
Therapeutic and Toxic Effects of Valproic Acid Metabolites

 ,
,  ,
,  ,
,  and
and
Abstract
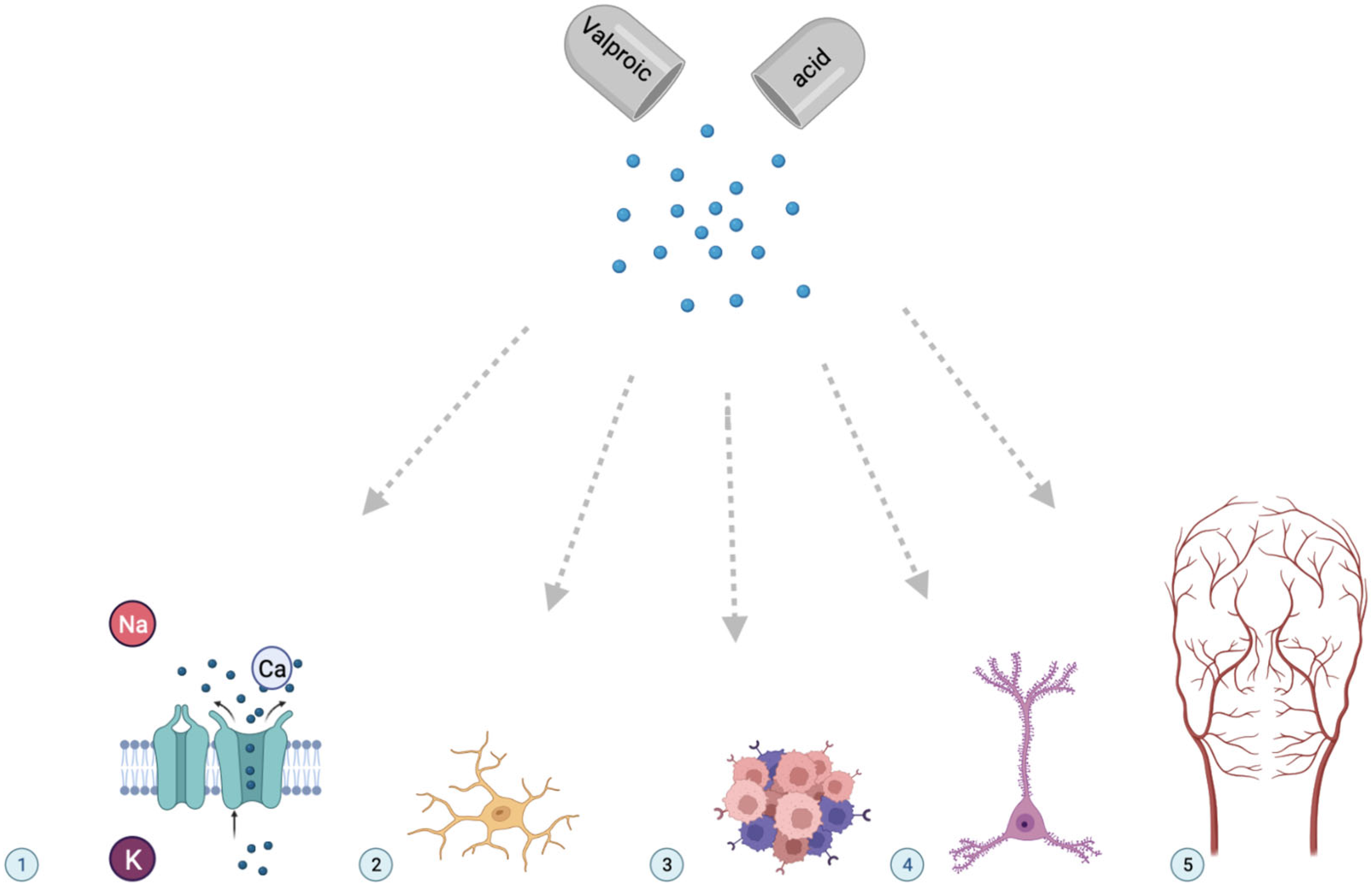
:1. Introduction
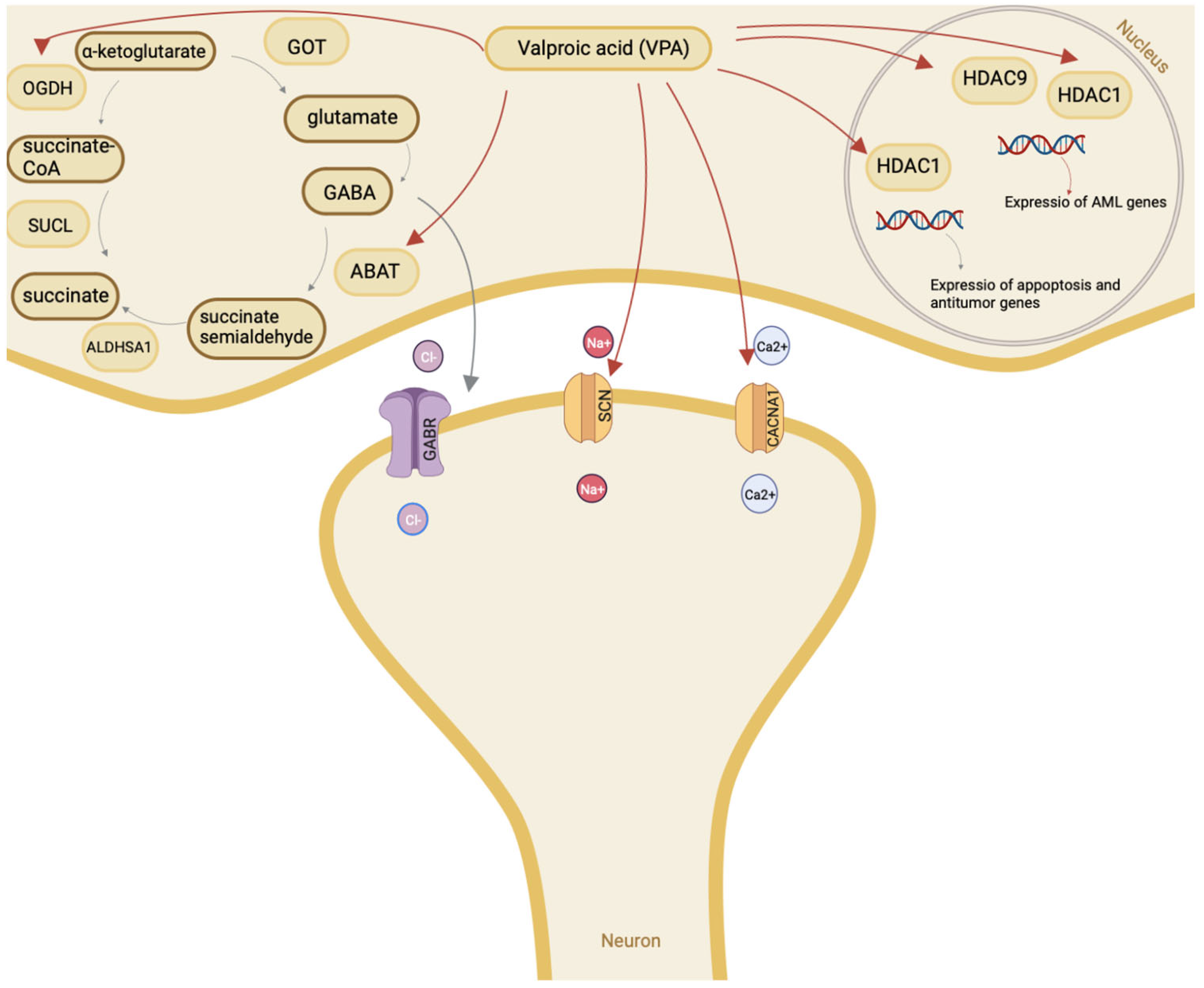
2. Mechanism of Valproic Acid Action
3. Pharmacometabolomics and Pharmacogenomics of Valproic Acid
4. Therapeutic Metabolites of Valproic Acid
4.1. Valproate Acid Glucuronide
4.2. 2-N-Propyl-2-Pentenoic Acid
4.3. Valproyl-CoA
4.4. 4-Ene-Valproic Acid
4.5. 2-Ene-Valproic Acid
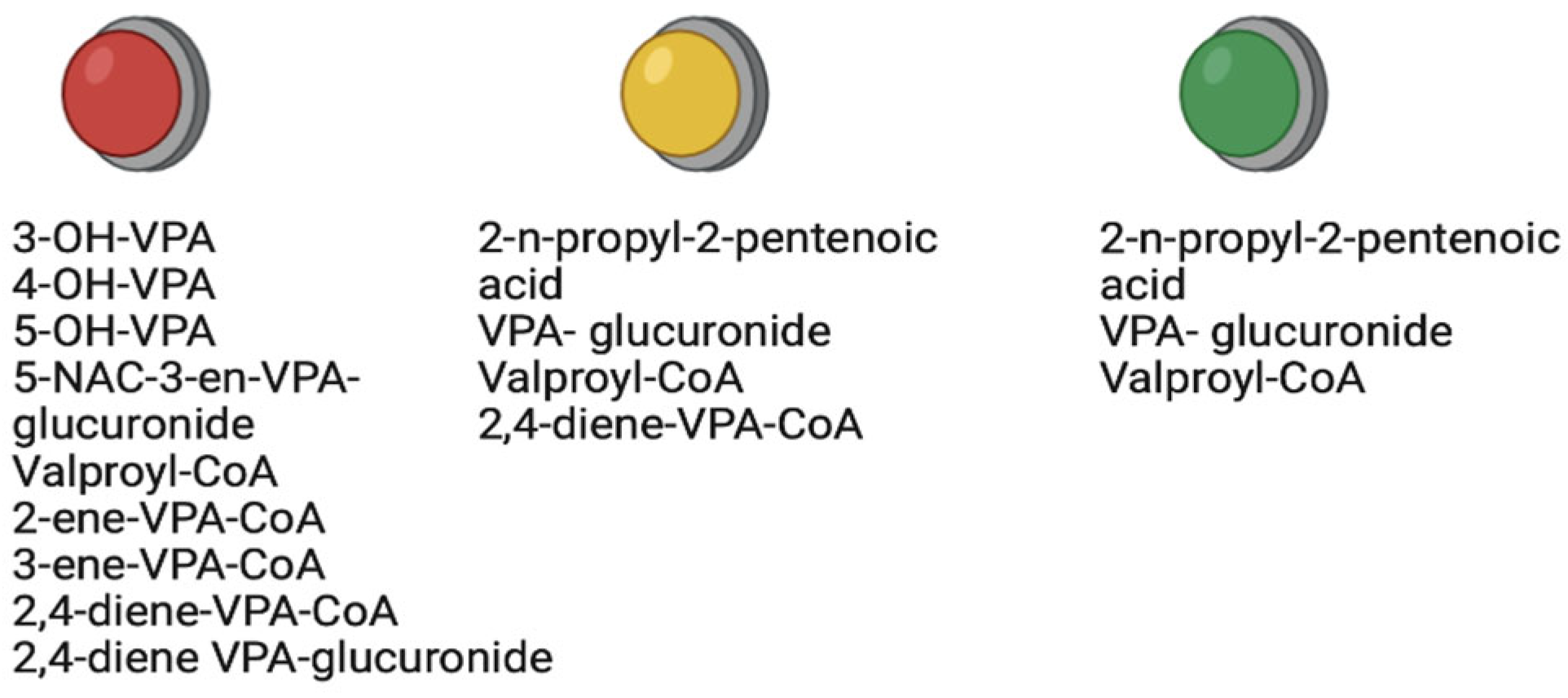
5. Role of Therapeutic Metabolites of Valproic Acid
6. Toxic Metabolites of Valproic Acid
6.1. 3-Hydroxyvalproic Acid
6.2. 4-Hydroxyvalproic Acid
6.3. 5-Hydroxyvalproic Acid
6.4. 3-Oxovalproic Acid
6.5. Valproyl-CoenzymeA
6.6. 2-N-Propyl-4-Oxopentanoic Acid
6.7. 4-Ene-Valproic Acid
6.8. 2-Propyl-2,4-Pentadienoic Acid
6.9. 2-Ene-Valproic Acid
6.10. Valproylcarnitine
7. Role of Toxic Metabolites of Valproic Acid
8. Discussion
9. Limitations
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (OMIM Number) | Chromosomal Location | Protein | SNV (RSID) | References |
|---|---|---|---|---|
| Oxidation | ||||
| CYP2A6 (122720) | 19q13.2 | Isoenzyme 2A6 of cytochrome P450 | *2 (rs1801272) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. |
| *5 (rs5031017) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. The heterozygous GT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| rs (111033610) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| rs28399447 | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *6 (rs4986891) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *7 (rs5031016) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *9A (s28399433) | The homozygous GG genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *11 (rs28399447) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *17 (rs28399454) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *18 (rs1809810) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *19 (rs1809810 ) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *23 (rs56256500) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *24A (rs143731390) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *24A (rs72549435) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *26 (rs59552350) | The homozygous GG genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *26 (rs4986891) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *27 (rs2839944) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *27 (rs28399445) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *35 (rs143731390) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *41 (rs140471703 7) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| CYP2C9 (601130) | 10q23.33 | Isoenzyme 2A6 of cytochrome P450 | *2 (rs1799853) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. |
| *3 (rs1057910) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *4 (rs56165452) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *5 (rs28371686) | The homozygous GG genotype– a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *8 (rs7900194) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *11 (rs28371685) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *13 (rs72558187) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *15 (rs72558190) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| CYP2B6 (123930) | 19q13.2 | Isoenzyme 2A6 of cytochrome P450 | *5A (rs3211371) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. |
| *8 (rs12721655) | The homozygous GG genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *18 (rs28399499) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *27 (rs36079186) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *28 (rs34097093) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| CYP3A4 (124010) | 7q22.1 | Isoenzyme 2A6 of cytochrome P450 | *3 (rs4986910) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. |
| *17 (rs4987161) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *18 (rs28371759) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *20 (rs67666821) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| CYP2C19 (124020) | 10q23.33 | Isoenzyme 2A6 of cytochrome P450 | *2 (rs4244285) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. |
| *4 (rs28399504) | The homozygous GG genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *5 (rs56337013) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *8 (rs41291556) | The homozygous CC genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| CYP2D6 (124030) | 22q13.2 | Isoenzyme 2A6 of cytochrome P450 | *4 (rs3892097) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. |
| *4F (rs3892097) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *4G (rs3892097) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *4H (rs3892097) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *8 (rs5030865) | The homozygous AA genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *10 (rs1065852) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| *17 (rs28371706) | The homozygous TT genotype—a biomarker for a high risk of impaired VPA oxidation and increased levels of toxic metabolites. | |||
| Glucuronization | ||||
| UGT1A1 (191740) | 2q37.1 | UDP-glucuronosyltransferase | rs111033539 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs111033540 | The homozygous GG genotype (Crigler Najjar syndrome, Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs111033541 | The homozygous GG genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT1A3 (606428) | 2q37.1 | UDP-glucuronosyltransferase | rs111033541 | The homozygous GG genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs10929302 | A biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs111033539 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| 191740 | rs111033540 | The homozygous GG genotype (Crigler-Najjar syndrome, Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | ||
| rs11891311 | Allele G—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites (встрeчaeтся чащe у азиатoв). | |||
