
Chromatomass-Spectrometric Method for the Quantitative Determination of Amino- and Carboxylic Acids in Biological Samples
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Analytical Equipment
2.3. Standards
2.4. Esterification of Amino Acids
2.5. Esterification of Carboxylic Acids
2.6. Biological Samples
2.7. Conditions for Chromatographic Separation and Triple Quadrupole Mass Spectrometric Signal Detection
2.8. Conditions for Chromatographic Separation and Quadrupole Time-of-Flight Mass Spectrometric Signal Detection
3. Results
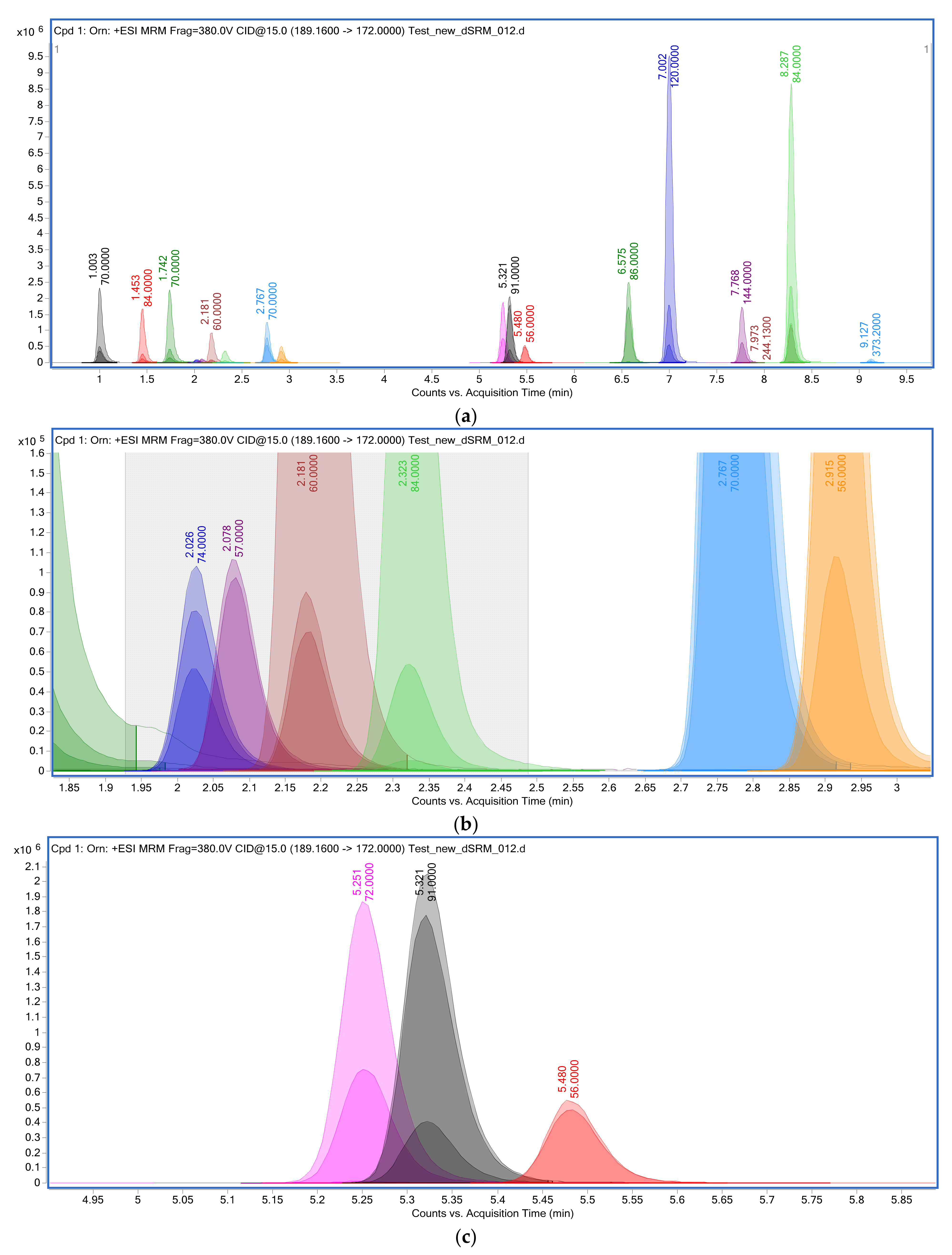
3.1. HPLC-MS/MS Measurements of Amino Acids and Their Derivatized Forms
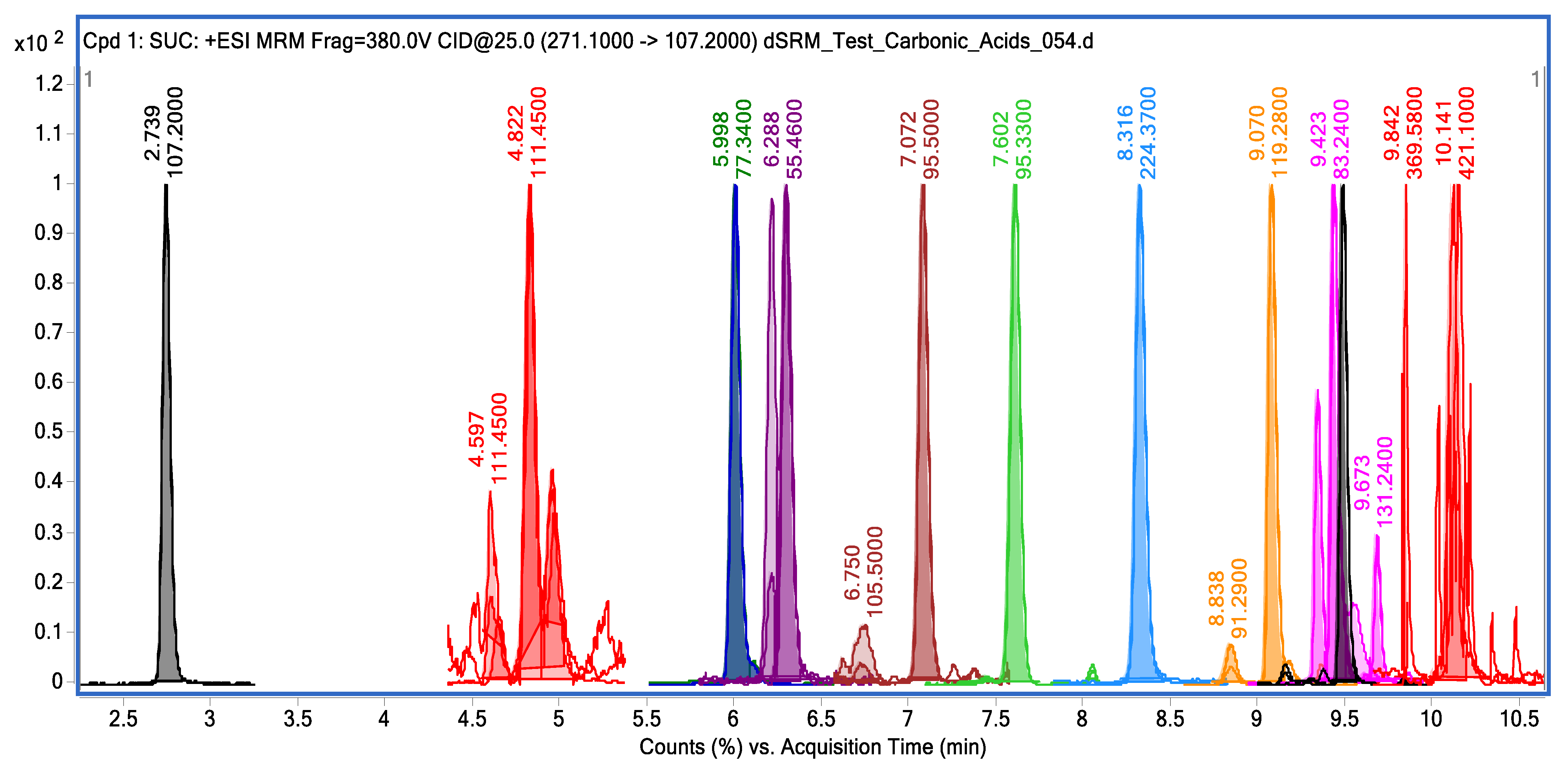
3.2. HPLC-MS/MS Measurements of Carboxylic Acids and Their Derivatized Forms
3.3. Development of Multi-Parameter Methods for Quantitative HPLC-MS/MS Monitoring of Multiple Reactions Analysis of Carboxylic Acids in Solution
3.4. HPLC-MS Analysis of Metabolites in Cell Culture Samples
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chace, D.H.; Lim, T.; Hansen, C.R.; Adam, B.W.; Hannon, W.H. Quantification of Malonylcarnitine in Dried Blood Spots by Use of MS/MS Varies by Stable Isotope Internal Standard Composition. Clin. Chim. Acta Int. J. Clin. Chem. 2009, 402, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Chace, D.H. Mass Spectrometry in Newborn and Metabolic Screening: Historical Perspective and Future Directions. J. Mass Spectrom. JMS 2009, 44, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Chace, D.H.; Millington, D.S.; Terada, N.; Kahler, S.G.; Roe, C.R.; Hofman, L.F. Rapid Diagnosis of Phenylketonuria by Quantitative Analysis for Phenylalanine and Tyrosine in Neonatal Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 1993, 39, 66–71. [Google Scholar] [CrossRef]
- Chace, D.H.; Hillman, S.L.; Van Hove, J.L.; Naylor, E.W. Rapid Diagnosis of MCAD Deficiency: Quantitative Analysis of Octanoylcarnitine and Other Acylcarnitines in Newborn Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 1997, 43, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Chace, D.H.; Pons, R.; Chiriboga, C.A.; McMahon, D.J.; Tein, I.; Naylor, E.W.; De Vivo, D.C. Neonatal Blood Carnitine Concentrations: Normative Data by Electrospray Tandem Mass Spectometry. Pediatr. Res. 2003, 53, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American College of Medical Genetics Newborn Screening Expert Group. Newborn Screening: Toward a Uniform Screening Panel and System—Executive Summary. Pediatrics 2006, 117 Pt 2, S296–S307. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, R.A.; Lawson, A.M. Organic Acids in Man: Analytical Chemistry, Biochemistry, and Diagnosis of the Organic Acidurias; Chapman and Hall: London, UK; New York, NY, USA, 1982. [Google Scholar]
- Lehotay, D.C.; Clarke, J.T. Organic Acidurias and Related Abnormalities. Crit. Rev. Clin. Lab. Sci. 1995, 32, 377–429. [Google Scholar] [CrossRef]
- APHL. Guidance for Laboratory Biomonitoring Programs; APHL: Silver Spring, MD, USA; p. 103.
- Blau, N.; Duran, M.; Blaskovics, M.E.; Gibson, K.M. (Eds.) Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Søvik, O. Inborn Errors of Amino Acid and Fatty Acid Metabolism with Hypoglycemia as a Major Clinical Manifestation. Acta Paediatr. Scand. 1989, 78, 161–170. [Google Scholar] [CrossRef]
- Gallagher, R.C.; Pollard, L.; Scott, A.I.; Huguenin, S.; Goodman, S.; Sun, Q. Laboratory Analysis of Organic Acids, 2018 Update: A Technical Standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 683–691. [Google Scholar] [CrossRef]
