 2021, 148, 104799. [Google Scholar] [CrossRef] [PubMed]
- Teles, H.L.; Hemerly, J.P.; Pauletti, P.M.; Pandolfi, J.R.; Araújo, A.R.; Valentini, S.R.; Young, M.C.M.; Bolzani, V.D.S.; Silva, D. Cytotoxic lignans from the stems of Styrax camporum (Styracaceae). Nat. Prod. Res. 2005, 19, 319–323. [Google Scholar] [CrossRef]
- Chang, F.-P.; Chao, W.; Wang, S.-Y.; Huang, H.-C.; Sung, P.-J.; Chen, J.-J.; Cheng, M.-J.; Huang, G.-J.; Kuo, Y.-H. Three new iridoid derivatives have been isolated from the stems of Neonauclea reticulata (Havil.) Merr. with cytotoxic activity on hepatocellular carcinoma cells. Molecules 2018, 23, 2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G.J.A. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Park, B.-Y.; Oh, S.-R.; Ahn, K.-S.; Kwon, O.-K.; Lee, H.-K. (–)-Syringaresinol inhibits proliferation of human promyelocytic HL-60 leukemia cells via G1 arrest and apoptosis. Int. Immunopharmacol. 2008, 8, 967–973. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | CID | Score | RMSD | Binding Site Residual Interaction | Hydrogen Bonds |
|---|---|---|---|---|---|
| Secoisolariciresinol | 65,373 | −12.9507 | 1.4987 | Arg288, Lys184, Gly130, Ser129 | 3 |
| Syringaresinol | 100,067 | −10.3479 | 1.6195 | Tyr192, Arg288, Lys184, Ala258, Gly130 | 5 |
| Cleomiscosin A | 442,510 | −10.3156 | 1.0467 | Arg288, Glu191, Lys184 | 4 |
| Tubulosine | 72,341 | −10.0725 | 1.7187 | Tyr192, Lys184 | 2 |
| Terrestriamide | 5,321,824 | −10.0456 | 1.7236 | Arg288, Lys184 | 2 |
| 3-Phosphoglyceric acid | Natural inhibitor | −8.4342 | 1.3230 | Tyr192, Arg288, Glu191 | 3 |
| Compounds | Lipinski’s RO5 | ||||||
|---|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | Number of HBA | Number of HBD | MlogP | Veber’s Rule | Violations | ||
| TPSA (Å) | RB | ||||||
| Syringaresinol | 418 | 8 | 2 | 2.62 | 95.86 | 6 | 0 |
| Cleomiscosin A | 386.3 | 8 | 2 | 1.96 | 107.60 | 4 | 0 |
| Secoisolariciresinol | 362.4 | 6 | 4 | 2.8 | 99.3 | 9 | 0 |
| Terrestriamide | 327.3 | 6 | 3 | 1.78 | 95.6 | 6 | 0 |
| Tubulosine | 475.6 | 6 | 3 | 4.86 | 69.8 | 5 | 0 |
| Compounds | |||||
|---|---|---|---|---|---|
| Parameters | Syringaresinol | Tubulosine | Secoisolariciresinol | Terrestriamide | Cleomiscosin A |
| Absorption | |||||
| GI absorption | High | High | High | High | High |
| Water solubility (log mol/L) | −3.92 | −3.41 | −3.76 | −3.13 | −3.64 |
| Skin permeability (cm/s) | −7.27 | −5.93 | −6.72 | −6.65 | −7.15 |
| P-gp substrate | No | Yes | No | No | No |
| P-gp inhibitor | Yes | Yes | No | No | Yes |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Distribution | |||||
| BBB | No | Yes | No | No | No |
| Subcellular localization | Mitochondria | Mitochondria | Mitochondria | Mitochondria | Mitochondria |
| Metabolism | |||||
| CYP1A2 inhibitor | No | No | Yes | No | No |
| CYP2C19 inhibitor | Yes | No | Yes | No | No |