| rs281865418 | The homozygous GG; AA genotypes (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs28934877 | The homozygous GG; CC genotypes (Crigler-Najjar syndrome Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs3064744 | The homozygous TA; TA genotypes—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs35003977 | The homozygous GG genotype (Hyperbilirubinemia Gilbert’s syndrome; Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs34993780 | The homozygous AA; CC; GG genotypes (Lucey-Driscoll syndrome; Crigler-Najjar syndrome; Hyperbilirubinemia)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs35350960 | The homozygous AA; TT genotypes—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs3755319 | The homozygous GG genotype (Lucey-Driscoll syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs386576623 | The homozygous GG genotype (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs4124874 | The homozygous CC genotype (Gilbert syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs4148323 | The homozygous AA genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776761 | The homozygous AA genotype (Gilbert syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776763 | The homozygous TT genotype (Gilbert syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776764 | The homozygous CC genotype (Gilbert syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776765 | The homozygous TT genotype (Crigler-Najjar syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776766 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587784535 | The homozygous TT genotype (Hyperbilirubinemia) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs62625011 | The homozygous AA genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs6742078 | The homozygous TT genotype (16% bilirubin levels increased risk of gallstones) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551341 | The homozygous AA genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551348 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551349 | The homozygous TT genotype (Crigler Najjar syndrome Gilbert’s syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551350 | The homozygous TT genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551351 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551353 | The homozygous TT genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs755218546 | The homozygous AA genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs886039771 | The homozygous AA genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT1A4 (606429) | 2q37.1 | UDP-glucuronosyltransferase | rs111033539 | The homozygous TT genotype (Crigler-Najjar syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs111033540 | The homozygous AA genotype (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs111033541 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs2011425 | The homozygous GG; AA genotypes—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs281865418 | The homozygous GG; AA genotypes (Crigler-Najjar syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs28934877 | The homozygous GG; CC genotypes (Crigler-Najjar syndrome; Gilbert’s syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs34993780 | The homozygous GG; AA; CC genotypes (Lucey-Driscoll syndrome Crigler-Najjar syndrome Hyperbilirubinemia) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs35003977 | The homozygous GG genotype (Hyperbilirubinemia Gilbert’s syndrome; Crigler-Najjar syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs3755319 | The homozygous GG genotype (Lucey-Driscoll syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs386576623 | The homozygous GG genotype (Crigler-Najjar syndrome Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs4124874 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776761 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776763 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776764 | The homozygous CC genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776765 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776766 | The homozygous GG genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587784535 | The homozygous TT genotype (Hyperbilirubinemia) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs62625011 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs6742078 | The homozygous TT genotype (+16% bilirubin levels increased risk of gallstones) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551341 | The homozygous AA genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551348 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551349 | The homozygous TT genotype (Crigler Najjar syndrome Gilbert’s syndrome) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551350 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551351 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551353 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs755218546 | The homozygous AA genotype —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs797046090 | Genotypes CGTA;CGTA (Hyperbilirubinemia) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs886039771 | The homozygous AA genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT1A6 (606431) | 2q37.1 | UDP-glucuronosyltransferase | rs111033539 | The homozygous TT genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs111033540 | The homozygous GG genotype (Crigler-Najjar syndrome Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs111033541 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs2011425 | The homozygous AA; GG genotypes—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs281865418 | The homozygous AA; GG genotypes (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs28934877 | The homozygous CC; GG genotypes (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs34993780 | The homozygous AA; GG genotypes (Lucey-Driscoll syndrome; Crigler-Najjar syndrome Hyperbilirubinemia)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs35003977 | The homozygous GG genotype (Hyperbilirubinemia Gilbert’s syndrome; Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs3755319 | The homozygous GG genotype (Lucey-Driscoll syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs386576623 | The homozygous GG genotype (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs4124874 | The homozygous GG genotype (Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776761 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776763 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776764 | The homozygous CC genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776765 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776766 | The homozygous GG genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587784535 | The homozygous TT genotype (Hyperbilirubinemia)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs62625011 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs6742078 | The homozygous TT genotype (+16% bilirubin levels increased risk of gallstones) —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551341 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs72551348 | The homozygous GG genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT1A8 (606433) | 2q37.1 | UDP-glucuronosyltransferase | rs111033539 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs111033540 | The homozygous GG genotype (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs111033541 | The homozygous GG genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs2011425 | The homozygous GG; AA genotypes—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs281865418 | The homozygous GG; AA genotypes (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs28934877 | The homozygous GG; CC genotypes (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs34993780 | The homozygous AA; GG; CC genotypes (Lucey-Driscoll syndrome; Crigler-Najjar syndrome Hyperbilirubinemia)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs35003977 | The homozygous GG genotype (Hyperbilirubinemia Gilbert’s syndrome; Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs3755319 | The homozygous GG genotype (Lucey-Driscoll syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs386576623 | The homozygous GG genotype (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs4124874 | The homozygous CC genotype (Gilbert syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs587776761 | The homozygous AA genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT1A9 (606434) | 2q37.1 | UDP-glucuronosyltransferase | rs111033539 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs111033540 | The homozygous GG genotype (Crigler-Najjar syndrome; Gilbert’s syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs111033541 | The homozygous GG genotype (Crigler-Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT1A10 (606435) | 2q37.1 | UDP-glucuronosyltransferase | rs72551350 | The homozygous TT genotype (Crigler Najjar syndrome)—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs17863783 | The homozygous TT genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT2B7 (600068) | 4q13.2 | UDP-glucuronosyltransferase | rs12233719 | The homozygous TT genotype —a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| rs7438135 | The homozygous GG genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs7439366 | The homozygous TT genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| rs7668258 | The homozygous TT genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. | |||
| UGT2B15 (600069) | 4q13.2 | UDP-glucuronosyltransferase | rs1902023 | The homozygous GG genotype—a biomarker for a high risk of impaired VPA glucuronization and increased levels of toxic metabolites. |
| Acetylation | ||||
| ACADSB (600301) | 10q26.13 | Acyl-CoA Dehydrogenase | rs58639322 | The homozygous TT genotype —a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. |
| rs142095937 | The homozygous GG genotype —a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. | |||
| rs137852649 | The homozygous TT genotype—a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. | |||
| rs760791287 | The homozygous TT genotype—a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. | |||
| rs147936696 | The homozygous AA genotype —a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. | |||
| rs387906409 | The homozygous AA genotype—a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. | |||
| ACSM1 (614357) | 16p12.3 | Acyl-CoA Synthetase 1 | rs163234 | The homozygous AA genotype —a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. |
| rs151222 | The homozygous CC genotype —a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. | |||
| rs433598 | The homozygous CC genotype—a biomarker for a high risk of impaired VPA acetylation and increased levels of toxic metabolites. |
References
- Woolley, B.; Mills, J. Versatile Valproic Acid. Issues Ment. Health Nurs. 2022, 43, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic acid and epilepsy: From molecular mechanisms to clinical evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.M.; Naunton, M. Valproate: A simple chemical with so much to offer. J. Clin. Pharm. Ther. 2005, 30, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Macritchie, K.A.; Geddes, J.R.; Scott, J.; Haslam, D.R.; Goodwin, G.M. Valproic acid, valproate and divalproex in the maintenance treatment of bipolar disorder. Cochrane Database Syst. Rev. 2001, 3, CD003196. [Google Scholar] [CrossRef]
- Iannaccone, T.; Sellitto, C.; Manzo, V.; Colucci, F.; Giudice, V.; Stefanelli, B.; Iuliano, A.; Corrivetti, G.; Filippelli, A. Pharmacogenetics of Carbamazepine and Valproate: Focus on Polymorphisms of Drug Metabolizing Enzymes and Transporters. Pharmaceuticals 2021, 14, 204. [Google Scholar] [CrossRef]
- Linde, M.; Mulleners, W.M.; Chronicle, E.P.; McCrory, D.C. Valproate (valproic acid or sodium valproate or a combination of the two) for the prophylaxis of episodic migraine in adults. Cochrane Database Syst. Rev. 2013, 6, CD010611. [Google Scholar] [CrossRef] [Green Version]
- Guo, A.; Li, J.; Luo, L.; Chen, C.; Lu, Q.; Ke, J.; Feng, X. Valproic acid mitigates spinal nerve ligation-induced neuropathic pain in rats by modulating microglial function and inhibiting neuroinflammatory response. Int. Immunopharmacol. 2021, 92, 107332. [Google Scholar] [CrossRef]
- Wang, Y.; Xia, J.; Helfer, B.; Li, C.; Leucht, S. Valproate for schizophrenia. Cochrane Database Syst. Rev. 2016, 11. [Google Scholar] [CrossRef]
- Lambie, D.G.; Johnson, R.H.; Vijayasenan, M.E.; Whiteside, E.A. Sodium valproate in the treatment of the alcohol withdrawal syndrome. Aust. N. Z. J. Psychiatry 1980, 14, 213–215. [Google Scholar] [CrossRef]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef]
- Kristensen, Ø.; Lølandsmo, T.; Isaksen, A.; Vederhus, J.K.; Clausen, T. Treatment of polydrug-using opiate dependents during withdrawal: Towards a standardisation of treatment. BMC Psychiatry 2006, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Kraus, S.W.; Voon, V.; Potenza, M.N. Should compulsive sexual behavior be considered an addiction? Addiction 2016, 111, 2097–2106. [Google Scholar] [CrossRef] [Green Version]