- Iwase, H.; Ozawa, S.; Ikuta, M.; Ono, I. Determination of Amino Acids in Human Plasma by Liquid Chromatography with Postcolumn Ninhydrin Derivatization Using a Hydroxyapatite Cartridge for Precolumn Deproteination. J. Chromatogr. B Biomed. Appl. 1995, 663, 15–24. [Google Scholar] [CrossRef]
- Armstrong, M.; Jonscher, K.; Reisdorph, N.A. Analysis of 25 Underivatized Amino Acids in Human Plasma Using Ion-Pairing Reversed-Phase Liquid Chromatography/Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. RCM 2007, 21, 2717–2726. [Google Scholar] [CrossRef] [PubMed]
- Dietzen, D.J.; Weindel, A.L.; Carayannopoulos, M.O.; Landt, M.; Normansell, E.T.; Reimschisel, T.E.; Smith, C.H. Rapid Comprehensive Amino Acid Analysis by Liquid Chromatography/Tandem Mass Spectrometry: Comparison to Cation Exchange with Post-Column Ninhydrin Detection. Rapid Commun. Mass Spectrom. RCM 2008, 22, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
- Casetta, B.; Tagliacozzi, D.; Shushan, B.; Federici, G. Development of a Method for Rapid Quantitation of Amino Acids by Liquid Chromatography-Tandem Mass Spectrometry (LC-MSMS) in Plasma. Clin. Chem. Lab. Med. 2000, 38, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, H.; Dettmer, K.; Gronwald, W.; Oefner, P.J. Advances in Amino Acid Analysis. Anal. Bioanal. Chem. 2009, 393, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Meininger, C.J. Analysis of Citrulline, Arginine, and Methylarginines Using High-Performance Liquid Chromatography. Methods Enzymol. 2008, 440, 177–189. [Google Scholar] [CrossRef]
- Shurubor, Y.I.; Cooper, A.J.L.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Krasnikov, B.F. Simultaneous Determination of Tricarboxylic Acid Cycle Metabolites by High-Performance Liquid Chromatography with Ultraviolet Detection. Anal. Biochem. 2016, 503, 8–10. [Google Scholar] [CrossRef]
- Le, A.; Ng, A.; Kwan, T.; Cusmano-Ozog, K.; Cowan, T.M. A Rapid, Sensitive Method for Quantitative Analysis of Underivatized Amino Acids by Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS). J. Chromatogr. B 2014, 944, 166–174. [Google Scholar] [CrossRef]
- Kambhampati, S.; Li, J.; Evans, B.S.; Allen, D.K. Accurate and Efficient Amino Acid Analysis for Protein Quantification Using Hydrophilic Interaction Chromatography Coupled Tandem Mass Spectrometry. Plant Methods 2019, 15, 46. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Tu, M.-J.; Zhang, C.; Jilek, J.L.; Zhang, Q.-Y.; Yu, A.-M. A Reliable LC-MS/MS Method for the Quantification of Natural Amino Acids in Mouse Plasma: Method Validation and Application to a Study on Amino Acid Dynamics during Hepatocellular Carcinoma Progression. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2019, 1124, 72–81. [Google Scholar] [CrossRef]
- Phipps, W.S.; Crossley, E.; Boriack, R.; Jones, P.M.; Patel, K. Quantitative Amino Acid Analysis by Liquid Chromatography-tandem Mass Spectrometry Using Low Cost Derivatization and an Automated Liquid Handler. JIMD Rep. 2019, 51, 62–69. [Google Scholar] [CrossRef]