| CYP2C9 inhibitor | Yes | No | No | No | No |
| CYP2D6 inhibitor | No | Yes | No | No | No |
| CYP3A4 inhibitor | Yes | No | No | Yes | No |
| Excretion | |||||
| Total clearance (log mL/min/kg) | 0.255 | 1.082 | 0.248 | 0.211 | 0.394 |
| Toxicity | |||||
| Carcinogenicity | No | No | No | No | No |
| Toxicity [36] | No | No | No | No | No |
| Hepatotoxicity | No | No | No | No | No |
| Oral rat acute toxicity (LD50 mol/kg) | 2.59 | 2.76 | 2.03 | 2.11 | 2.7031 |
| Synthetic accessibility | 4.36 | 4.95 | 3.21 | 2.55 | 4.47 |
| Compounds | Cancer Cell Line | Pa | Pi | Tumor Type |
|---|---|---|---|---|
| Syringaresinol | HOP-18 | 0.559 | 0.006 | Non-small-cell lung carcinoma |
| PC-6 | 0.549 | 0.020 | --- | |
| A549 | 0.561 | 0.047 | Small epithelial cell lung carcinoma | |
| NCI-H187 | 0.434 | 0.043 | Non-small-cell lung carcinoma | |
| MDA-MB-453 | 0.426 | 0.053 | Breast adenocarcinoma | |
| NCI-H838 | 0.452 | 0.083 | Small cell lung carcinoma | |
| HCC-2998 | 0.404 | 0.035 | Colon adenocarcinoma | |
| OVCAR-4 | 0.407 | 0.046 | Ovarian adenocarcinoma | |
| H9 | 0.358 | 0.030 | Leukemia | |
| NALM-6 | 0.383 | 0.061 | Leukemia | |
| DMS-114 | 0.421 | 0.104 | --- | |
| CFPAC-1 | 0.357 | 0.071 | Pancreatic carcinoma | |
| NCI-H322M | 0.349 | 0.067 | Non-small-cell lung carcinoma | |
| OVCAR-5 | 0.347 | 0.067 | Ovarian adenocarcinoma | |
| MKN-7 | 0.301 | 0.047 | Gastric carcinoma | |
| HCT-15 | 0.311 | 0.068 | Colon adenocarcinoma | |
| HL-60 | 0.303 | 0.085 | Leukemia | |
| Cleomiscosin A | HL-60 | 0.642 | 0.013 | Leukemia |
| MCF-7 | 0.567 | 0.038 | Breast carcinoma | |
| PC-6 | 0.407 | 0.039 | Non-small-cell lung carcinoma | |
| NALM-6 | 0.388 | 0.051 | Leukemia | |
| A549 | 0.358 | 0.103 | Small epithelial cell lung carcinoma | |
| Secoisolariciresinol | MDA-MB-453 | 0.443 | 0.033 | Breast adenocarcinoma |
| PC-6 | 0.389 | 0.044 | Small cell lung carcinoma | |
| HOP-18 | 0.339 | 0.053 | Non-small-cell lung carcinoma | |
| HCT-15 | 0.341 | 0.056 | Adenocarcinoma | |
| SK-MEL-2 | 0.306 | 0.078 | Melanoma | |
| A549 | 0.250 | 0.166 | Small epithelial cell lung carcinoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, G.B.; Qasim, M.; Rasul, A.; Ashfaq, U.A.; Alnuqaydan, A.M. Identification of Lignan Compounds as New 6-Phosphogluconate Dehydrogenase Inhibitors for Lung Cancer. Metabolites 2023, 13, 34. https://doi.org/10.3390/metabo13010034
Khan GB, Qasim M, Rasul A, Ashfaq UA, Alnuqaydan AM. Identification of Lignan Compounds as New 6-Phosphogluconate Dehydrogenase Inhibitors for Lung Cancer. Metabolites. 2023; 13(1):34. https://doi.org/10.3390/metabo13010034
Chicago/Turabian StyleKhan, Gul Bushra, Muhammad Qasim, Azhar Rasul, Usman Ali Ashfaq, and Abdullah M. Alnuqaydan. 2023. "Identification of Lignan Compounds as New 6-Phosphogluconate Dehydrogenase Inhibitors for Lung Cancer" Metabolites 13, no. 1: 34. https://doi.org/10.3390/metabo13010034