- Chateauvieux, S.; Morceau, F.; Dicato, M.; Diederich, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010, 479364. [Google Scholar] [CrossRef] [Green Version]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Gurpur, P.B.; Liu, J.; Burkin, D.J.; Kaufman, S.J. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am. J. Pathol. 2009, 174, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Duenas-Gonzalez, A.; Candelaria, M.; Perez-Plascencia, C.; Perez-Cardenas, E.; de la Cruz-Hernandez, E.; Herrera, L.A. Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 2008, 34, 206–222. [Google Scholar] [CrossRef]
- Zhu, M.M.; Li, H.L.; Shi, L.H.; Chen, X.P.; Luo, J.; Zhang, Z.L. The pharmacogenomics of valproic acid. J. Hum. Genet. 2017, 62, 1009–1014. [Google Scholar] [CrossRef]
- Perucca, E. Pharmacological and therapeutic properties of valproate: A summary after 35 years of clinical experience. CNS Drugs. 2002, 16, 695–714. [Google Scholar] [CrossRef]
- Silva, M.F.; Aires, C.C.; Luis, P.B.; Ruiter, J.P.; IJlst, L.; Duran, M.; Wanders, R.J.; de Almeida, I.T. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J. Inherit. Metab. Dis. 2008, 31, 205–216. [Google Scholar] [CrossRef]
- Benjamin, R.J.; Piorczynski, T.В.; Hansen, J.M. 285—VPA Inhibits P19 Neural Differentiation through Redox Dysregulation and Oxidative Stress. Free. Radic. Biol. Med. 2017, 112, 190. [Google Scholar] [CrossRef]
- Sepahi, S.; Riahi-Zanjani, B.; Ghorani-Azam, A. Effect of valproic acid on metabolic status and endocrine system in pediatric patients with epilepsy: Systematic literature review. Rev. Clin. Med. 2017, 4, 7–13. [Google Scholar] [CrossRef]
- Chateauvieux, S.; Eifes, S.; Morceau, F.; Grigorakaki, C.; Schnekenburger, M.; Henry, E.; Dicato, M.; Diederich, M. Valproic acid perturbs hematopoietic homeostasis by inhibition of erythroid differentiation and activation of the myelo-monocytic pathway. Biochem. Pharmacol. 2011, 81, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Abbott, F.S. Bioactivation of a toxic metabolite of valproic acid, (E)-2-propyl-2,4-pentadienoic acid, via glucuronidation. LC/MS/MS characterization of the GSH-glucuronide diconjugates. Chem. Res. Toxicol. 1996, 9, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Mawasi, H.; Eisenkraft, A.; Elger, C.E.; Bialer, M.; Kunz, W.S. Mitochondrial Liver Toxicity of Valproic Acid and Its Acid Derivatives Is Related to Inhibition of α-Lipoamide Dehydrogenase. Int. J. Mol. Sci. 2017, 18, 1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, M.J.; Yuan, P.; Chen, G.; Manji, H.K. Neuronal peroxisome proliferator-activated receptor gamma signaling: Regulation by mood-stabilizer valproate. J. Mol. Neurosci. 2008, 35, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Togi, S.; Kamitani, S.; Kawakami, S.; Ikeda, O.; Muromoto, R.; Nanbo, A.; Matsuda, T. HDAC3 influences phosphorylation of STAT3 at serine 727 by interacting with PP2A. Biochem. Biophys. Res. Commun. 2009, 379, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Dodurga, Y.; Gundogdu, G.; Tekin, V.; Koc, T.; Satiroglu-Tufan, N.L.; Bagci, G.; Kucukatay, V. Valproic acid inhibits the proliferation of SHSY5Y neuroblastoma cancer cells by downregulating URG4/URGCP and CCND1 gene expression. Mol. Biol. Rep. 2014, 41, 4595–5599. [Google Scholar] [CrossRef]
- Li, Z.; Yang, L.; Zhang, S.; Song, J.; Sun, H.; Shan, C.; Wang, D.; Liu, S. Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect. Cell Transplant. 2021, 30, 9636897211027524. [Google Scholar] [CrossRef]
- Ebmeyer, J.; Braeuning, A.; Glatt, H.; These, A.; Hessel-Pras, S.; Lampen, A. Human CYP3A4-mediated toxification of the pyrrolizidine alkaloid lasiocarpine. Food Chem. Toxicol. 2019, 130, 79–88. [Google Scholar] [CrossRef]
- Pal, R.; Singh, K.; Khan, S.A.; Chawla, P.; Kumar, B.; Akhtar, M.J. Reactive metabolites of the anticonvulsant drugs and approaches to minimize the adverse drug reaction. Eur. J. Med. Chem. 2021, 226, 113890. [Google Scholar] [CrossRef]
- Surendradoss, J.; Chang, T.K.; Abbott, F.S. Evaluation of in situ generated valproyl 1-O-β-acyl glucuronide in valproic acid toxicity in sandwich-cultured rat hepatocytes. Drug Metab. Dispos. 2014, 42, 1834–1842. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Lv, H.; Guo, Y.; Teka, T.; Wang, X.; Huang, Y.; Han, L.; Pan, G. Structure-Based Reactivity Profiles of Reactive Metabolites with Glutathione. Chem. Res. Toxicol. 2020, 33, 1579–1593. [Google Scholar] [CrossRef]
- Jovanović-Stević, S.; Radisavljević, S.; Scheurer, A.; Ćoćić, D.; Šmit, B.; Petković, M.; Živanović, M.N.; Virijević, K.; Petrović, B. Bis(triazinyl)pyridine complexes of Pt(II) and Pd(II): Studies of the nucleophilic substitution reactions, DNA/HSA interactions, molecular docking and biological activity. J. Biol. Inorg. Chem. 2021, 26, 625–637. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar] [CrossRef]
- Leone, A.; Nie, A.; Parker, J.B.; Sawant, S.; Piechta, L.A.; Kelley, M.F.; Kao, L.M.; Jim Proctor, S.; Verheyen, G.; Johnson, M.D.; et al. Oxidative stress/reactive metabolite gene expression signature in rat liver detects idiosyncratic hepatotoxicants. Toxicol. Appl. Pharmacol. 2014, 275, 189–197. [Google Scholar] [CrossRef]
- Iyanagi, T. Molecular mechanism of phase I and phase II drug-metabolizing enzymes: Implications for detoxification. Int. Rev. Cytol. 2007, 260, 35–112. [Google Scholar] [CrossRef]
- Shnaider, N.A.; Dmitrenko, D.V. Chronic valproic acid intoxication in epileptology: Diagnosis and treatment. Neurol. Neuropsychiatry Psychosom. 2016, 8, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.L.; Jing, X.; Sun, J.Y.; Hu, Y.H.; Xu, Z.J.; Ni, M.M.; Chen, F.; Lu, X.P.; Qiu, J.C.; Wang, T. Valproic Acid and the Liver Injury in Patients with Epilepsy: An Update. Curr. Pharm. Des. 2019, 25, 343–351. [Google Scholar] [CrossRef]
- Aly, M.I.; Abdel-Latif, A.A. Studies on distribution and metabolism of valproate in rat brain, liver, and kidney. Neurochem. Res. 1980, 5, 1231–1242. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, D.; Wang, C.; An, G.; Zhu, L.; Cui, C. Comprehensive Analysis of Metabolic Changes in Male Mice Exposed to Sodium Valproate Based on GC-MS Analysis. Drug Des. Devel. Ther. 2022, 16, 1915–1930. [Google Scholar] [CrossRef]