- Song, C.; Zhang, S.; Ji, Z.; Li, Y.; You, J. Accurate Determination of Amino Acids in Serum Samples by Liquid Chromatography–Tandem Mass Spectrometry Using a Stable Isotope Labeling Strategy. J. Chromatogr. Sci. 2015, 53, 1536–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.M.; Linforth, R.S.; Wilkinson, S.J.; Smart, K.A.; Cook, D.J. Rapid Analysis of Formic Acid, Acetic Acid, and Furfural in Pretreated Wheat Straw Hydrolysates and Ethanol in a Bioethanol Fermentation Using Atmospheric Pressure Chemical Ionisation Mass Spectrometry. Biotechnol. Biofuels 2011, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, L.N.; Becker, C.; Chambliss, C.K. Analytical Characterization of Fermentation Inhibitors in Biomass Pretreatment Samples Using Liquid Chromatography, UV-Visible Spectroscopy, and Tandem Mass Spectrometry. Methods Mol. Biol. Clifton NJ 2009, 581, 125–143. [Google Scholar] [CrossRef]
- Kerem, Z.; Bravdo, B.-A.; Shoseyov, O.; Tugendhaft, Y. Rapid Liquid Chromatography-Ultraviolet Determination of Organic Acids and Phenolic Compounds in Red Wine and Must. J. Chromatogr. A 2004, 1052, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Scarlata, C.J.; Hyman, D.A. Development and Validation of a Fast High Pressure Liquid Chromatography Method for the Analysis of Lignocellulosic Biomass Hydrolysis and Fermentation Products. J. Chromatogr. A 2010, 1217, 2082–2087. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass. 2012. Available online: http://www.nrel.gov/biomass/analytical_procedures.html (accessed on 13 September 2022).
- Chen, Z.; Kim, K.-R.; Owens, G.; Naidu, R. Determination of Carboxylic Acids from Plant Root Exudates by Ion Exclusion Chromatography with ESI-MS. Chromatographia 2008, 67, 113–117. [Google Scholar] [CrossRef]
- Gamoh, K.; Saitoh, H.; Wada, H. Improved Liquid Chromatography/Mass Spectrometric Analysis of Low Molecular Weight Carboxylic Acids by Ion Exclusion Separation with Electrospray Ionization. Rapid Commun. Mass Spectrom. RCM 2003, 17, 685–689. [Google Scholar] [CrossRef]
- Flores, P.; Hellín, P.; Fenoll, J. Determination of Organic Acids in Fruits and Vegetables by Liquid Chromatography with Tandem-Mass Spectrometr. Food Chem. 2012, 132, 1049–1054. [Google Scholar] [CrossRef]
- Olson, K.C.; Chen, G.; Lynch, C.J. Quantification of Branched-Chain Keto Acids in Tissue by Ultra Fast Liquid Chromatography–Mass Spectrometry. Anal. Biochem. 2013, 439, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Michopoulos, F.; Whalley, N.; Theodoridis, G.; Wilson, I.D.; Dunkley, T.P.J.; Critchlow, S.E. Targeted Profiling of Polar Intracellular Metabolites Using Ion-Pair-High Performance Liquid Chromatography and -Ultra High Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry: Applications to Serum, Urine and Tissue Extracts. J. Chromatogr. A 2014, 1349, 60–68. [Google Scholar] [CrossRef]
- Guo, L.; Worth, A.J.; Mesaros, C.; Snyder, N.W.; Glickson, J.D.; Blair, I.A. Diisopropylethylamine/Hexafluoroisopropanol-Mediated Ion-Pairing UHPLC-MS for Phosphate and Carboxylate Metabolite Analysis: Utility for Studying Cellular Metabolism. Rapid Commun. Mass Spectrom. RCM 2016, 30, 1835–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemkov, T.; Sun, K.; Reisz, J.A.; Yoshida, T.; Dunham, A.; Wen, E.Y.; Wen, A.Q.; Roach, R.C.; Hansen, K.C.; Xia, Y.; et al. Metabolism of Citrate and Other Carboxylic Acids in Erythrocytes As a Function of Oxygen Saturation and Refrigerated Storage. Front. Med. 2017, 4, 175. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Fast and Robust Quantification of Anionic and Aromatic Metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef] [PubMed]
- Development of a New LC-MS/MS Method for the Quantification of Keto Acids. Available online: https://www.jstage.jst.go.jp/article/jpchrom/35/3/35_2014.017/_article (accessed on 13 September 2022).
- Dietzen, D.J.; Rinaldo, P.; Whitley, R.J.; Rhead, W.J.; Hannon, W.H.; Garg, U.C.; Lo, S.F.; Bennett, M.J. National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines: Follow-up Testing for Metabolic Disease Identified by Expanded Newborn Screening Using Tandem Mass Spectrometry; Executive Summary. Clin. Chem. 2009, 55, 1615–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, V.; Garbade, S.F.; Langhans, C.D.; Hoffmann, G.F.; Pollitt, R.J.; Downing, M.; Bonham, J.R. Qualitative Urinary Organic Acid Analysis: Methodological Approaches and Performance. J. Inherit. Metab. Dis. 2008, 31, 690–696. [Google Scholar] [CrossRef]
- Hoffmann, G.; Aramaki, S.; Blum-Hoffmann, E.; Nyhan, W.; Sweetman, L.; Hoffmann, G.; Aramaki, S.; Blum-Hoffmann, E.; Nyhan, W.L.; Sweetman, L. Quantitative Analysis for Organic Acids in Biological Samples: Batch Isolation Followed by Gas Chromatographic-Mass Spectrometric Analysis. Clin Chem 35: 587-595. Clin. Chem. 1989, 35, 587–595. [Google Scholar] [CrossRef]
- Chen, S.-F.; Mowery, R.A.; Castleberry, V.A.; van Walsum, G.P.; Chambliss, C.K. High-Performance Liquid Chromatography Method for Simultaneous Determination of Aliphatic Acid, Aromatic Acid and Neutral Degradation Products in Biomass Pretreatment Hydrolysates. J. Chromatogr. A 2006, 1104, 54–61. [Google Scholar] [CrossRef]
- Du, B.; Sharma, L.N.; Becker, C.; Chen, S.-F.; Mowery, R.A.; van Walsum, G.P.; Chambliss, C.K. Effect of Varying Feedstock-Pretreatment Chemistry Combinations on the Formation and Accumulation of Potentially Inhibitory Degradation Products in Biomass Hydrolysates. Biotechnol. Bioeng. 2010, 107, 430–440. [Google Scholar] [CrossRef]
- Goodman, S.I.; Markey, S.P. Diagnosis of Organic Acidemias by Gas Chromatography--Mass Spectrometry. Lab. Res. Methods Biol. Med. 1981, 6, 1–158. [Google Scholar]
- Peters, V.; Bonham, J.R.; Hoffmann, G.F.; Scott, C.; Langhans, C.-D. Qualitative Urinary Organic Acid Analysis: 10 Years of Quality Assurance. J. Inherit. Metab. Dis. 2016, 39, 683–687. [Google Scholar] [CrossRef]
- Turgeon, C.; Magera, M.J.; Allard, P.; Tortorelli, S.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Matern, D. Combined Newborn Screening for Succinylacetone, Amino Acids, and Acylcarnitines in Dried Blood Spots. Clin. Chem. 2008, 54, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.S.; Ozand, P.T.; Bucknall, M.P.; Little, D. Diagnosis of Inborn Errors of Metabolism from Blood Spots by Acylcarnitines and Amino Acids Profiling Using Automated Electrospray Tandem Mass Spectrometry. Pediatr. Res. 1995, 38, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shurubor, Y.I.; Matson, W.R.; Willett, W.C.; Hankinson, S.E.; Kristal, B.S. Biological Variability Dominates and Influences Analytical Variance in HPLC-ECD Studies of the Human Plasma Metabolome. BMC Clin. Pathol. 2007, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegner, H.M.; Mechtel, N.; Heidenreich, E.; Wirth, A.; Cortizo, F.G.; Bennewitz, K.; Fleming, T.; Andresen, C.; Freichel, M.; Teleman, A.A.; et al. Deep Metabolic Profiling Assessment of Tissue Extraction Protocols for Three Model Organisms. Front. Chem. 2022, 10, 869732. [Google Scholar] [CrossRef] [PubMed]
- Chilakala, S.; Mehtab, V.; Tallapally, M.; Vemula, M.; Shaikh, A.S.; Chenna, S.; Upadhyayula, V. SEC-MS/MS Determination of Amino Acids from Mango Fruits and Application of the Method for Studying Amino Acid Perturbations Due to Post Harvest Ripening. LWT 2021, 138, 110680. [Google Scholar] [CrossRef]
- Piraud, M.; Vianey-Saban, C.; Petritis, K.; Elfakir, C.; Steghens, J.-P.; Bouchu, D. Ion-Pairing Reversed-Phase Liquid Chromatography/Electrospray Ionization Mass Spectrometric Analysis of 76 Underivatized Amino Acids of Biological Interest: A New Tool for the Diagnosis of Inherited Disorders of Amino Acid Metabolism. Rapid Commun. Mass Spectrom. 2005, 19, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Browne, R.; Whitcomb, B. Procedures for Determination of Detection Limits: Application to High-Performance Liquid Chromatography Analysis of Fat-Soluble Vitamins in Human Serum. Epidemiology 2010. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Kuhara, T. α-Ketoglutaramate: An Overlooked Metabolite of Glutamine and a Biomarker for Hepatic Encephalopathy and Inborn Errors of the Urea Cycle. Metab. Brain Dis. 2014, 29, 991–1006. [Google Scholar] [CrossRef] [Green Version]