- Ferguson, C.S.; Tyndale, R.F. Cytochrome P450 enzymes in the brain: Emerging evidence of biological significance. Trends Pharmacol. Sci. 2011, 32, 708–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knights, M.; Finlay, E. The effects of sodium valproate on the renal function of children with epilepsy. Pediatr. Nephrol. 2014, 29, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Darius, J.; Meyer, F.P.; Sabel, B.A.; Schroeder, U. Influence of nanoparticles on the brain-to-serum distribution and the metabolism of valproic acid in mice. J. Pharm. Pharmacol. 2000, 52, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Mancl, E.E.; Gidal, B.E. The effect of carbapenem antibiotics on plasma concentrations of valproic acid. Ann. Pharmacother. 2009, 43, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Graf, R.; Gossrau, R.; Merker, H.J.; Schwabe, R.; Stahlmann, R.; Nau, H. Enzyme cytochemistry combined with electron microscopy, pharmacokinetics, and clinical chemistry for the evaluation of the effects of steady-state valproic acid concentrations on the mouse. Histochemistry 1985, 83, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Melegh, B.; Pap, M.; Morava, E.; Molnar, D.; Dani, M.; Kurucz, J. Carnitine-dependent changes of metabolic fuel consumption during long-term treatment with valproic acid. J. Pediatr. 1994, 125, 317–321. [Google Scholar] [CrossRef]
- Ma, L.; Wang, Y.; Chen, X.; Zhao, L.; Guo, Y. Involvement of CYP2E1-ROS-CD36/DGAT2 axis in the pathogenesis of VPA-induced hepatic steatosis in vivo and in vitro. Toxicology 2020, 445, 152585. [Google Scholar] [CrossRef]
- Zaccara, G.; Lattanzi, S. A review of pharmacokinetic drug interactions between antimicrobial and antiseizure medications in children. Epileptic Disord. 2021, 23, 229–256. [Google Scholar] [CrossRef]
- Vanadia, E.; Gibson, K.M.; Pearl, P.L.; Trapolino, E.; Mangano, S.; Vanadia, F. Therapeutic efficacy of magnesium valproate in succinic semialdehyde dehydrogenase deficiency. JIMD Rep. 2013, 8, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan, A.; Rahman, J.; Banerjee, D.; Mohammed, A.R.H.; Malik, B.H. Parkinsonism: A Rare Adverse Effect of Valproic Acid. Cureus 2020, 12, e8782. [Google Scholar] [CrossRef]
- Lunke, S.; Maxwell, S.; Khurana, I.; KN, H.; Okabe, J.; Al-Hasani, K.; El-Osta, A. Epigenetic evidence of an Ac/Dc axis by VPA and SAHA. Clin. Epigenetics 2021, 13, 58. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Kojima, A.; Asai, Y.; Nadai, M.; Katoh, M. Changes in uridine 5’-diphospho-glucuronosyltransferase 1A6 expression by histone deacetylase inhibitor valproic acid. Biopharm. Drug Dispos. 2022, 43, 175–182. [Google Scholar] [CrossRef]
- Yamada, M.K.; Nakanishi, K.; Ohba, S.; Nakamura, T.; Ikegaya, Y.; Nishiyama, N.; Matsuki, N. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J. Neurosci. 2002, 22, 7580–7585. [Google Scholar] [CrossRef] [Green Version]
- Shaltiel, G.; Mark, S.; Kofman, O.; Belmaker, R.H.; Agam, G. Effect of valproate derivatives on human brain myo-inositol-1-phosphate (MIP) synthase activity and amphetamine-induced rearing. Pharmacol. Rep. 2007, 59, 402–407. [Google Scholar]
- Watterson, J.M.; Watson, D.G.; Meyer, E.M.; Lenox, R.H. A role for protein kinase C and its substrates in the action of valproic acid in the brain: Implications for neural plasticity. Brain Res. 2002, 934, 69–80. [Google Scholar] [CrossRef]
- Parikh, S.K.; Silberstein, S.D. Current Status of Antiepileptic Drugs as Preventive Migraine Therapy. Curr. Treat. Options Neurol. 2019, 21, 16. [Google Scholar] [CrossRef]
- Traetta, M.E.; Codagnone, M.G.; Uccelli, N.A.; Ramos, A.J.; Zárate, S.; Reinés, A. Hippocampal neurons isolated from rats subjected to the valproic acid model mimic in vivo synaptic pattern: Evidence of neuronal priming during early development in autism spectrum disorders. Mol. Autism. 2021, 12, 23. [Google Scholar] [CrossRef]
- Rosenberg, G. The mechanisms of action of valproate in neuropsychiatric disorders: Can we see the forest for the trees? Cell Mol. Life Sci. 2007, 64, 2090–2103. [Google Scholar] [CrossRef]
- Becker, W.J. Acute Migraine Treatment in Adults. Headache 2015, 55, 778–793. [Google Scholar] [CrossRef]
- Sharma, R.P.; Rosen, C.; Kartan, S.; Guidotti, A.; Costa, E.; Grayson, D.R.; Chase, K. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: Preliminary results from a clinical population. Schizophr. Res. 2006, 88, 227–231. [Google Scholar] [CrossRef]
- Bahna, S.G.; Niles, L.P. Epigenetic regulation of melatonin receptors in neuropsychiatric disorders. Br. J. Pharmacol. 2018, 175, 3209–3219. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.Y.; Ahn, M.H.; Yee, J.; Lee, N.; Han, J.M.; Gwak, H.S. Influence of CYP2C9 and CYP2A6 on plasma concentrations of valproic acid: A meta-analysis. Eur. J. Clin. Pharmacol. 2020, 76, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Nasyrova, R.F.; Sivakova, N.А.; Lipatova, L.V.; Ivashchenko, D.V.; Sosina, K.А.; Drokov, А.P.; Shnayder, N.А. Biological Markers Of The Antiepileptic Drugs Efficacy And Safety: Pharmacogenetics And Pharmacokinetics. Sib. Med. Rev. 2017, 1, 18–25. [Google Scholar] [CrossRef]
- Kiang, T.K.; Ho, P.C.; Anari, M.R.; Tong, V.; Abbott, F.S.; Chang, T.K. Contribution of CYP2C9, CYP2A6, and CYP2B6 to valproic acid metabolism in hepatic microsomes from individuals with the CYP2C9*1/*1 genotype. Toxicol. Sci. 2006, 94, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Yu, J.T.; Sun, Y.P.; Ou, J.R.; Song, J.H.; Yu, Y. The influence of cytochrome oxidase CYP2A6, CYP2B6, and CYP2C9 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. Clin. Neurol. Neurosurg. 2010, 112, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, X.; Mao, P.; Song, W.; Liu, L.; Zhang, Y. Impact of CYP2C19 and CYP2C9 gene polymorphisms on sodium valproate plasma concentration in patients with epilepsy. Eur. J. Hosp. Pharm. 2022, 29, 198–201. [Google Scholar] [CrossRef]
- Chen, Y.B.; Liang, C.S.; Wang, L.J.; Hung, K.C.; Carvalho, A.F.; Solmi, M.; Vieta, E.; Tseng, P.T.; Lin, P.Y.; Tu, Y.K.; et al. Comparative effectiveness of valproic acid in different serum concentrations for maintenance treatment of bipolar disorder: A retrospective cohort study using target trial emulation framework. EClinicalMedicine 2022, 54, 101678. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, T.; Li, G.; Qiu, F.; Sun, Y.; Zhao, L. Associations of CYP2C9 and CYP2A6 Polymorphisms with the Concentrations of Valproate and its Hepatotoxin Metabolites and Valproate-Induced Hepatotoxicity. Basic Clin. Pharmacol. Toxicol. 2017, 121, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Shnayder, N.А.; Dmitrenko, D.V.; Bochanova, E.N. Pharmacogenetics. valproic acid. In Clinical Psychopharmacogenetics: Monograph. Ad.; Nasyrova, R.F., Nesnanov, N.G., Eds.; Publisher House DEAN: St. Petersburg, Russia, 2019; pp. 265–278. [Google Scholar]