- Kuhara, T.; Akiyama, T.; Ohse, M.; Koike, T.; Shibasaki, J.; Imai, K.; Cooper, A.L. Identification of New Biomarkers of Pyridoxine-Dependent Epilepsy by GC/MS-Based Urine Metabolomics. Anal. Biochem. 2020, 604, 113739. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No. | Number of Compounds | Type of Sample * | Method | Analysis, Min | Method Features | LLOD (μM) | LLOQ (μM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | 33 | Plasma | LC-QQQ ** | 15 | – | 0.01-2.5 | 0.01–10 | [20] |
| 2 | 20 | Protein components of plant | LC-QLIT *** | 8 | 13C/15N-labeled standard | 0.1–11 | 0.12–35 | [21] |
| 3 | 28 | Serum, plasma, urine, cerebral spinal fluid, and dried blood spot | 2D-LC-MS/MS | 15 | PGC-LC **** | ≤0.1 | – | [20] |
| 4 | 20 | Mouse plasma | LC-QQQ | 13 | 13C/15N-labeled standard | 0.1–3 | – | [22] |
| 5 | 26 | Plasma, urine, cerebral spinal fluid | LC-QQQ | 11 | ATRAQ ***** | – | 2.5–10 | [23] |
| 6 | 6 | Serum | LC-QQQ | 11 | – | 0.001–0.0025 | 0.003–0.007 | [24] |
| No. | Number of Compounds | Type of Sample * | Method | Analysis, Min | Method Features | LLOD (μM) | LLOQ (μM) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | 3 | Mouse tissue | LC-QTOF ** | 6 | 13C-labeled standard | 0.005 | – | [33] |
| 2 | 30 | Cell culture media, cell extracts, tissue extracts, blood, urine | LC-QQQ *** | 21 | HILIC **** | – | <5 | [34] |
| 3 | 59 | Human melanoma cells | LC-QQQ | 20 | 13C/15N-labeled standard | – | – | [35] |
| 4 | 7 | Human red blood cell | LC-Orbitrap | >10 | 13C-labeled standard | – | – | [36] |
| 5 | 10 (total 138 metabolites) | Yeast | LC-QQQ | 36 | – | 0.001–3.7 | – | [37] |
| 6 | 10 | Rat plasma | LC-QQQ | 21.5 | – | 0.01–0.25 | 0.01–0.1 | [38] |
| Time, Min | % A | % B | Flow Rate, µL/Min |
|---|---|---|---|
| 00.0 | 98 | 2 | 0.30 |
| 01.7 | 98 | 2 | 0.30 |
| 02.2 | 85 | 12 | 0.30 |
| 06.0 | 73 | 27 | 0.30 |
| 07.8 | 55 | 45 | 0.30 |
| 09.0 | 55 | 45 | 0.30 |
| 11.0 | 2 | 98 | 0.30 |
| 12.5 | 2 | 98 | 0.30 |
| 12.7 | 30 | 70 | 0.30 |
| 13.0 | 98 | 2 | 0.30 |
| 13.5 | 98 | 2 | 0.30 |
| AA | Chromatographic Characteristics * | m/z | |||
|---|---|---|---|---|---|
| RT, Min | FWHM, s | As | A, cnts × s | ||
| Orn | 1.003 | 4.61 | 1.13 | 666771 | 222 |
| Ala | 1.091 | 3.12 | 1.08 | 189398 | 146 |
| Lys | 1.548 | 4.12 | 1.06 | 1195030 | 146 |
| Arg | 1.846 | 6.15 | 0.87 | 1109451 | 260 |
| Gly | 2.128 | 7.39 | 1.89 | 1515129 | 212 |
| Asn | 2.138 | 5.47 | 0.91 | 286435 | 162 |
| Ser | 2.299 | 6.39 | 0.75 | 832514 | 174 |
| Gln | 2.468 | 3.12 | 1.08 | 1054774 | 132 |
| Cit | 2.747 | 7.19 | 0.83 | 894130 | 188 |
| Thr | 3.027 | 6.24 | 1.19 | 1604746 | 206 |
| Val | 5.416 | 8.73 | 0.91 | 1391872 | 246 |
| Tyr | 5.521 | 5.91 | 1.09 | 972054 | 203 |
| Met | 5.637 | 6.39 | 1.23 | 366957 | 231 |
| Ile | 6.499 | 6.38 | 0.91 | 7682444 | 188 |
| Leu | 6.611 | 8.06 | 0.84 | 276501 | 176 |
| Phe | 6.996 | 5.15 | 1.01 | 1180098 | 172 |
| Asp | 7.884 | 4.89 | 1.21 | 1168386 | 189 |
| Trp | 8.278 | 4.22 | 0.88 | 410052 | 238 |
| Glu | 8.352 | 5.69 | 1.69 | 695427 | 203 |
| AA | T = 24 h | T = 72 h | T = 96 h | Degradation, % |
|---|---|---|---|---|
| Ala | 0.87 | 0.81 | 0.76 | 24 |
| Lys | 1.02 | 1 | 0.99 | 1 |
| Arg | 1.06 | 0.97 | 0.94 | 6 |
| Asn | 1 | 0.94 | 0.91 | 9 |
| Gly | 1 | 0.98 | 0.95 | 5 |
| Ser | 1.03 | 0.99 | 0.98 | 2 |
| Gln | 0.97 | 0.96 | 0.93 | 7 |
| Thr | 0.94 | 0.89 | 0.83 | 17 |
| Val | 0.99 | 0.98 | 0.95 | 5 |
| Tyr | 0.95 | 0.92 | 0.9 | 10 |
| Met | 0.9 | 0.74 | 0.66 | 34 |
| Leu | 1.01 | 0.92 | 0.94 | 6 |
| Phe | 1.09 | 1.02 | 1 | 0 |
| Asp | 0.97 | 0.94 | 0.88 | 12 |
| Trp | 0.92 | 0.88 | 0.83 | 17 |
| Glu | 0.97 | 0.92 | 0.9 | 10 |
| His | 0.93 | 0.92 | 0.94 | 6 |
| Pro | 1.1 | 0.97 | 0.97 | 3 |
| AA | Parent Ion, m/z | Retention Time, Min | Chromatographic Peak Height | Peak Width at Half Maximum, Min | Chromatographic Peak Area |
|---|---|---|---|---|---|
| Orn | 189.16 | 1.003 | 2,328,761 | 0.060 | 9,761,179 |
| Ala | 146.10 | 1.089 | 2,134,894 | 0.067 | 8,632,169 |
| Lys | 203.17 | 1.453 | 1,660,029 | 0.057 | 6,460,292 |
| Arg | 231.18 | 1.742 | 2,262,781 | 0.061 | 9,666,924 |
| Asn | 189.12 | 2.026 | 103,235 | 0.058 | 408,153 |
| Gly | 132 | 2.078 | 105,593 | 0.061 | 427,161 |
| Ser | 162.11 | 2.181 | 925,478 | 0.059 | 3,700,371 |
| Gln | 203.14 | 2.323 | 369,800 | 0.065 | 1,619,872 |
| Cit | 232.16 | 2.767 | 1,250,933 | 0.059 | 5,037,170 |
| Thr | 176.13 | 2.915 | 512,854 | 0.060 | 2,088,165 |
| Val | 174.15 | 5.251 | 1,864,356 | 0.061 | 7,702,974 |
| Tyr | 238.14 | 5.321 | 2,051,976 | 0.062 | 8,525,253 |
| Met | 206.12 | 5.480 | 546,499 | 0.065 | 2,375,335 |
| Leu | 188.16 | 6.575 | 2,486,617 | 0.064 | 10,641,134 |
| Phe | 222.14 | 7.002 | 9,538,221 | 0.074 | 46,234,568 |
| Asp | 246.17 | 7.768 | 1,722,700 | 0.064 | 7,551,718 |
| Trp | 261.16 | 8.285 | 1,146,979 | 0.070 | 5,360,110 |
| Glu | 260.18 | 8.287 | 8,682,020 | 0.072 | 41,459,023 |
| Bet | 393.21 | 9.127 | 107,109 | 0.066 | 464,904 |
| AA | k Slope Tangent | b Vertical Offset | r2 Correlation Coefficient | w Weight Factor | p Number of Points | L Number of Concentration Levels |
|---|---|---|---|---|---|---|
| Orn | 0.011669 | 0.002859 | 0.9975 | - | 21 | 7 |
| Ala | 0.087133 | 0.025649 | 0.9931 | - | 21 | 7 |
| Lys | 0.264227 | 0.054483 | 0.9987 | 1/x | 21 | 7 |
| Arg | 0.301652 | 0.589995 | 0.9876 | 1/x | 21 | 7 |
| Asn | 0.018444 | 0.002325 | 0.9994 | - | 21 | 7 |
| Gly | 0.036322 | 0.013624 | 0.9731 | 1/x2 | 21 | 7 |
| Ser | 0.212549 | 0.151202 | 0.9989 | - | 21 | 7 |
| Gln | 0.261127 | 0.135219 | 0.9985 | - | 21 | 7 |
| Cit | 0.024455 | 0.042102 | 0.9965 | 1/x | 21 | 7 |
| Thr | 0.102644 | 0.059708 | 0.9943 | 1/x | 21 | 7 |
| Val | 0.373563 | 0.277502 | 0.9830 | 1/x | 21 | 7 |
| Tyr | 0.291899 | 0.260901 | 0.9883 | 1/x | 21 | 7 |
| Met | 0.093068 | 0.073779 | 0.9918 | 1/x | 21 | 7 |
| Leu | 0.457484 | 0.768680 | 0.9513 | 1/x | 21 | 7 |
| Phe | 0.963174 | 6.473994 | 0.8625 | 1/x2 | 21 | 7 |
| Asp | 0.358717 | 0.312286 | 0.9796 | 1/x | 21 | 7 |
| Trp | 0.000045 | 0.000440 | 0.9687 | - | 21 | 7 |
| Glu | 2.399192 | 0.921144 | 0.9734 | 1/x2 | 15 | 5 |
| Carboxylic Acid | Chromatographic Characteristics * | m/z | |||
|---|---|---|---|---|---|
| RT, Min | FWHM, s | As | A, cnts × s | ||
| Succinic acid | 2.7 | 2.94 | 0.88 | 55,540 | 165.05 |
| Lactic acid | 4.8 | 4.08 | 1.11 | 14,045 | 225.05 |
| Pyruvic acid | 6.0 | 3.42 | 0.93 | 1,152,626 | 202.05 |
| Oxaloacetic acid | 6.3 | 3.72 | 0.94 | 31,212 | 285.1 |
| Isocitric acid | 7.1 | 3.60 | 1.12 | 32,330 | 421.15 |
| Taurine | 7.6 | 3.78 | 1.08 | 228,750 | 271.1 |
| Citric acid | 8.3 | 3.72 | 1.07 | 277,775 | 269.05 |
| α-Ketomethylbutyric acid | 9.1 | 2.88 | 0.97 | 1,226,6593 | 179.05 |
| α-Ketomethythiolbutyric acid | 9.4 | 2.58 | 1.09 | 473,609 | 193.28 |
| Malic acid | 9.5 | 2.58 | 1.01 | 278,520 | 167.05 |
| Fumaric acid | 10.1 | 3.42 | 1.11 | 126,475 | 165.05 |
| Carboxylic Acid | k Slope Tangent | b Vertical Offset | r2 Correlation Coefficient | w Weight Factor | p Number of Points | L Number of Concentration Levels |
|---|---|---|---|---|---|---|
| Succinic acid | 302.98 | 1654.2 | 0.9915 | - | 18 | 6 |
| Lactic acid | 73.312 | 1298.8 | 0.9667 | - | 18 | 6 |
| Pyruvic acid | 693.44 | 5815.8 | 0.9964 | - | 21 | 7 |
| Oxaloacetic acid | 156.49 | 2739.6 | 0.9866 | - | 21 | 7 |
| Isocitric acid | 145.06 | 18160 | 0.9602 | 1/x | 21 | 7 |
| Taurine | 124.72 | 33.636 | 0.9554 | 1/x | 18 | 6 |
| Citric acid | 165.17 | 1424.2 | 0.9990 | - | 21 | 7 |
| α-Ketobutyric acid | 217.54 | 1266.1 | 0.9987 | - | 21 | 7 |
| α-Ketomethylbutyric acid | 640.31 | 16027 | 0.9781 | - | 21 | 7 |
| α-Ketomethythiolbutyric acid | 265.23 | 1639.4 | 0.9976 | - | 21 | 7 |
| Malic acid | 155.42 | 1391.5 | 0.9820 | - | 18 | 6 |
| Fumaric acid | 561.54 | 4880.0 | 0.9959 | - | 21 | 7 |