- Wang, C.; Wang, P.; Yang, L.P.; Pan, J.; Yang, X.; Ma, H.Y. Association of CYP2C9, CYP2A6, ACSM2A, and CPT1A gene polymorphisms with adverse effects of valproic acid in Chinese patients with epilepsy. Epilepsy Res. 2017, 132, 64–69. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, C.; He, X.; Qiu, F.; Zhao, L. Effects of UGT1A6, UGT2B7, and CYP2C9 genotypes on plasma concentrations of valproic acid in Chinese children with epilepsy. Drug Metab. Pharmacokinet. 2012, 27, 536–542. [Google Scholar] [CrossRef]
- Hung, C.C.; Ho, J.L.; Chang, W.L.; Tai, J.J.; Hsieh, T.J.; Hsieh, Y.W.; Hsieh, Y.W.; Liou, H.H. Association of genetic variants in six candidate genes with valproic acid therapy optimization. Pharmacogenomics 2011, 12, 1107–1117. [Google Scholar] [CrossRef]
- Marahatta, A.; Bhandary, B.; Jeong, S.K.; Kim, H.R.; Chae, H.J. Soybean greatly reduces valproic acid plasma concentrations: A food-drug interaction study. Sci. Rep. 2014, 4, 4362. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.M.; Zhang, L.F.; Wang, G.J.; Zhang, S.N.; Zhou, J.H.; Hao, H.P. Influence of UDP-glucuronosyltransferase polymorphisms on valproic acid pharmacokinetics in Chinese epilepsy patients. Eur. J. Clin. Pharmacol. 2012, 68, 1395–1401. [Google Scholar] [CrossRef]
- Stępień, K.M.; Tomaszewski, M.; Tomaszewska, J.; Czuczwar, S.J. The multidrug transporter P-glycoprotein in pharmacoresistance to antiepileptic drugs. Pharmacol. Rep. 2012, 64, 1011–1019. [Google Scholar] [CrossRef]
- Hung, C.C.; Chen, C.C.; Lin, C.J.; Liou, H.H. Functional evaluation of polymorphisms in the human ABCB1 gene and the impact on clinical responses of antiepileptic drugs. Pharmacogenet. Genomics. 2008, 18, 390–402. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. J. Am. Soc. Exp. Neurother. 2005, 2, 86–98. [Google Scholar] [CrossRef]
- Sabin, O.; Pop, R.; Trifa, A.; Buzoianu, A.D. The influence of CYP2C9, CYP2C19 and ABCB1 polymorphisms on the plasma concentrations of valproic acid in epileptic patients. HVM Bioflux. 2016, 8, 29–33. [Google Scholar]
- Siddiqui, A.; Kerb, R.; Weale, M.E.; Brinkmann, U.; Smith, A.; Goldstein, D.B.; Wood, N.W.; Sisodiya, S.M. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N. Engl. J. Med. 2003, 348, 1442–1448. [Google Scholar] [CrossRef] [Green Version]
- Ajmi, M.; Boujaafar, S.; Zouari, N.; Amor, D.; Nasr, A.; Rejeb, N.B.; Amor, S.B.; Omezzine, A.; Benammou, S.; Bouslama, A. Association between ABCB1 polymorphisms and response to first-generation antiepileptic drugs in a Tunisian epileptic population. Int. J. Neurosci. 2018, 128, 705–714. [Google Scholar] [CrossRef]
- Yu, L.; Liao, W.P.; Yi, Y.H.; Qiu, G. ABCB1 G2677T/A polymorphism is associated with the risk of drug-resistant epilepsy in Asians. Epilepsy Res. 2015, 115, 100–108. [Google Scholar] [CrossRef]
- Yagi, M.; Nakamura, T.; Okizuka, Y.; Oyazato, Y.; Kawasaki, Y.; Tsuneishi, S.; Sakaeda, T.; Matsuo, M.; Okumura, K.; Okamura, N. Effect of CPS14217C>A genotype on valproic-acid-induced hyperammonemia. Pediatr. Int. 2010, 52, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tian, Q.; Zhang, M.; Chen, D.; Gao, X.; Yang, H.; Li, H.; Li, C.; Wen, J.; Li, Y.; et al. CPS1 T1405N polymorphism, HDL cholesterol, homocysteine and renal function are risk factors of VPA induced hyperammonemia among epilepsy patients. Epilepsy Res. 2019, 154, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Chinnery, P.F. Mitochondrial DNA polymerase-gamma and human disease. Hum. Mol. Genet. 2006, 15, R244–R252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilkina, O.S.; Shnayder, N.A.; Artyukhovm, I.P.; Moskaleva, P.V.; Panina, Y.S. Problems of differential diagnosis of myoclon us-epilepsy associated with the mutation of the POLG gene and juvenile myoclonic epilepsy: A clinical case. Russ. J. Child Neurol. 2018, 13, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.D.; Horvath, R.; Baruffini, E.; Ferrero, I.; Bulst, S.; Watkins, P.B.; Fontana, R.J.; Day, C.P.; Chinnery, P.F. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology 2010, 52, 1791–1796. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.F.; Meng, L.Y.; Zhang, Y.F.; Yang, Y.C.; Cheng, H.Y.; Jiang, Z.H.; Zhang, H.; Chen, Y.C. POLG Mutations Are Probably Rare in the Han Chinese Population. Chin. Med. Sci. J. 2020, 35, 350–356. [Google Scholar] [CrossRef]
- Saruwatari, J.; Deguchi, M.; Yoshimori, Y.; Noai, M.; Yoshida, S.; Ogusu, N.; Oniki, K.; Yoshida, S.; Yasui-Furukori, N.; Kaneko, S.; et al. Superoxide dismutase 2 Val16Ala polymorphism is a risk factor for the valproic acid-related elevation of serum aminotransferases. Epilepsy Res. 2012, 99, 183–186. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Chen, K.C.; Ding, C.Y.; Tsai, W.J.; Wu, J.F.; Peng, C.C. Valproic acid substantially downregulated genes folr1, IGF2R, RGS2, COL6A3, EDNRB, KLF6, and pax-3, N-acetylcysteine alleviated most of the induced gene alterations in chicken embryo model. Rom. J. Morphol. Embryol. 2013, 54, 993–1004. [Google Scholar]
- Kim, B.; Kim, C.Y.; Lee, M.J.; Joo, Y.H. Preliminary evidence on the association between XBP1-116C/G polymorphism and response to prophylactic treatment with valproate in bipolar disorders. Psychiatry Res. 2009, 168, 209–212. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Zhou, Y.; Ni, G.; Su, Q.; Chen, Z.; Chen, Z.; Li, J.; Chen, X.; Hou, X.; et al. Association of LEPR and ANKK1 Gene Polymorphisms with Weight Gain in Epilepsy Patients Receiving Valproic Acid. Int. J. Neuropsychopharmacol. 2015, 18, pyv021. [Google Scholar] [CrossRef] [Green Version]
- Drokov, A.P.; Lipatova, L.V.; Shnaĭder, N.A.; Nasyrova, R.F. Pharmacogenetic markers of metabolic disorders in the treatment with valproic acid. Zhurnal Nevrol. Psikhiatrii Im. S.S. Korsakova 2018, 118, 82–89. [Google Scholar] [CrossRef]
- Song, A.; Cho, G.-W.; Vijayakumar, K.A.; Moon, C.; Ang, M.J.; Kim, J.; Park, I.; Jang, C.H. Neuroprotective Effect of Valproic Acid on Salicylate-Induced Tinnitus. Int. J. Mol. Sci. 2021, 23, 23. [Google Scholar] [CrossRef]