| Primary Human Lung Fibroblast Cell Cultures | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Metabolite | 0 h | 24 h | 48 h | 72 h | 1 Week | 2 Weeks | ||||||
| Mean ± SD | CV,% | Mean ± SD | CV,% | Mean ± SD | CV,% | Mean ± SD | CV,% | Mean ± SD | CV,% | Mean ± SD | CV,% | |
| Amino acids | ||||||||||||
| Ornithine | 1.19 ± 0.00 | 0.24 | 10.49 ± 0.37 | 3.55 | 3.74 ± 0.02 | 0.59 | 1.29 ± 0.02 | 1.27 | 2.04 ± 0.08 | 3.84 | 7.44 ± 0.27 | 3.58 |
| Alanine | 2.10 ± 0.03 | 1.45 | 2.79 ± 0.10 | 3.45 | 1.25 ± 0.00 | 0.17 | 3.69 ± 0.04 | 1.02 | 1.36 ± 0.02 | 1.45 | 1.25 ± 0.02 | 1.64 |
| Lysine | 12.24 ± 0.52 | 4.27 | 15.18 ± 0.09 | 0.58 | 14.11 ± 0.00 | 0.03 | 17.47 ± 1.48 | 8.44 | 13.72 ± 0.80 | 5.83 | 6.25 ± 0.05 | 0.80 |
| Arginine | 19.25 ± 0.20 | 1.05 | 23.52 ± 0.05 | 0.20 | 20.34 ± 0.06 | 0.29 | 16.84 ± 0.06 | 0.38 | 17.07 ± 0.43 | 2.54 | 20.97 ± 0.49 | 2.35 |
| Asparagine | 18.67 ± 0.23 | 1.22 | 23.75 ± 2.28 | 9.59 | 20.21 ± 0.69 | 3.43 | 16.76 ± 0.47 | 2.79 | 16.83 ± 1.27 | 7.52 | 19.06 ± 0.25 | 1.31 |
| Glycine | 11.13 ± 0.03 | 0.24 | 12.88 ± 0.68 | 5.27 | 5.45 ± 0.13 | 2.39 | 7.65 ± 0.28 | 3.64 | 5.13 ± 0.09 | 1.68 | 1.13 ± 0.07 | 5.90 |
| Serine | 9.64 ± 0.75 | 7.81 | 10.62 ± 0.49 | 4.66 | 9.16 ± 0.17 | 1.84 | 6.72 ± 0.25 | 3.76 | 5.97 ± 0.02 | 0.36 | 5.25 ± 0.39 | 7.47 |
| Glutamine | 54.14 ± 2.31 | 4.27 | 50.12 ± 2.23 | 4.45 | 57.38 ± 4.00 | 6.98 | 44.28 ± 1.89 | 4.28 | 49.33 ± 3.70 | 7.49 | 49.34 ± 3.95 | 8.01 |
| Citrulline | 0.03 ± 0.00 | 6.53 | 0.06 ± 0.01 | 8.84 | 0.05 ± 0.00 | 6.87 | 0.02 ± 0.00 | 3.01 | 0.05 ± 0.00 | 8.16 | 0.10 ± 0.01 | 8.49 |
| Threonine | 11.58 ± 1.08 | 9.32 | 13.51 ± 0.90 | 6.63 | 13.86 ± 0.56 | 4.05 | 13.28 ± 0.15 | 1.15 | 14.27 ± 0.33 | 2.34 | 14.87 ± 0.08 | 0.56 |
| Valine | 1.45 ± 0.03 | 2.15 | 1.72 ± 0.04 | 2.22 | 1.71 ± 0.02 | 1.20 | 1.59 ± 0.03 | 2.18 | 1.56 ± 0.03 | 2.17 | 1.47 ± 0.06 | 3.84 |
| Tyrosine | 3.24 ± 0.09 | 2.82 | 3.77 ± 0.20 | 5.37 | 4.11 ± 0.08 | 1.98 | 4.71 ± 0.04 | 0.80 | 4.64 ± 0.01 | 0.15 | 4.41 ± 0.27 | 6.04 |
| Methionine (sulfoxide) | 1.39 ± 0.03 | 2.04 | 1.22 ± 0.03 | 2.27 | 0.91 ± 0.02 | 2.49 | 1.00 ± 0.01 | 0.78 | 5.93 ± 0.34 | 5.70 | 3.84 ± 0.03 | 0.88 |
| Selenomethionine | 0.71 ± 0.02 | 2.18 | 2.49 ± 0.06 | 2.50 | 3.03 ± 0.14 | 4.48 | 5.62 ± 0.10 | 1.77 | 6.37 ± 0.00 | 0.02 | 3.99 ± 0.13 | 3.14 |
| Leucine | 10.02 ± 0.34 | 3.37 | 10.31 ± 0.65 | 6.33 | 10.28 ± 0.18 | 1.78 | 10.10 ± 0.02 | 0.24 | 8.99 ± 0.60 | 6.72 | 8.26 ± 0.40 | 4.88 |
| Isoleucine | 10.21 ± 0.08 | 0.75 | 11.20 ± 0.61 | 5.44 | 10.86 ± 0.64 | 5.86 | 10.09 ± 0.00 | 0.02 | 9.80 ± 0.53 | 5.46 | 8.74 ± 0.28 | 3.15 |
| Phenylalanine | 2.13 ± 0.19 | 8.99 | 2.48 ± 0.09 | 3.62 | 2.44 ± 0.05 | 2.06 | 2.70 ± 0.14 | 5.15 | 2.71 ± 0.15 | 5.70 | 2.60 ± 0.10 | 3.84 |
| Aspartate | 7.79 ± 0.65 | 8.32 | 8.00 ± 0.25 | 3.17 | 5.83 ± 0.14 | 2.41 | 5.45 ± 0.36 | 6.60 | 4.49 ± 0.36 | 8.04 | 5.39 ± 0.16 | 3.03 |
| Tryptophane | 0.60 ± 0.00 | 0.82 | 0.59 ± 0.01 | 1.81 | 0.73 ± 0.02 | 2.15 | 0.62 ± 0.03 | 4.46 | 0.73 ± 0.06 | 8.46 | 0.75 ± 0.04 | 4.92 |
| Glutamate | 5.46 ± 0.24 | 4.39 | 11.50 ± 0.20 | 1.76 | 9.63 ± 0.19 | 2.00 | 8.93 ± 0.05 | 0.52 | 8.72 ± 0.45 | 5.19 | 8.73 ± 0.19 | 2.15 |
| Betaine | 1.46 ± 0.04 | 2.86 | 1.67 ± 0.10 | 5.96 | 1.66 ± 0.05 | 3.19 | 1.49 ± 0.11 | 7.22 | 1.51 ± 0.04 | 2.82 | 1.37 ± 0.08 | 6.02 |
| Cysteine (sulfonate) | 4.91 ± 0.28 | 5.63 | 3.86 ± 0.19 | 5.00 | 5.01 ± 0.11 | 2.18 | 3.57 ± 0.03 | 0.89 | 3.12 ± 0.15 | 4.72 | 3.27 ± 0.26 | 8.02 |
| Glutathione (oxidized) | 0.07 ± 0.00 | 1.08 | 0.59 ± 0.03 | 5.37 | 0.66 ± 0.01 | 1.83 | 0.98 ± 0.03 | 3.52 | 0.22 ± 0.00 | 0.98 | 0.65 ± 0.02 | 2.51 |
| Proline | 5.65 ± 0.01 | 0.16 | 6.79 ± 0.31 | 4.50 | 5.58 ± 0.30 | 5.40 | 8.75 ± 0.61 | 6.94 | 6.30 ± 0.09 | 1.41 | 7.97 ± 0.14 | 1.70 |
| Histidine (2-oxo-) | 1.93 ± 0.07 | 3.88 | 2.61 ± 0.16 | 5.98 | 2.38 ± 0.07 | 2.96 | 2.54 ± 0.10 | 3.80 | 2.00 ± 0.04 | 2.18 | 1.47 ± 0.07 | 4.43 |
| Carboxylic acids | ||||||||||||
| Succinic acid | 0.46 ± 0.01 | 2.62 | 0.79 ± 0.01 | 1.08 | 0.18 ± 0.00 | 2.82 | 0.14 ± 0.00 | 3.14 | 1.62 ± 0.03 | 1.62 | 0.25 ± 0.00 | 2.00 |
| Lactic acid | 20.02 ± 0.37 | 1.87 | 7.92 ± 0.21 | 2.68 | 5.83 ± 0.06 | 1.08 | 6.47 ± 0.08 | 1.27 | 18.45 ± 0.59 | 3.22 | 8.43 ± 0.28 | 3.36 |
| Pyruvic acid | 1.09 ± 0.02 | 1.43 | 0.16 ± 0.01 | 5.24 | 0.18 ± 0.00 | 1.62 | 0.05 ± 0.00 | 1.40 | 2.13 ± 0.01 | 0.37 | 0.17 ± 0.00 | 0.42 |
| Oxaloacetic acid | 1.84 ± 0.01 | 0.58 | 1.68 ± 0.03 | 1.81 | 0.41 ± 0.00 | 0.00 | 0.07 ± 0.00 | 3.14 | 3.07 ± 0.06 | 2.00 | 0.20 ± 0.00 | 0.00 |
| Isocitric acid | 0.003 ± 0.00 | 0.00 | 0.01 ± 0.00 | 0.00 | 0.001 ± 0.00 | 0.00 | 0.001 ± 0.00 | 0.00 | 0.11 ± 0.00 | 1.25 | 0.001 ± 0.00 | 0.00 |
| Taurine | 0.02 ± 0.00 | 2.89 | 0.03 ± 0.00 | 0.00 | 0.09 ± 0.00 | 4.88 | 0.07 ± 0.00 | 0.00 | 0.07 ± 0.00 | 2.08 | 0.12 ± 0.00 | 3.03 |
| Citric acid | 0.02 ± 0.00 | 0.00 | N/D | N/D | N/D | N/D | 0.001 ± 0.00 | 0.00 | 0.39 ± 0.00 | 0.73 | N/D | N/D |
| α-Ketobutyric acid | 0.45 ± 0.00 | 0.63 | 0.31 ± 0.00 | 0.23 | 0.33 ± 0.01 | 1.93 | 0.36 ± 0.01 | 1.95 | 0.75 ± 0.01 | 1.33 | 0.57 ± 0.00 | 0.37 |
| α-Ketomethylbutyric acid | 0.34 ± 0.02 | 4.56 | 0.21 ± 0.01 | 6.06 | 0.15 ± 0.00 | 2.29 | 0.04 ± 0.00 | 1.84 | 0.71 ± 0.01 | 1.70 | 0.05 ± 0.00 | 4.29 |
| α-Ketomethylselenobutyric acid | N/D | N/D | 0.06 ± 0.00 | 3.45 | 0.18 ± 0.01 | 4.43 | 0.28 ± 0.01 | 3.30 | 0.07 ± 0.00 | 3.87 | 0.19 ± 0.01 | 4.19 |
| α-Ketoglutaramic acid | 0.18 ± 0.00 | 2.77 | 0.03 ± 0.00 | 2.11 | 0.42 ± 0.02 | 4.71 | 2.53 ± 0.15 | 5.92 | 0.01 ± 0.00 | 0.00 | 1.74 ± 0.04 | 2.12 |
| α-Ketoglutaric acid | 0.001 ± 0.00 | 0.00 | 0.00 ± 0.00 | 0.00 | 0.03 ± 0.00 | 4.88 | 0.002 ± 0.00 | 0.00 | 0.001 ± 0.00 | 0.00 | 0.001 ± 0.00 | 0.00 |
| Malic acid | 0.21 ± 0.01 | 3.02 | 0.40 ± 0.00 | 0.88 | 0.02 ± 0.00 | 3.63 | 0.01 ± 0.00 | 0.00 | 2.08 ± 0.08 | 3.94 | 0.01 ± 0.00 | 0.00 |
| Fumaric acid | 1.93 ± 0.02 | 1.10 | 0.58 ± 0.01 | 1.33 | 0.13 ± 0.00 | 0.00 | 0.10 ± 0.00 | 3.63 | 1.50 ± 0.00 | 0.28 | 0.53 ± 0.01 | 1.74 |
| cis-Aconitic acid | 0.004 ± 0.00 | 0.00 | 0.03 ± 0.00 | 2.32 | 0.03 ± 0.00 | 0.00 | 0.08 ± 0.00 | 0.94 | 0.02 ± 0.00 | 0.00 | 0.07 ± 0.00 | 1.99 |
| CoA | 4.11 ± 0.03 | 0.69 | 4.55 ± 0.01 | 0.22 | 4.83 ± 0.02 | 0.48 | 3.54 ± 0.15 | 4.24 | 4.46 ± 0.05 | 1.17 | 4.59 ± 0.02 | 0.35 |
| acetyl-CoA | 0.04 ± 0.00 | 1.75 | 0.78 ± 0.01 | 1.91 | 0.90 ± 0.00 | 0.32 | 1.06 ± 0.01 | 0.80 | 0.83 ± 0.03 | 3.25 | 1.68 ± 0.07 | 4.09 |
| Average CV, % | 0.45 ± 0.00 | 0.63 | 0.31 ± 0.00 | 0.23 | 0.33 ± 0.01 | 1.93 | 0.36 ± 0.01 | 1.95 | 0.75 ± 0.01 | 1.33 | 0.57 ± 0.00 | 0.37 |
| CV, ≥10% | 0 | 0 | 0 | 0 | 0 | 0 | ||||||
| CV, 5–10% | 15 | 29 | 10 | 14 | 26 | 17 | ||||||
| CV, 0–5% | 85 | 71 | 90 | 86 | 74 | 83 | ||||||
| Parameters | Amino Acids | Carboxylic Acids | |
|---|---|---|---|
| Peak asymmetry | 1.096 ± 0.277 | 1.028 ± 0.065 | |
| Peak width at half height, s | 5.601 ± 1.526 | 3.338 ± 0.513 | |
| Retention time deviation, % | L01 * 0.08 µM | 99.28 ± 3.19 | 108.81 ± 2.50 |
| L04 0.63 µM | 103.22 ± 4.07 | 101.42 ± 3.12 | |
| L07 5.00 µM | 97.58 ± 2.65 | 98.16 ± 2.19 | |
| Minimum SNR for L01 | 4437 | 279.84 | |
| Detection limit ** | <4.8 nM | <0.5 μM | |
| Estimated detection limit ** | 0.17 nM | 7 nM | |
| Analysis time, minutes | 13.5 | 12 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaysheva, A.L.; Kopylov, A.T.; Stepanov, A.A.; Malsagova, K.A.; Izotov, A.A.; Shurubor, Y.I.; Krasnikov, B.F. Chromatomass-Spectrometric Method for the Quantitative Determination of Amino- and Carboxylic Acids in Biological Samples. Metabolites 2023, 13, 16. https://doi.org/10.3390/metabo13010016
Kaysheva AL, Kopylov AT, Stepanov AA, Malsagova KA, Izotov AA, Shurubor YI, Krasnikov BF. Chromatomass-Spectrometric Method for the Quantitative Determination of Amino- and Carboxylic Acids in Biological Samples. Metabolites. 2023; 13(1):16. https://doi.org/10.3390/metabo13010016
Chicago/Turabian StyleKaysheva, Anna L., Arthur T. Kopylov, Alexander A. Stepanov, Kristina A. Malsagova, Alexander A. Izotov, Yevgeniya I. Shurubor, and Boris F. Krasnikov. 2023. "Chromatomass-Spectrometric Method for the Quantitative Determination of Amino- and Carboxylic Acids in Biological Samples" Metabolites 13, no. 1: 16. https://doi.org/10.3390/metabo13010016