- Siemes, H.; Nau, H.; Schultze, K.; Wittfoht, W.; Drews, E.; Penzien, J.; Seidel, U. Valproate (VPA) metabolites in various clinical conditions of probable VPA-associated hepatotoxicity. Epilepsia 1993, 34, 332–346. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Beger, R.D.; Schmidt, M.A.; Kaddurah-Daouk, R. Current Concepts in Pharmacometabolomics, Biomarker Discovery, and Precision Medicine. Metabolites 2020, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Suzuki, E.; Yamamura, N.; Ogura, Y.; Nakai, D.; Kubota, K.; Kobayashi, N.; Miura, S.; Okazaki, O. Identification of valproic acid glucuronide hydrolase as a key enzyme for the interaction of valproic acid with carbapenem antibiotics. Drug Metab. Dispos. 2010, 38, 1538–1544. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.F.; Ruiter, J.P.; IJlst, L.; Jakobs, C.; Duran, M.; de Almeida, I.T.; Wanders, R.J. Differential effect of valproate and its Delta2- and Delta4-unsaturated metabolites, on the beta-oxidation rate of long-chain and medium-chain fatty acids. Chem. Biol. Interact. 2001, 137, 203–212. [Google Scholar] [CrossRef]
- Friel, P. Valproyl CoA: An active metabolite of valproate? Med. Hypotheses 1990, 31, 31–32. [Google Scholar] [CrossRef]
- Eyal, S.; Yagen, B.; Shimshoni, J.; Bialer, M. Histone deacetylases inhibition and tumor cells cytotoxicity by CNS-active VPA constitutional isomers and derivatives. Biochem. Pharmacol. 2005, 69, 1501–1508. [Google Scholar] [CrossRef]
- Ehlers, K.; Stürje, H.; Merker, H.J.; Nau, H. The valproic acid metabolite E-2-n-propyl-2-pentenoic acid does not induce spina bifida in the mouse. Dev. Pharmacol. Ther. 1992, 19, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Song, S.; Park, Y.; Kang, S.; Seo, H. HDAC Inhibition by Valproic Acid Induces Neuroprotection and Improvement of PD-like Behaviors in LRRK2 R1441G Transgenic Mice. Exp. Neurobiol. 2019, 28, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zádori, D.; Geisz, A.; Vámos, E.; Vécsei, L.; Klivényi, P. Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol. Biochem. Behav. 2009, 94, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target Against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef]
- Scheuing, L.; Chiu, C.T.; Liao, H.M.; Linares, G.R.; Chuang, D.M. Preclinical and clinical investigations of mood stabilizers for Huntington’s disease: What have we learned? Int. J. Biol. Sci. 2014, 10, 1024–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Castro, R.; Schcolnik-Cabrera, A.; Rodríguez-López, G.; Campillo-Navarro, M.; Puebla-Osorio, N.; Estrada-Parra, S.; Estrada-García, I.; Chacón-Salinas, R.; Chávez-Blanco, A.D. Exploring the Drug Repurposing Versatility of Valproic Acid as a Multifunctional Regulator of Innate and Adaptive Immune Cells. J. Immunol. Res. 2019, 2019, 9678098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, K.P.; Sperling, L.E.; Teixeira, C.; Sommer, L.; Colombo, M.; Koester, L.S.; Pranke, P. VPA/PLGA microfibers produced by coaxial electrospinning for the treatment of central nervous system injury. Braz. J. Med. Biol. Res. 2020, 53, e8993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Ye, J.; Chen, X.; Shi, J.; Wu, W.; Lin, W.; Lin, W.; Li, Y.; Fu, H.; Li, S. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-κB pathway dependent of HDAC3. J. Neuroinflammation. 2018, 15, 150. [Google Scholar] [CrossRef]
- Heers, H.; Stanislaw, J.; Harrelson, J.; Lee, M.W. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur. J. Pharmacol. 2018, 835, 61–74. [Google Scholar] [CrossRef]
- Scholz, B.; Schulte, J.S.; Hamer, S.; Himmler, K.; Pluteanu, F.; Seidl, M.D.; Stein, J.; Wardelmann, E.; Hammer, E.; Völker, U.; et al. HDAC (Histone Deacetylase) Inhibitor Valproic Acid Attenuates Atrial Remodeling and Delays the Onset of Atrial Fibrillation in Mice. Circ. Arrhythm. Electrophysiol. 2019, 12, e007071. [Google Scholar] [CrossRef]
- Abbott, F.S.; Kassam, J.; Orr, J.M.; Farrell, K. The effect of aspirin on valproic acid metabolism. Clin. Pharmacol. Ther. 1986, 40, 94–100. [Google Scholar] [CrossRef]
- Ohnishi, S.; Okamura, N.; Sakamoto, S.; Hasegawa, H.; Norikura, R.; Kanaoka, E.; Takahashi, K.; Horie, K.; Sakamoto, K.; Baba, T. Role of Na+/L-carnitine transporter (OCTN2) in renal handling of pivaloylcarnitine and valproylcarnitine formed during pivalic acid-containing prodrugs and valproic acid treatment. Drug Metab. Pharmacokinet. 2008, 23, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Sztajnkrycer, M.D. Valproic acid toxicity: Overview and management. J. Toxicol. Clin. Toxicol. 2002, 40, 789–801. [Google Scholar] [CrossRef]
- Luís, P.B.; Ruiter, J.; IJlst, L.; de Almeida, I.T.; Duran, M.; Wanders, R.J.; Silva, M.F. Valproyl-CoA inhibits the activity of ATP- and GTP-dependent succinate:CoA ligases. J. Inherit. Metab. Dis. 2014, 37, 353–357. [Google Scholar] [CrossRef]
- PubChem. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004-. PubChem Compound Summary for CID 125849, Valproylcarnitine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Valproylcarnitine (accessed on 21 December 2022).
- Lheureux, P.E.; Hantson, P. Carnitine in the treatment of valproic acid-induced toxicity. Clin. Toxicol. 2009, 47, 101–111. [Google Scholar] [CrossRef]
- Millington, D.S.; Bohan, T.P.; Roe, C.R.; Yergey, A.L.; Liberato, D.J. Valproylcarnitine: A novel drug metabolite identified by fast atom bombardment and thermospray liquid chromatography-mass spectrometry. Clin. Chim. Acta. 1985, 145, 69–76. [Google Scholar] [CrossRef]
- Torres, S.; Baulies, A.; Insausti-Urkia, N.; Alarcón-Vila, C.; Fucho, R.; Solsona-Vilarrasa, E.; Núñez, S.; Robles, D.; Ribas, V.; Wakefield, L.; et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019, 157, 552–568. [Google Scholar] [CrossRef] [Green Version]
- Dmitrenko, D.V.; Shnaider, N.A.; Strotskaya, I.G.; Kichkaylo, A.S.; Zobova, S.N. Mechanisms of valproate-induced teratogenesis. Neurol. Neuropsychiatry Psychosom. 2017, 9, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Doré, M.; San Juan, A.E.; Frenette, A.J.; Williamson, D. Clinical Importance of Monitoring Unbound Valproic Acid Concentration in Patients with Hypoalbuminemia. Pharmacotherapy 2017, 37, 900–907. [Google Scholar] [CrossRef]
- Patel, A.R.; Nagalli, S. Valproate Toxicity; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560898/ (accessed on 27 November 2022).
- Becker, C.M.; Harris, R.A. Influence of valproic acid on hepatic carbohydrate and lipid metabolism. Arch. Biochem. Biophys. 1983, 223, 381–392. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Y.; He, H.; Lou, L.; Ye, W.; Chen, Y.; Wang, J. Synergistically killing activity of aspirin and histone deacetylase inhibitor valproic acid (VPA) on hepatocellular cancer cells. Biochem. Biophys. Res. Commun. 2013, 436, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Vaiman, E.E.; Shnaider, N.A.; Nesnanov, N.G.; Nasyrova, R.F. Drug-induced Parkinsonism. Soc. Clin. Psychiatry 2021, 13, 96–103. [Google Scholar]
- Carriere, C.H.; Kang, N.H.; Niles, L.P. Neuroprotection by valproic acid in an intrastriatal rotenone model of Parkinson’s disease. Neuroscience 2014, 267, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, F.; Tampi, R.R. Valproic acid-induced parkinsonism in the elderly: A comprehensive review of the literature. Am. J. Geriatr. Pharmacother. 2011, 9, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Neavin, D.; Kaddurah-Daouk, R.; Weinshilboum, R. Pharmacometabolomics informs Pharmacogenomics. Metabolomics 2016, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Ellero-Simatos, S.; Lewis, J.P.; Georgiades, A.; Yerges-Armstrong, L.M.; Beitelshees, A.L.; Horenstein, R.B.; Dane, A.; Harms, A.C.; Ramaker, R.; Vreeken, R.J.; et al. Pharmacometabolomics reveals that serotonin is implicated in aspirin response variability. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e125. [Google Scholar] [CrossRef]
- Wang, L.; McLeod, H.L.; Weinshilboum, R.M. Genomics and drug response. N. Engl. J. Med. 2011, 364, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Nasyrova, R.F.; Neznanov, N.G. Clinical Psychopharmacogenetics; Publisher DEAN: St. Petersburg, Russia, 2019; p. 405. [Google Scholar]
- Kaddurah-Daouk, R.; Weinshilboum, R.M. Pharmacometabolomics Research Network. Pharmacometabolomics: Implications for clinical pharmacology and systems pharmacology. Clin. Pharmacol. Ther. 2014, 95, 154–167. [Google Scholar] [CrossRef]
- Okumura, A.; Takagi, M.; Numoto, S.; Iwayama, H.; Azuma, Y.; Kurahashi, H. Effects of l-carnitine supplementation in patients with childhood-onset epilepsy prescribed valproate. Epilepsy Behav. 2021, 122, 108220. [Google Scholar] [CrossRef]
- Poupon, R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: An overview of their mechanisms of action. Clin. Res. Hepatol. Gastroenterol. 2012, 36 (Suppl. 1), S3–S12. [Google Scholar] [CrossRef]
- Goossens, J.F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, H.; Ji, S.C.; Yoon, S.H.; Cho, J.Y.; Chung, J.Y. Pharmacokinetics and Pharmacodynamics of Ursodeoxycholic Acid in an Overweight Population with Abnormal Liver Function. Clin. Pharmacol. Drug Dev. 2021, 10, 68–77. [Google Scholar] [CrossRef]
- Asgarshirazi, M.; Shariat, M.; Dalili, H.; Keihanidoost, Z. Ursodeoxycholic Acid Can Improve Liver Transaminase Quantities in Children with Anticonvulsant Drugs Hepatotoxicity: A Pilot Study. Acta Med. Iran. 2015, 53, 351–355. [Google Scholar]
- Plummer, S.; Beaumont, B.; Elcombe, M.; Wallace, S.; Wright, J.; Mcinnes, E.F.; Currie, R.A.; Cowie, D. Species differences in phenobarbital-mediated UGT gene induction in rat and human liver microtissues. Toxicol. Rep. 2020, 8, 155–161. [Google Scholar] [CrossRef]
- Tutty, M.A.; Movia, D.; Prina-Mello, A. Three-dimensional (3D) liver cell models—A tool for bridging the gap between animal studies and clinical trials when screening liver accumulation and toxicity of nanobiomaterials. Drug Deliv. Transl. Res. 2022, 12, 2048–2074. [Google Scholar] [CrossRef]
- Ravi, N.V.; Maany, I.; Burke, W.M.; Dhopesh, V.; Woody, G.E. Detoxification with phenobarbital of alprazolam-dependent polysubstance abusers. J. Subst. Abuse Treat. 1990, 7, 55–58. [Google Scholar] [CrossRef]
- Bochanova, E.N.; Snaider, N.A.; Dmitrenco, D.V.; Artukhov, I.P.; Gusev, S.D.; Yurjieva, E.A.; Shilkina, O.S. Process of Personalized Prescription of Valproic Acid as the Main Element of the Management of Epilepsy. Int. J. Biomed. 2018, 8, 26–32. [Google Scholar] [CrossRef]
- Ji, Y.; Hebbring, S.; Zhu, H.; Jenkins, G.D.; Biernacka, J.; Snyder, K.; Drews, M.; Fiehn, O.; Zeng, Z.; Schaid, D.; et al. Glycine and a glycine dehydrogenase (GLDC) SNP as citalopram/escitalopram response biomarkers in depression: Pharmacometabolomics-informed pharmacogenomics. Clin. Pharmacol. Ther. 2011, 89, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Sychev, D.А.; Shuev, G.N.; Torbenkov, Е.S.; Adriyanova, М.А. Personalized medicine: The view of a clinical pharmacologist. Cons. Med. 2017, 19, 61–68. [Google Scholar] [CrossRef]








| Metabolite | HMDB Number | Clinical Effect | References |
|---|---|---|---|
| VPA-glucuronide | 0000901 | Therapeutic Neutral | [18,98] |
| 2-n-propyl-2-pentenoic acid | 0013902 | Therapeutic | [99] |
| Valproyl-CoA | 0013115 | Therapeutic | [100] |
| 4-ene-VPA | 0013897 | Therapeutic | [101] |
| 2-ene-VPA | 0013902 | Therapeutic | [101] |
| Metabolite | HMDB Number | Clinical Effect | References |
|---|---|---|---|
| VPA-glucuronide | 0000901 | Therapeutic Neutral | [18,98] |
| 2-n-propyl-2-pentenoic acid | 0013902 | Therapeutic | [99] |
| 4-ene-VPA | 0013897 | Therapeutic | [100] |
| 2-ene-VPA | 0013902 | Therapeutic | [101] |
| Metabolite | HMDB Number | Clinical Effect | References |
|---|---|---|---|
| 3-hydroxy valproic acid | 0013899 | Toxic | [95] |
| 4-hydroxy valproic acid | 0013900 | Toxic | [112] |
| 5-hydroxy valproic acid | 0013898 | Toxic | [95] |
| Valproyl Coenzyme A | 0013115 | Toxic | [95] |
| Valproylcarnitine | 0259757 | Toxic | [113] |
| 2-n-propyl-2-pentenoic acid | 0013902 | Toxic | [114] |
| 2-n-propyl-4-oxopentanoic acid | 0060683 | Toxic | [114] |
| 2-Propyl-2,4-pentadienoic acid | 0060682 | Toxic | [23] |
| 3-oxovalproic acid | 0060685 | Toxic | [95] |
| 4-ene-valproic acid | 0013897 | Toxic | [95] |
| 2-ene-valproic acid | 0013902 | Toxic | [114] |
| 2-ene-valproic acid Coenzyme A | 0060714 | Toxic | [38] |
| 3-ene-valproic acid Coenzyme A | 0060740 | Toxic | [95] |
| 2-diene-valproic acid glucuronide | Toxic | [23] |
| Metabolite | HMDB Number | Clinical Effect | References |
|---|---|---|---|
| 3-hydroxy valproic acid | 0013899 | Toxic | [95] |
| 4-hydroxy valproic acid | 0013900 | Toxic | [112] |
| 5-hydroxy valproic acid | 0013898 | Toxic | [95] |
| 3-oxovalproic acid | 0060685 | Toxic | [95] |
| 2-ene-valproic acid | 0013902 | Toxic | [114] |
| 2-ene-valproic acid Coenzyme A | 0060714 | Toxic | [38] |
| 3-ene-valproic acid Coenzyme A | 0060740 | Toxic | [95] |
| 2-propyl-2,4-pentadienoic acid | 0060682 | Toxic | [23] |
| 2-n-propyl-4-oxopentanoic acid | 0060683 | Toxic | [114] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnayder, N.A.; Grechkina, V.V.; Khasanova, A.K.; Bochanova, E.N.; Dontceva, E.A.; Petrova, M.M.; Asadullin, A.R.; Shipulin, G.A.; Altynbekov, K.S.; Al-Zamil, M.; et al. Therapeutic and Toxic Effects of Valproic Acid Metabolites. Metabolites 2023, 13, 134. https://doi.org/10.3390/metabo13010134
Shnayder NA, Grechkina VV, Khasanova AK, Bochanova EN, Dontceva EA, Petrova MM, Asadullin AR, Shipulin GA, Altynbekov KS, Al-Zamil M, et al. Therapeutic and Toxic Effects of Valproic Acid Metabolites. Metabolites. 2023; 13(1):134. https://doi.org/10.3390/metabo13010134
Chicago/Turabian StyleShnayder, Natalia A., Violetta V. Grechkina, Aiperi K. Khasanova, Elena N. Bochanova, Evgenia A. Dontceva, Marina M. Petrova, Azat R. Asadullin, German A. Shipulin, Kuanysh S. Altynbekov, Mustafa Al-Zamil, and et al. 2023. "Therapeutic and Toxic Effects of Valproic Acid Metabolites" Metabolites 13, no. 1: 134. https://doi.org/10.3390/metabo13010134